


Abstract
We developed a new technique to immobilize a set of molecular beacons on an agarose film-coated slide and found that it has the ability to identify a single nucleotide difference in label-free DNA targets. The annealing properties, specificity and hybridization dynamics of the present technique were compared with those of the conventional technique that directly immobilizes molecular beacons on a planar glass slide. It is demonstrated that the molecular beacon array on an agarose film has high quench efficiency, an excellent discrimination ratio for single nucleotide mismatches and a short detection time. We hypothesize that such a low fluorescence background and high specificity molecular beacon array will find practical applications in label-free, high-throughput mutation analysis and disease diagnosis.
INTRODUCTION
Molecular beacons are oligonucleotide probes that can rapidly report the presence of label-free targets in homogeneous solutions (1). They are hairpin-shaped molecules with an internally quenched fluorophore whose fluorescence is restored when they bind to a target nucleic acid. Because they have excellent specificity to single nucleotide differences and can directly detect unlabeled targets, molecular beacons have been attracting increasing interest in single nucleotide polymorphism (SNP) and mutation detection (2,3), pathogenic detection (4,5) and RNA detection within living cells (6) since Tyagi and Kramer firstly introduced them in 1996 (1). In order to detect multiple targets simultaneously with molecular beacons in one test tube, multicolor molecular beacons and wavelength-shifting molecular beacons have been developed (7,8). But the number of detected targets is strictly limited due to the limitation of suitable fluorescent labels.
Recently, several groups have immobilized molecular beacons on a solid surface to construct a molecular beacon array to resolve the target DNA sequences spatially (9–12). Tan and colleagues first immobilized a set of molecular beacons on a silica surface through biotin–avidin binding to construct a micrometer DNA biosensor (9,10). They achieved a rapid response and stable and reproducible results with such DNA biosensors, which make it possible to detect a large number of targets simultaneously. Steemers et al. (11) immobilized molecular beacons on a randomly ordered optic fiber to construct gene arrays. They greatly decreased the feature size to construct a miniaturized array capable of detecting unlabeled DNA targets at subnanomolar concentrations. Taking advantage of low diffusion limitations and high local concentrations of sensing beads on the distal end of the fiber, they improved the signal-to-background ratio. The above works opened an important area of label-free, large-scale and high-throughout detection of DNA sequence information.
However, there are still several bottlenecks unsolved for the molecular beacon array technology in practical applications. First, a high fluorescent background is generated by incomplete quenching of the molecular beacons, which greatly decreases the signal-to-background ratio. Secondly, the electrostatic properties at the solid–liquid interface and the local ionic strength of the immobilized molecular beacons are greatly different from that in the bulk solution. It is difficult to counteract the interfacial effect even if high ionic strength hybridization buffer is used. Presently, the fluorescence increment ratio of the complementary immobilized molecular beacon probes to that of the non-complementary ones is only about 1–2 after hybridization with targets, while this ratio of the same molecular beacons is tens to hundreds in homogeneous solutions (9,10).
Our group tried to overcome the above-mentioned obstacles by improving the technique of immobilizing molecular beacons. We compared several different immobilization methods and different modified substrates. We synthesized amino-modified molecular beacon probes that can be covalently immobilized on slides via the Schiff base aldehyde–amine chemistry. During our investigation, we realized that it is important to improve the local environment of immobilized molecular beacons at the solid–liquid interface. Three-dimensional functionalized hydrophilic gel films, such as polyacrylamide and agarose, are used more and more as support substrates of microarrays because they combine the advantages of a porous structure and a planar surface (13,14). These films provide high binding capacity and a solution-like environment in which hybridization and other processes resemble a homogeneous liquid phase reaction rather than a heterogeneous liquid–solid interface reaction.
In this paper, we report a technique to immobilize a set of molecular beacons on an agarose film-coated glass slide and compare the results with those immobilized on a glutaraldehyde-derived glass slide. The annealing properties, specificity and hybridization dynamics of molecular beacon arrays immobilized on an agarose film are greatly improved, compared with on a glutaraldehyde-derived glass slide. The molecular beacons array has the ability to identify a single nucleotide difference in the target DNA sequence.
MATERIALS AND METHODS
Molecular beacons
The molecular beacon probes and oligonucleotide target used in this study were synthesized and purified by Shengyou Inc. (Shanghai, China) and are summarized in Table 1. These molecular beacons are specially designed to detect a SNP in codon 158 of the human apoE gene.
Table 1. Molecular beacon probes and target sequence.

Underlining indicates the stem sequence of the molecular beacon. Bold underlining indicates the single nucleotide difference between sequences. MB1 is non-complementary to T2. MB2 is perfectly matched with T2. MB3, MB4 and MB5 are 1 base mismatched with T2.
Instrumentation
The hybridization fluorescence images were collected with the standard FITC filter by laser scanning confocal microscope (Leica TCS SP) employing a 488 nm Ar ion laser. Images were analyzed by ImageJ (NIH, http://rsb.info.nih.gov/nih-image/index.html). A fluidic sample cell made of anodized aluminum was fabricated, on which a molecular beacon array was mounted. The cell can be directly mounted on the microscope stage for real-time fluorescence observation during the hybridization process. Hybridization buffer and target solution were pumped into the fluidic cell with a peristaltic pump.
Preparing activated agarose film-coated glass slides
Preparation of the agarose film was introduced elsewhere (14). A 1% agarose solution was prepared by adding 100 mg agarose to 10 ml deionized distilled water, mixing and boiling for 5 min. Then 2 ml of the agarose solution was poured over each of the aminosilane-derived glass slides (Dako, catalog no. S3003) which were prewarmed at 60°C. After gelation of the agarose, the slides were dried at 37°C in a dryer overnight. The dried slides can be stored at 4°C for future use. Before immobilization of the molecular beacon probes, the agarose films were activated by immersion in 20 mM NaIO4 in 0.1 M phosphate-buffered saline (PBS), pH 7.2, for 30 min at room temperature, then thoroughly rinsed with deionized distilled water and dried.
Preparing glutaraldehyde-derived glass slides
The aminosilane-derived glass slides were cleaned with deionized distilled water and incubated in 5% glutaraldehyde in 0.1 M PBS, pH 7.4, for 2 h. Then the slides were thoroughly washed twice with methanol, acetone and deionized distilled water, and dried.
Manufacture of the molecular beacon array
Spotting solutions were obtained by dissolving molecular beacon probes in sodium carbonate buffer (0.1 M, pH 9.0) at a concentration of 10 µM. A pin-based spotting robot PixSys5500 (Cartesian Tech. Inc.) with a CMP3 pin was used to perform molecular beacon array spotting. About 500 pl spotting solution was spotted on the activated agarose film-coated glass slide or glutaraldehyde-derived glass slide with 120 µm diameter and 200 µm spacing. After spotting, the agarose film-coated glass slides were incubated in a humid chamber at room temperature overnight and washed with 0.1% Tween in deionized distilled water and dried. Glutaraldehyde-derived glass slides were incubated at room temperature for 2 h and at 37°C for 2 h. Then the slides were washed thoroughly in 0.1% Tween. The molecular beacon array can be used for immediate hybridization or stored at 4°C for future use.
Hybridization of the molecular beacon array
Before hybridization, the slides were incubated in different hybridization buffers for 30 min at room temperature to facilitate the annealing of the stem of molecular beacon probes. Oligonucleotide targets in different ionic strength hybridization buffers were used to investigate the hybridization properties of the molecular beacon array.
Hybridization of molecular beacons with target in solution
An aliquot of 50 nM molecular beacon probes and 250 nM targets in different hybridization buffers were used to investigate the hybridization properties in solution. The fluorescence intensity increment was measured with a RF-5000 spectrophotometer (Shimadzu). The excitation wavelength was 488 nm and the emission intensity was recorded at 512 nm.
Hybridization of PCR products to the molecular beacon array
Human genomic DNA was extracted from whole blood cells by the phenol/chloroform method. A 218 bp DNA fragment of the apoE gene was amplified with the primer sequences 5′-TCCAAGGAGCTGCAGGCGGCGCA (forward) and 5′-GCCCCGGCCTGGTACACTGCCA (reverse) as previously described (15). The symmetric PCRs were performed in a total volume of 25 µl containing 75 mM Tris–HCl pH 9.0, 20 mM ammonium sulfate, 0.1 ml/l Tween, 1.5 mM MgCl2, 500 nM each primer, 200 µM dNTPs, 100 ml/l DMSO and 1 U Taq polymerase (Takara Shuzo Co. Ltd). Amplification conditions consisted of an initial 10 min denaturation at 94°C followed by 40 cycles of 30 s denaturation at 94°C, 30 s annealing at 65°C and 30 s extension at 70°C. The mixture for asymmetric PCR contained all the reaction components in identical amounts as in the symmetric PCR except that the forward primer concentration was 50 nM. Fragmented PCR products were prepared as symmetric PCRs described above with the exception of using 160 µM dTTP and 40 µM dUTP instead of 200 µM dTTP and annealing at 55°C for 30 s. The PCR products were fragmented by adding 2 U uracil N-glycosylase (UNG) (Gibco) and incubating at 37°C for 60 min, followed by heating the solution to 95°C for 5 min to inactivate the enzyme. The PCR products were analyzed by 2% agarose gel electrophoresis.
The apoE genotype was determined by DNA sequencing (Shengyou Inc.) and PCR–RFLP. PCR–RFLP was carried out by adding 2 U HaeII (Sibenzyme) to 20 µl symmetric PCR products for 2 h at 65°C and analyzed by 4% agarose gel electrophoresis (Agarose MS; Roche) (15).
Aliquots of 20 µl asymmetric and fragmented PCR products were added to 80 µl hybridization buffers (20 mM Tris–HCl pH 8.0, 10 mM MgCl2) and pumped into the hybridization cell for 30 min.
RESULTS AND DISCUSSION
Design of molecular beacon array
Molecular beacons are oligonucleotide probes with a hairpin-shaped structure in which the 5′ and 3′ ends are self-complementary, bearing a fluorophore on one end and a quencher on the other. In the absence of the target, the self-complementary stem structure holds the fluorophore so close to the quencher that fluorescence does not occur. When binding to the target, the rigidity of the probe–target duplex forces the stem to unwind, causing separation of the fluorophore and the quencher and the restoration of fluorescence (1). In this work, the molecular beacons designed for immobilization contain three parts. First, a single strand hairpin structure. Most studies have indicated that a 15–25 base loop together with a 5–7 bp stem will provide an appropriate balance to form a hairpin structure (1–8). We chose a 16 base loop and 6 bp stem in our design. Five molecular beacon probes were carefully selected (Table 1): four of them with 1 base difference at the central position in the loop sequence (MB2–MB5) to demonstrate the single nucleotide discrimination capability and one with a non-related loop sequence (MB1) used as a negative control. Second, fluorescein in an internal location within the 5′ arm was used as the fluorophore and 4-(4-dimethylaminopherylazo)benzoic acid (DABCYL) at the 3′ end was used as the quencher. Fluorescein and DABCYL are the most widely used fluorophore and quencher pair because DABCYL is non-fluorescent and can quench fluorescein extremely well. Third, a 20 base thymine spacer with an amino group linked to the 5′ end extends past the position of the fluorescein in the 5′ arm of our molecular beacons. The 20 base thymine was used to minimize destabilization caused by 5′ end immobilization. The 5′ end amino-modified molecular beacon probes can be covalently immobilized on the activated agarose film and the glutaraldehyde-derived glass slides via the Schiff base aldehyde–amine chemistry. We have checked the features of these molecular beacons in solution by hybridizing with the complementary target sequence and a >50-fold fluorescence intensity change was obtained, which proved efficient quenching and restoration of the molecular beacon probes.
The scheme of the molecular beacon array before and after hybridization is illustrated in Figure 1. Like conventional molecular beacons in solution, the immobilized molecular beacons MB1 and MB2 assume a hairpin structure before binding to the complementary targets, emitting little fluorescence. When a hybridization solution of the target T2 complementary to MB2 is applied, the corresponding molecular beacon probe MB2 forms a rigid probe–target duplex, the fluorescein and quencher are forced to separate and fluorescence is restored, while MB1 maintains the hairpin structure. The fluorescence signals are spatially resolved to determine the target sequence.
Figure 1.
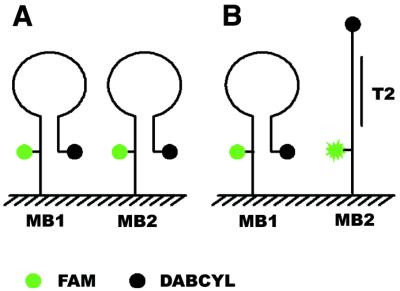
Scheme of immobilized molecular beacon array. MB1 and MB2 are two different immobilized molecular beacons. Target T2 is complementary to MB2 and non-complementary to MB1. (A) Before hybridization. (B) After hybridization with T2. MB2 forms a rigid probe–target duplex with T2 and the fluorescein and the quencher are forced to separate, while MB1 maintains the hairpin structure.
Annealing of immobilized molecular beacon probes
In order to get a high signal-to-background ratio, the molecular beacon should form a stable stem structure to efficiently quench the fluorescence before it is used for hybridization. Because the negatively charged phosphate backbone of the oligonucleotide would hinder the formation of the stem structure of the molecular beacon probes, it is important to select the ionic strength, especially of divalent cations, to ensure high quench efficiency of the molecular beacon (9,10).
We investigated the influence of MgCl2 concentration on the annealing properties of immobilized molecular beacon arrays. When the molecular beacon array was allowed to dry or incubated in 20 mM Tris–HCl pH 8.0, buffer containing no cation, quench efficiency was low and a high fluorescence background image of the molecular beacon array could be registered. With an increase in ionic strength, the fluorescence background decreased. The relationship between the normalized fluorescence intensity and MgCl2 concentration is illustrated in Figure 2. In the case of the glutaraldehyde-derived glass slide, the fluorescence background decreased with an increase in MgCl2 concentration. When the concentration of MgCl2 reached 100 mM, no significant changes could be observed after incubation. The fluorescence intensity of molecular beacon arrays in hybridization buffer containing 500 nM MgCl2 was ∼60% of the initial intensity. In the case of the agarose film-immobilized molecular beacon array, a low concentration of MgCl2 could effectively maintain the stem structure and quench the fluorescence. The fluorescence intensity was only ∼20% of the initial intensity in hybridization buffer containing 10 mM MgCl2. Further increasing the MgCl2 concentration had little effect in improving the quench efficiency. The quench efficiency could be higher than 99% in bulk solutions containing 1–5 mM MgCl2 (1,7). There are two effects that could contribute to the decrease in quenching efficiency of the agarose film-immobilized molecular beacon array. First, there are still some surface effects which destabilize the stem structure of the molecular beacon. Secondly, steric effects caused by high immobilization densities retard the formation of the stem structure and lower the quench efficiency.
Figure 2.

Normalized fluorescence intensity of the immobilized molecular beacons. The fluorescence intensity was the average of all molecular beacon probes on a glutaraldehyde-derived glass slide and an agarose film after annealing in different concentrations of MgCl2 for 30 min.
Specificity in detecting 1 base mismatched label-free targets
In order to investigate the specificity of the immobilized molecular beacons, 10 nM oligonucleotide target T2 in different hybridization buffers (20 mM Tris–HCl pH 8.0, different concentration of MgCl2) was applied to the molecular beacon array. During the hybridization process at room temperature, serial images were collected every minute for 20 min. The background before hybridization and the last hybridization image collected are shown in Figures 3 and 4. In these images, the molecular beacon probes were spotted on the slides in triplet format. From left to right, the probes are MB1 (non-complementary to T2), MB2 (perfectly matched with T2) and MB3, MB4 and MB5 (one central base mismatched with T2), respectively. The background and hybridization fluorescence intensities were extracted with ImageJ software and the fluorescence intensity increase was calculated.
Figure 3.

Images and fluorescence intensities of the molecular beacon array on a glutaraldehyde-derived glass slide. The molecular beacons were spotted on the slides in triplet format. From left to right, the probes are MB1 (non-complementary to T2), MB2 (perfectly matched with T2) and MB3, MB4 and MB5 (one central base mismatch with T2). Hybridization buffer: 10 nM T2 in 20 mM Tris–HCl (pH 8.0) containing (1) 5, (2) 50 and (3) 500 mM MgCl2. Images of (A) and (B) are fluorescence images before and after hybridization, respectively. The plots of (C) and (D) are fluorescence intensity before and after hybridization and fluorescence intensity increment, respectively.
Figure 4.

Images and fluorescence intensities of the molecular beacon array on an agarose film. The molecular beacons were spotted on the slides in triplet format. From left to right, the probes are MB1 (non-complementary to T2), MB2 (perfectly matched with T2) and MB3, MB4 and MB5 (one central base mismatch with T2). Hybridization buffer: 10 nM T2 in 20 mM Tris–HCl (pH 8.0) containing (1) 5, (2) 50 and (3) 500 mM MgCl2. Images of (A) and (B) are fluorescence images before and after hybridization, respectively. The plots of (C) and (D) are fluorescence intensity before and after hybridization and fluorescence intensity increment, respectively.
Figure 3 displays the hybridization results for the glutaraldehyde-derived glass slide-immobilized molecular beacon array in hybridization buffers containing 5, 50 and 500 mM MgCl2, respectively. When the hybridization buffer containing 5 mM MgCl2 was applied, the fluorescence intensity decreased. We attributed it to photobleaching by continuous laser scanning. For hybridization buffer containing 50 and 500 mM MgCl2, the fluorescence intensity increase of perfectly matched probes was more than that of 1 base mismatched probes. The non-complementary molecular beacon probe MB1 used as a negative control also showed a little decrease in fluorescence.
Figure 4 displays the corresponding results for the agarose film-immobilized molecular beacon array. The hybridization results in 5 mM MgCl2 hybridization buffer showed that the fluorescence intensity increment of the perfectly matched probes was >2-fold that of the 1 base mismatched probes. The perfectly matched probes and 1 base mismatched probes could be easily distinguished from the image. With an increment in MgCl2 concentration, the ratio of fluorescence intensity increment of the perfectly matched probes to that of the 1 base mismatched probes also increased, even if the increase rate was low when the MgCl2 concentration was >10 mM.
The mismatch discrimination ratio, which can be used to evaluate the ability to identify a single nucleotide mismatch, is defined as (PM – MM)/PM. PM is the fluorescence increment of perfectly matched probes and MM is the average fluorescence increment of 1 base mismatched probes. The mismatch discrimination ratio versus MgCl2 concentration is plotted in Figure 5. For the glutaraldehyde-derived glass slide-immobilized array, the mismatch discrimination ratio increased from 0.09 (10 mM MgCl2) to 0.3 (500 mM MgCl2). This ratio was 0.67 in 5 mM MgCl2, increasing to 0.81 in 10 mM MgCl2, with little more increment with higher concentrations of MgCl2, for the agarose film-immobilized molecular beacon array. For solution data, the discrimination ratio was 0.98 in 1 mM MgCl2 and increased to 0.99 in higher concentrations of MgCl2. From this plot, we can see that the mismatch discrimination ratio in agarose film is similar to that in homogeneous solution. Considering the mismatch discrimination ratio for immobilized linear probes is ∼0.3–0.7 (13), the results indicate that the molecular beacon in solution and immobilized on agarose film holds high specificity while the glass slide-immobilized one does not.
Figure 5.

Mismatch discrimination ratios of the different molecular beacons in bulk solution, immobilized on a glutaraldehyde-derived glass slide and on an agarose film, respectively. Discrimination ratio is defined as (PM – MM)/PM. PM is the fluorescence increment of perfectly matched probes and MM is the average fluorescence increment of three 1 base mismatched probes. The hybridization solution was 10 nM target T2 in 20 mM Tris–HCl pH 8.0, with different concentrations of MgCl2.
The above results can be explained by the following. First, it is due to incomplete quenching of the molecular beacon. From the results of annealing of the molecular beacon array, we find that a large number of the glass-immobilized molecular beacons are not quenched. These unquenched molecular beacon probes contribute to a high background and greatly reduce the number of probes available for conformation change and fluorescence restoration. High quench efficiency of the agarose film-immobilized molecular beacons contributes to the high mismatch discrimination ratio. Secondly, immobilization of the molecular beacon changes the electrostatic properties and the local ionic strength. The environment of the glass-immobilized molecular beacon probes is quite different from that they experience in bulk solution. Even when high concentrations of hybridization buffer are used, it is difficult to counteract this interfacial effect. Agarose films can provide a favorable solution-like environment and hybridization in low ionic strength hybridization buffer can produce reliable results. High ionic strength hybridization buffer will facilitate the formation of secondary structure of the PCR products and hinder the hybridization with immobilized probes in practical use.
Hybridization dynamics of molecular beacon array
The real-time hybridization process at room temperature was investigated. After the target solution was pumped into the hybridization well, images were collected every minute for 20 min. Time serial images of 10 nM target in 500 mM MgCl2 hybridization buffer are shown in Figure 6. The corresponding fluorescence increment versus time is plotted in Figure 7. These figures indicate that the hybridization process was so fast that 90% of the reaction was completed in 3 min for glass slide-immobilized molecular beacon probes and in 7 min for agarose-immobilized ones. For glass slide-immobilized probes, a smaller fluorescence increment for mismatched probes was observed in the first minute. Though the fluorescence increment for the perfectly matched probe was 1.4 times that of 1 base mismatched ones at steady state, the ratio was 4 times in the first minute. The mismatch discrimination ratio also decreased from 0.75 in the first minute to 0.3 at steady state. This result is similar to that reported by Liu and Tan (9,10). We should get more information from the hybridization dynamic plot for the glass slide-immobilized molecular beacon array. A short reaction time is one of the main advantages of microarray technology over traditional filter hybridization. The hybridization process in agarose film was a little slower than that on the glass slide, which can be explained by mass transport-limited diffusion of the target molecule in the agarose film. However, the agarose film-immobilized molecular beacon array can give a higher mismatch discrimination ratio and more reliable results in a really short time.
Figure 6.

Hybridization dynamics images of molecular beacon arrays immobilized on (A) a glutaraldehyde-derived glass slide and (B) an agarose film. The hybridization solution was 10 nM target T2 in 20 mM Tris–HCl pH 8.0 and 500 mM MgCl2.
Figure 7.

Fluorescence intensity increment versus time of molecular beacon arrays immobilized on (A) a glutaraldehyde-derived glass slide and (B) an agarose film coated on a glass slide. The hybridization solution was 10 nM target T2 in 20 mM Tris–HCl pH 8.0 and 500 mM MgCl2.
Hybridization of PCR products to the molecular beacon array
To investigate the performance of the molecular beacon arrays in complex environments, PCR products of the apoE gene were used as targets. Figure 8A and B shows DNA sequencing profile and PCR–RFLP results for the PCR products. The genotype of apoE codon 158 of the PCR products was determined to be the CGC/TGC heterozygote (15). Electrophoretic images of symmetric and asymmetric PCR products and PCR products before and after UNG digestion are illustrated in Figure 8C. The hybridization images of asymmetric and fragmented PCR products are displayed in Figure 8D and E, respectively. The fluorescence intensity increments are shown in Figure 8F and the discrimination ratios were calculated to be 0.61 and 0.64. Though the discrimination ratio is lower than that of the oligonucleotide targets at the same concentration of MgCl2, it is good enough for practical application.
Figure 8.

(A) Sequencing result of the PCR product with an ABI Prism 377. The arrow indicates the C/T heterozygote of apoE codon 158. (B) Electrophoretic image. Lanes 1 and 2 depict the 218 bp PCR product before and after HaeII digestion, respectively. Mutation from C to T causes loss of the restriction site. Lane 2 depicts a C/T heterozygote (15). (C) Electrophoretic image. Lanes 1 and 2 are symmetric and asymmetric PCR products, respectively. Lanes 3 and 4 are PCR products before and after fragmentation with UNG, respectively. (D) and (E) are the fluorescence images before and after hybridization with asymmetric and fragmented PCR products, respectively. The hybridization solutions containing 20 µl PCR products and 80 µl hybridization buffers (20 mM Tris–HCl pH 8.0 and 10 mM MgCl2). (F) The fluorescence intensity increments after hybridization with asymmetric and fragmented PCR products.
CONCLUSION
In this paper, we report a technique to immobilize molecular beacon arrays on an agarose film and glass slide, and demonstrate the excellent performance of agarose film as an immobilization substrate. Molecular beacon arrays have shown many advantages over conventional microarrays. First, no target labeling is needed. Labeling is an important step in most of the microarray-based target preparation protocols. It is not only time consuming and rather expensive, but can also change the levels of targets originally present in the sample. The use of molecular beacons allows one to eliminate target labeling. Secondly, no washing step is required. Since the unlabeled targets will not contribute to the fluorescence background and specificity of the molecular beacon probes is guaranteed by the thermodynamic properties of the hairpin structure (16), a washing step is not necessary. The high background caused by the washing problem in conventional porous film microarrays is resolved. It will also simplify miniature device, such as lab-on-a-chip, design without having to consider washing problems in small volumes. Moreover, the hybridization process can be easily monitored in real time and more reliable results can be obtained from the hybridization dynamics curves. Thirdly, the high specificity of molecular beacons ensures that a single oligonucleotide mismatch can be easily detected. The presence of a hairpin structure maximizes the specificity of molecular beacon probes. Furthermore, unlike linear probes, molecular beacon probes are insensitive to mismatch type and position, which greatly simplifies probe design.
We investigated the annealing and hybridization processes of molecular beacon arrays on different substrates in different hybridization buffers. For glass slides, interfacial effects destabilize the hairpin structure of molecular beacons and lead to a high background. Agarose film, combining the fast reaction speed of glass slides and a solution-like environment, provides an ideal support for molecular beacon arrays. A low fluorescence background after annealing and a high mismatch discrimination ratio promise great capacities for practical applications of agarose film-immobilized molecular beacon arrays. The instrumentation for fabricating these new microarrays and signal detection is compatible with state-of-the-art microarray techniques. Other high density microarray fabrication techniques, such as light-directed synthesis (17), the liquid dispersing method (18) and molecular stamping (19,20), allow fabrication of high density molecular beacon microarrays economically. It is expected that agarose film-immobilized molecular beacon microarrays can perform high-throughput mutation analysis and disease diagnosis in a parallel, cost-saving and label-free way.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by National Outstanding Youth Foundation grant 61525102, National Key Fundamental Research Project 973-13-01-06 and National Natural Science Foundation grant 60071001.
REFERENCES
- 1.Tyagi S. and Kramer,F.R. (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol., 14, 303–308. [DOI] [PubMed] [Google Scholar]
- 2.Piatek A.S., Tyagi,S., Pol,A.C., Telenti,A., Miller,L.P., Kramer,F.R. and Alland,D. (1998) Molecular beacon sequence analysis for detecting drug resistance in Mycobacterium tuberculosis. Nat. Biotechnol., 16, 359–363. [DOI] [PubMed] [Google Scholar]
- 3.Kostrikis L.G., Tyagi,S., Mhlanga,M.M., Ho,D.D. and Kramer,F.R. (1998) Molecular beacons—spectral genotyping of human alleles. Science, 279, 1228–1229. [DOI] [PubMed] [Google Scholar]
- 4.Chen W., Martinez,G. and Mulchandani,A. (2000) Molecular beacons: a real-time polymerase chain reaction assay for detecting Salmonella. Anal. Biochem., 280, 166–172. [DOI] [PubMed] [Google Scholar]
- 5.Vet J.A.M., Majithia,A.R., Marras,S.A.E., Tyagi,S., Dube,S., Poiesz,B.J. and Kramer,F.R. (1999) Multiplex detection of four pathogenic retroviruses using molecular beacons. Proc. Natl Acad. Sci. USA, 96, 6394–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sokol D.L., Zhang,X.L., Lu,P.Z. and Gewitz,A.M. (1998) Real time detection of DNA RNA hybridization in living cells. Proc. Natl Acad. Sci. USA, 95, 11538–11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tyagi S., Bratu,D.P. and Kramer,F.R. (1998) Multicolor molecular beacons for allele discrimination. Nat. Biotechnol., 16, 49–53. [DOI] [PubMed] [Google Scholar]
- 8.Tyagi S., Marras,S.A.E. and Kramer,F.R. (2000) Wavelength-shifting molecular beacons. Nat. Biotechnol., 18, 1191–1196. [DOI] [PubMed] [Google Scholar]
- 9.Liu X.J. and Tan,W.H. (1999) A fiber-optic evanescent wave DNA biosensor based on novel molecular beacons. Anal. Chem., 71, 5054–5059. [DOI] [PubMed] [Google Scholar]
- 10.Liu X.J., Farmerie,W., Schuster,S. and Tan,W.H. (2000) Molecular beacons for DNA biosensors with micrometer to submicrometer dimensions. Anal. Biochem., 283, 56–63. [DOI] [PubMed] [Google Scholar]
- 11.Steemers F.J., Ferguson,J.A. and Walt,D.R. (2000) Screening unlabeled DNA targets with randomly ordered fiber-optic gene arrays. Nat. Biotechnol., 18, 91–94. [DOI] [PubMed] [Google Scholar]
- 12.Brown L.J., Cummins,J., Hamilton,A. and Brown,T. (2000) Molecular beacons attached to glass beads fluoresce upon hybridisation to target DNA. Chem. Commun., 621–622. [Google Scholar]
- 13.Guschin D., Yershov,G., Zaslavsky,A., Gemmell,A., Shick,V., Proudnikov,D., Arenkov,P. and Mirzabekov,A. (1997) Manual manufacturing of oligonucleotide, DNA and protein microchips. Anal. Biochem., 250, 203–211. [DOI] [PubMed] [Google Scholar]
- 14.Afanassiev V., Hanemann,V. and Wolfl,S. (2000) Preparation of DNA and protein micro arrays on glass slides coated with an agarose film. Nucleic Acids Res., 28, e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zivelin A., Rosenberg,N., Peretz,H., Amit,Y., Kornbrot,N. and Seligsohn,U. (1997) Improved method for genotyping apolipoprotein E polymorphisms by a PCR-based assay simultaneously utilizing two distinct restriction enzymes. Clin. Chem., 43, 1657–1659. [PubMed] [Google Scholar]
- 16.Bonnet G., Tyagi,S., Libchaber,A. and Kramer,F.R. (1999) Thermodynamic basis of the enhanced specificity of structured DNA probes. Proc. Natl Acad. Sci. USA, 96, 6171–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chee M., Yang,R., Hubbell,E., Berno,A., Huang,X.C., Stern,D., Winkler,J., Lockhart,D.J., Morris,M.S. and Fodor,S.P.A. (1996) Accessing genetic information with high-density DNA arrays. Science, 274, 610–614. [DOI] [PubMed] [Google Scholar]
- 18.Hughes T.R., Mao,M., Jones,A.R., Burchard,J., Marton,M.J., Shannon,K.W., Lefkowitz,S.M., Ziman,M., Schelter,J.M., Meyer,M.R. et al. (2001) Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat. Biotechnol., 19, 342–347. [DOI] [PubMed] [Google Scholar]
- 19.Lu Z.H., Zhang,L.G., Chen,Y.L. and Ma,J.M. (1999) A method of preparation of compound micoarray chips by multi-stampings. China patent 98/1/11220.X, International patent PCT/CN99/00013, US patent 09/647,525 (to be issued).
- 20.Xiao P.F., He,N.Y., He,Q.G., Zhang,C.X., Wang,Y.W., Lu,Z.H. and Xu,J.Q. (2001) DNA microarray synthesis by using PDMS molecular stamp (II)—oligonucleotide on-chip synthesis using PDMS stamp. Sci. China Ser. B Chem., 44, 442–448. [Google Scholar]