

Abstract
Fanconi anemia (FA) is a congenital multisystem disorder characterized by early-onset bone marrow failure (BMF) and cancer susceptibility. While ex vivo gene addition and repair therapies are being considered as treatment options, depleted hematopoietic stem cell (HSC) pools, poor HSC mobilization, compromised survival during ex vivo transduction, and increased sensitivity to conventional conditioning strategies limit eligibility for FA patients to receive gene therapies. As an alternative approach, we explored in vivo protein replacement by mRNA delivery via lipid nanoparticles (LNPs). Our study aims to address several key obstacles to current mRNA-LNP treatment: access to the HSC niche, effective expression half-life, and potential mRNA LNP immunogenicity. Results demonstrate efficient in vivo LNP transfection of murine BM via intravenous or intrafemoral injections, yielding reporter expression across hematopoietic and non-hematopoietic BM niche populations. Functionally, LNP delivery of modified Fancc mRNA restored ex vivo expansion. In a proof of principle approach, LNP-treated murine Fancc−/− HSPCs engrafted with restored alkylator resistance up to 120 h post-treatment using circularized mRNA constructs. In vitro delivery of mRNA LNPs resulted in modest differences in innate immune target gene expression in both FA and wild-type HSPCs. Our results suggest that mRNA-LNP-based protein replacement therapy holds promise for clinical translation.
Keywords: MT: Delivery Strategies, lipid nanoparticles, Fanconi anemia, monogenic disorders, protein replacement therapy, in vivo delivery, mRNA therapy, intraosseous delivery, intrafemoral delivery, FANCC knockout mouse model
Graphical abstract
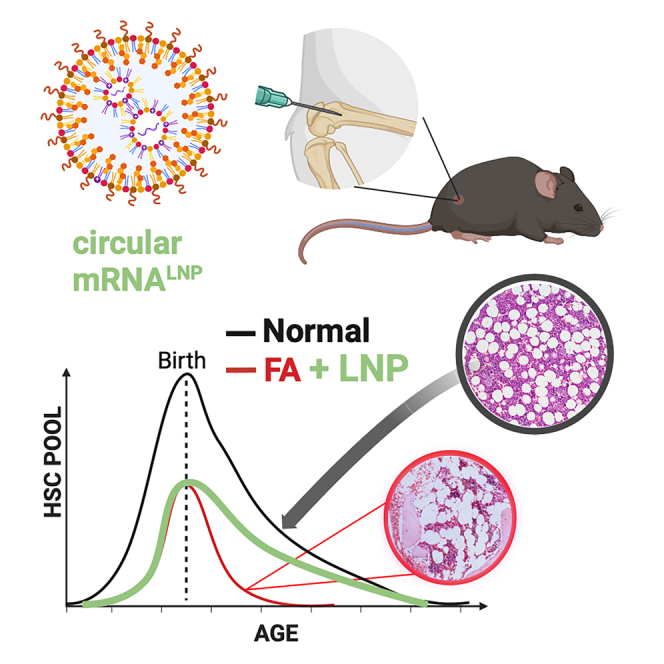
Kurre and colleagues report an approach to preserve regenerative reserve of the hematopoietic stem and progenitor cell pool in Fanconi anemia, a bone marrow failure disorder. The authors develop an in vivo treatment platform using lipid nanoparticle delivery of circular mRNA to ameliorate key phenotypic HSPC features in FA mice.
Introduction
Fanconi anemia (FA) is a congenital hematologic disease characterized by the early onset of bone marrow (BM) failure as well as an elevated predisposition to cancer, especially leukemia.1,2 To date, deleterious mutations in 22 genes have been definitively linked to the disorder.3,4 One common role of FA proteins encoded by these genes is the repair of DNA interstrand crosslinks. Loss of FA gene function also leads to the rapid depletion of hematopoietic stem cells (HSCs) and symptomatic BM failure in most patients.5 The only curative treatment is allogeneic BM transplant, albeit with demonstrated barriers in securing human leukocyte antigen-matched donors and inherent risks to the patient.6 These risks can be further compounded by secondary malignancies associated with pre-transplant conditioning. Thus, gene repair or addition therapies are attractive alternatives to explore.
Existing approaches to gene therapy for FA focus on restoring protein expression in multipotent HSC populations to preempt BM failure. Ex vivo lentiviral transduction of HSCs has demonstrated persistent gene marking and restoration of mitomycin C (MMC) tolerance in hematopoietic progenitor populations in several phase 1/2 pilot studies, but do not show definitive improvement in hematopoietic function.7,8 These studies have been critical, however, in identifying remaining barriers to clinical success. For example, the need to mobilize HSCs to the peripheral blood for apheresis and ex vivo gene transfer leaves adolescent patients with a priori reduced HSC pools ineligible for trial enrollment.8 Conceptually, in situ delivery of nucleic acids might overcome these barriers and avoid the loss of HSC engraftment potential due to ex vivo culture and improve treatment efficacy.9,10,11,12 Direct in vivo delivery also removes the need for the complex infrastructure and logistics associated with ex vivo modification, potentially leading to improved accessibility and cost-effective treatment options.13
Recent clinical trials for the treatment of monogenic metabolic diseases, including methylmalonic acidemia, acute intermittent porphyria, and ornithine transcarbamylase deficiency, have demonstrated the possibility of chronic in vivo replacement by messenger RNA (mRNA) delivered by lipid nanoparticles (LNPmRNA).14 While this approach exploits the tendency of systemically delivered LNPmRNA for sequestration in organs of high blood perfusion such as the liver, efficient transfection and mRNA expression have also been achieved in other tissues.15,16,17,18,19,20 These observations suggest that LNP-mediated delivery of mRNA for monogenic disorders may hold clinical utility more broadly. In the context of FA, mRNA delivery to HSCs may offer rescue of hematopoietic function and could support existing gene addition approaches for long-term correction.
Here, we set out to determine whether mRNA LNP can improve outcomes of HSCs in the context of FA. Our studies demonstrate that LNPs efficiently transfect murine BM HSCs in situ, after either intravenous (IV) or intrafemoral (IF) injection of LNPs. Additionally, LNPFancc delivery restored hallmark functions of FA hematopoietic stem and progenitor cells (HSPCs) in a murine FANCC-deficient model (Fancc−/−) both in vitro and in vivo, supporting further development of this approach for potential clinical applications in FA patients.
Results
LNPFancc restores alkylator resistance in Fancc−/− HSPCs
Whereas the physiologic triggers for loss of FA HSC remain to be defined, cellular sensitivity of FA cells to bifunctional alkylating agents such as MMC has been a mainstay of diagnosis, and its reversal is considered evidence of correction. Before producing Fancc mRNA to formulate LNPFancc, we first validated a candidate FANCC protein sequence by conferring MMC tolerance to Fancc-deficient murine cells using lentiviral vectors to deliver a constitutively expressed Fancc transgene. Fancc−/− Lin− (CD3ε−, Ly-6G/Ly-6C−, CD11b−, B220−, TER-119−), Sca1+, c-kit+ (LSK)-expressing cells in mice include the HSPC populations that are orthologs to HSPC populations exhausted by FA in humans. Like their human counterparts, murine Fancc−/− LSKs display high sensitivity to alkylating agents. LSKs receiving the Fancc transgene via lentiviral transduction displayed similar tolerance to MMC exposure as wild-type (WT) cells in a colony-forming assay, while untreated deficient cells did not proliferate or form colonies (Figure S1). Next, the FANCC mRNA sequence was codon optimized to decrease immunogenicity and improve mRNA yields, quality, and protein expression. The sequence was cloned into a custom in vitro transcription template for either linear or circular mRNA production prior to formulation into LNP (LNPLFancc or LNPCFancc). To verify FA pathway restoration in murine Fancc−/− cells by LNP, we challenged primary BM enriched for Fancc−/− Lin− populations with MMC after in vitro administration of LNPFancc. LNP treatment improved survival after 24 h of 20 nM MMC exposure by more than 2× (37.2% ± 11%) compared to the untreated cells (16% ± 9.1%) (Figures 1A–1C). Next, we isolated and expanded ex vivo cultures of LSKs, enriched for CD150+, CD48− signaling lymphocytic activation molecule (SLAM) immunophenotype, from normal and Fancc−/− mutant mice. These HSC populations typically serve as naive stem cell reserves in the BM and provide long-term (LT) broad repopulation capabilities in the BM. Gradual exhaustion of LT-HSC reserves drives FA disease progression toward BM failure.21 Pre-treatment with LNPLFancc increased survival rates of MMC-treated Fancc−/− HSCs up to 41% ± 18.5% of MMC-free controls, while LNPCFancc increased survival rates up to 48.7% ± 14.8% (Figures 1D–1F). Together, these experiments demonstrate the efficient recovery of MMC tolerance by transient expression of codon-optimized Fancc mRNA.
Figure 1.

In vitro delivery of FANCC LNPs to correct FA-deficient BM-derived HSPCs
(A) Transmitted images of Lin− whole bone marrow (BM) from Fancc−/− mutant mice in methylcellulose media after treatment with LNP and 20 nM mitomycin C (MMC). (B) Colony survival rates expressed as a percentage ratio of the number of colonies formed after exposure to MMC and of colonies formed without MMC treatment across a range of treatments. (C) Percentage ratios of the sum of colony spread area of colonies in (B). (D) Transmitted images of ex vivo expanded LT-HSCs from Fancc−/− mice in methylcellulose media after treatment with LNP and 30 nM MMC. (E and F) Colony survival rates of Fancc−/− or WT ex vivo expanded LT-HSCs after pre-treatment with 30 nM MMC and LNPLFancc or LNPCFancc. Data points represent biological replicates (p < 0.05).
In situ delivery of LNP to the BM broadly transfects BM populations
To develop an in situ strategy for FA protein replacement through mRNA delivery, we first sought to characterize LNP delivery to BM hematopoietic cells, comparing systemic IV injection with direct delivery of LNP to IF injection into the medullary space. For these studies, we delivered 3 μg mRNA-LNP encoding the Cre recombinase (LNPCre) to the tdTomato Ai14 reporter mouse line22 and measured transfection efficiency in SLAM immunophenotyped populations. These populations include partially committed multipotent progenitors (MPPs), such as MPP2 (LSK, CD48+, CD150+), MPP3 and MPP4 (LSK, CD48+, CD150−), and both LT and short-term naive HSCs (LSK, CD48−, CD150+ and LSK, CD48−, and CD150−, respectively) (Figure 2). Cells from injected femurs displayed increased tdTomato positivity compared to IV-injected mice. However, expression in contralateral (i.e., uninjected) femurs and spleens, representing systemic dissemination, did not change between modes of delivery (Figures 2D–2F). Transfection efficiency within IF or IV delivery did not appreciably differ between different progenitor cell populations. We also collected and analyzed residual non-hematopoietic populations (CD45−, Ter119−) (Figures 2G and 2H) from collagenase digested femurs. Expression of tdTomato in cells of non-hematopoietic origin, such as endothelial cells (CD31+) and mesenchymal stem (MSC) and progenitor cell populations (CD31−, CD51(+ or −), Sca-1(+ or −)), closely resembled that of hematopoietic populations, suggesting broad transfection across relevant BM niche cell populations.
Figure 2.

IF delivery of Cre-recombinase LNP to stably induce reporter expression
(A) Graphical representation highlighting the IF injection procedure for delivering LNP directly into the BM. (B and C) Widefield transmitted (B) and fluorescence (C) images of whole BM taken from Ai14 mice after IF LNPCre delivery. (D–H) FACS analysis of cells from IF- or IV-treated spleens (D) or femurs (E–H) comparing positivity rates between mice treated either IF or IV with LNPCre (p < 0.05).
Next, we tested a transient reporter system using LNPmCherry to resolve differences in relative LNP uptake per cell based on reporter fluorescence intensity. We compared reporter mean fluorescence intensity (MFI) and positivity rates of both IV- and IF-delivered mCherry mRNA in injected femurs, contralateral femurs (or corresponding femurs in IV administration), and the spleen (Figures 3A–3F). Fluorescence positivity thresholds for mCherry expression were set at the intensity value representing ≤0.5% of the positivity rate of untreated samples. In mice receiving IF injections, ipsilateral mCherry expression was significantly increased compared to the contralateral femurs in HSPCs, as well as terminally differentiated populations. Among specific HSPC populations, the proportion of mCherry-expressing cells did not vary between assayed whole BM or SLAM-expressing HSPC populations, suggesting similar transfection efficiencies across all measured cell types in the marrow compartment. In addition, HSPCs from the ipsilateral IF injection demonstrated significantly increased reporter expression (by both positivity rate and MFI) over HSPCs from the corresponding femurs in IV-treated mice (Figures 3A–3D). Contralateral femurs and spleens did not differ in expression between IF- and IV-treated mice (Figures 3B, 3C, 3E, and 3F). To determine whether gains in mCherry fluorescence resulted from increased uptake of LNPmRNA, we performed quantitative real-time PCR analysis to quantify those differences. Consistent with mCherry expression by fluorescence-activated cell sorting (FACS) analysis, relative mRNA transcript levels were also increased in injected femurs (Figures 3G–3I). Unlike the increase in injected femurs, mRNA levels of mCherry transcript in the spleen and contralateral femurs did not differ between IF- and IV- treated mice. We found that both reporter fluorescence and mRNA concentrations of BM populations from injected femurs reached levels found in the spleen, which is known to sequester LNPs in circulation.23,24,25
Figure 3.

IF vs. IV delivery of fluorescent reporter LNP
Comparisons of mCherry positivity rates (A–C) or mean fluorescence intensity (MFI; D–F) 24 h after either IF or IV delivery of mCherry LNP. (G) quantitative real-time PCR analysis of mCherry RNA isolated from whole BM 24 h post-injection. (H and I) Time series plots of relative mRNA concentrations in the femurs and spleen of IV-treated mice (H) or IF-treated mice (I). All mRNA concentrations are represented as the binary log of the relative fold change from femurs in IV-treated mice (p < 0.05).
Next, we administered 10 μg firefly luciferase (FLuc) linear mRNA in LNPs (LNPLFluc) to further resolve organ-level tropism differences between IF and IV modes of delivery by luciferase activity. After 24 h, luciferin was administered intraperitoneally, animals were sacrificed, organs of interest were explanted, and the radiance of individual extracted organs was measured. Consistent with fluorescence data, we observed elevated radiance specifically within injected femurs as compared to the contralateral counterparts. IV injection, by contrast, provided balanced luciferase signal between the two femurs (Figures 4A, 4B, and 4E–4H). A modest gain in fluorescence signals was seen in liver and spleen after IV injection, but differences in radiance between IF and IV delivery in any other organs did not reach significance (Figures 4C, 4D, 4I, and 4J). No differences between modes of injection were seen in biodistribution to heart, lungs, and kidneys (Figure S2).
Figure 4.

In vivo LNPLFluc expression after IF or IV delivery
(A–D) Graphs of the total luminescent flux measured after d-luciferin administration in LNPLFluc-treated mice. (E–J) Heatmaps of radiance from organs, corresponding to those in (A)–(D), reveal the distribution of radiance displayed as an overlay on reflected light images of the respective organ.
Expression lifetime of mRNA in vitro and in vivo is increased by circularization
One natural limitation of mRNA-based protein replacement therapeutics is the limited half-life of mRNA both in vitro and in vivo. To evaluate the expression lifetime of our mRNA constructs, we performed IF injections of LNPmCherry and measured reporter expression over time in BM populations. Mice were sacrificed at serial time points between 4 and 72 h post-injection, and harvested cell fractions were immunophenotyped (LSK, SLAM) and analyzed by flow cytometry (Figures 5A–5D). Expression of mCherry peaked between 6 and 24 h post-injection in all measured tissues (ipsilateral femur, contralateral femur, and spleen) and was detectable at least 72 h post-injection. In the context of FA, improving the lifetime of mRNA expression in vivo could provide a therapeutic bridge during ex vivo HSC manipulation. To that end, circular mRNA has previously been reported to maintain a longer in situ half-life due in part to the absence of a 5′ cap structure and 3′ poly-adenylated tails, bypassing enzymatic cleavage by endogenous exonucleases.26,27 To determine whether expression lifetime could be increased within hematopoietic cells using circularized constructs, we next formulated LNPs to deliver FLuc mRNA to ex vivo primary cultures as either a linear or circular mRNA construct (LNPLFLuc or LNPCFLuc, respectively). Compared with the mCherry reporter, luciferase enables improved sensitivity for measuring the impact of LNP dose and mRNA expression duration based on the total luminescence of luciferase activity. Consistent with previous reports demonstrating the increased expression lifetime of circular mRNA,28 we achieved longer luciferase expression lifetimes with LNPCFLuc in both ex vivo expanded LT-HSCs and primary mouse embryonic fibroblasts (MEFs) compared to LNPLFLuc (Figures 5E and 5F). When treated with LNPs for 24 h in vitro, the peak expression of cells treated with LNPCFLuc occurs at approximately 48- h post-exposure, while LNPLFLuc-treated cells peak shortly after LNP are cleared via media change (t = ∼24 h). In addition, we found that peak expression time and magnitude in these populations can be increased by extending cell-LNP exposure time (Figure S3), suggesting that the rate of particle uptake may be a rate-limiting factor of expression. To confirm the benefits of circularized mRNA Fancc constructs, we performed functional lifetime assessments of LNPLFancc and LNPCFancc. We measured survival rates of LNP-treated Fancc mutant ex vivo expanded LT-HSCs as a function of MMC exposure time (Figures 5G and 5H). Fancc-deficient ex vivo expanded HSCs exposed to 30 nM MMC for at least 24 h do not proliferate and will undergo apoptosis by day 6. In contrast, cells pre-treated with LNPLFancc or LNPCFancc for 24 h prior to MMC exposure both display varying levels of improved tolerance relative to WT cells for up to 48 h of total MMC exposure. LNPCFancc additionally displays significantly increased relative survival for up to 96 h of MMC exposure, suggesting a conferred MMC tolerance of up to 5 days post-LNP treatment (Figure 5H).
Figure 5.

Functional lifetime of LNP mRNAs
(A and B) Time series plots of positivity rates (A) or MFI (B) after LNPmCherry IF delivery measured up to 72 h post-injection. (C and D) Time series plots of luminescence as a function of time post-exposure to either linear or circular luciferase mRNA delivered with LNP in ex vivo expanded HSCs (C) or MEFs (D). (E) Graphical representation of the methodology employed to determine the functional lifetime of LNPLFancc and LNPCFancc. Briefly, ex vivo expanded cells were treated with LNP for 24 h, prior to a media change and addition of 30 nM MMC. (F) Relative survival, expressed as a ratio between FA and WT cells of the relative survival with and without MMC exposure, is plotted as a function of total exposure time to MMC over a 6-day period post-LNP treatment (p < 0.05). (G) Experimental schema for scoring MMC tolerance and survival of LT-HSC following a single LNP treatment. (H) Survival advantage of ex vivo expanded LT-HSC treated with LNP carrying circular Fancc mRNA.
LNPs are not potent immune activators of HSPCs
FA hematopoietic cells are particularly vulnerable to quiescence loss, DNA damage accumulation, and inflammation exhaustion.29,30,31 To investigate the impact of LNPs, which can be broadly inflammatory,32,33 on Fancc−/− HSCs, we initially screened WT and Fancc−/− ex vivo expanded LT-HSCs by array-based quantitative real-time PCR. Expression of innate and adaptive immune response genes was assayed after 6 or 24 h of exposure to 8.3 μg LNPLFancc or LNPCFancc per million cells and compared with respective untreated controls (Figure S4). In the context of LNP treatment, selective regulation of immunomodulatory genes was observed. Of the 84 genes screened, 15 genes in LNPLFancc-treated and 23 genes in LNPCFancc-treated cells were differentially expressed by a factor of at least 1.5 in both WT and mutant cells. To determine whether the ex vivo expanded populations were representative of inflammatory signaling in vivo, we next screened freshly isolated KSL populations from either WT or knockout (KO) mice treated with LNPCFancc in vitro for 24 h (Figure 6A). In the pooled KSL populations, baseline differences in expression between WT and KO were evidenced by a mild increase in inflammatory signaling in the mutant Fancc population. For validation, replicate tests in these populations were performed by assaying the most prominently differentially expressed genes by quantitative real-time PCR. Overall, while the trend of increased inflammation in KO populations held, we did not find consistent significant differences in expression between genotypes or between LNP treatment groups (Figures 6B and S5).
Figure 6.

Immune screening of HSPCs after LNP treatment
(A) KSL populations were pooled from three mice and screened for 84 innate and adaptive immune markers by quantitative real-time PCR array 24 h after treatment with LNPCFancc. Values are expressed as the binary logarithm of the fold change difference to the expression of the untreated WT population. (B) Nine targets representing either elevated expression in the screening array or commonly assayed targets in the context of LNP immunogenicity were analyzed in replicate (p < 0.05).
Repeat dosing of LNPCFancc increases HSPC clonogenicity in vitro
While MMC resistance faithfully represents the activity of FA proteins in DNA repair, we were looking for an improved metric of HSPC function unprovoked by alkylating agents or other experimental challenges. We showed that the duration of expression after a single dose of LNPCFancc is insufficient to show differences in progenitor clonogenicity between mutant and WT populations. Therefore, to determine whether the Fancc mutant deficit in clonogenicity could be improved by LNPCFancc, we explored the effects of repeat dosing of LNPCFancc in vitro. LT-HSCs isolated from Fancc mutant and WTmice were expanded over 14 days, and LNPCFancc was administered on days 0, 7, and 14 post-isolation. The total population doublings as a function of time were obtained from raw cell counts (Figure 7A). We found that multiple doses of LNPCFancc significantly improved proliferation and, additionally, brought the number of doublings in mutant HSCs up to that of the WT controls over a 14-day period. This contrasts with mutant Fancc LT-HSCs treated with a single dose of LNPs, which did not show significantly improved doubling over untreated controls (Figure S6). Next, expanded cells were treated with varying concentrations of MMC at day 15 of ex vivo culture for 24 h prior to plating into a colony-forming unit (CFU) assay at 500 cells/well (Figures 7B and 7C). When colonies were counted at day 7 post-plating, ex vivo LNPCFancc primed KO populations revealed an increase in colony counts and relative survival compared to untreated controls at both 10 and 100 nM MMC treatments. Additionally, expanded LT-HSCs were immunophenotyped to assess the relative proportions of HSPC populations (Figure 7D). Fancc mutant mice treated with LNPCFancc exhibited an increased proportion of stem cells retaining LT-HSC immunophenotyping relative untreated cells, and no difference was observed compared to WT mice.
Figure 7.

Impact of LNPCFancc on Fancc−/− HSPC in vitro proliferation and engraftment potential
(A) LT-HSCs isolated from WT or Fancc−/− mice were treated on days 0, 7, and 14 of ex vivo culture with LNPCFancc. Shown are population doublings calculated from cell counts at days 0, 7, 10, and 14. (B) After 15 days of ex vivo expansion, cells were incubated with MMC for 24 h prior to plating in CFU assay. Colonies were counted at 7 days post-plating. (C) Colony survival was determined as the number of colonies at a given condition relative to the respective cell population in MMC-free methylcellulose media. (D) LT-HSC populations were immunophenotyped and analyzed by FACS to determine relative proportions of HSPC subpopulations after 15 days of ex vivo expansion. (E) Ex vivo expanded LT-HSCs receiving a single dose of LNPCFancc at day 14 for 24 h were transplanted into lethally irradiated recipient mice either with (right) or without (left) 6 h of in vitro MMC exposure prior to transplantation.
Ex vivo treatment of Fancc−/− LT-HSC improves engraftment potential
Fancc−/− HSCs display reduced proliferation rates and decreased engraftment potential relative to their WT counterparts. To determine the impact of temporary restoration of the FA pathway on HSC behavior, we performed comparative studies of ex vivo expanded HSCs after treatment with Fancc mRNA LNP. When transplanted into lethally irradiated recipients in a competitive transplant setting, ex vivo expanded Fancc−/− LT-HSCs display decreased engraftment potential relative to WT controls based on day 14 donor chimerism post-transplant. Treatment with LNPCFancc prior to transplantation did not alter performance at this time point (Figure 7E), except when cells were first challenged by exposure to MMC for 6 h prior to transplantation. After MMC challenge, untreated Fancc−/− LT-HSCs displayed greatly diminished engraftment potential. Remarkably, this loss is completely protected against by pre-treatment selection using LNPCFancc. These data identify LNP treatment as a potential adjuvant therapeutic during ex vivo manipulation of Fancc−/− HSPCs.
Discussion
Ex vivo modification of HSPC in FA patients has provided strong proof of principle for the potential of protein replacement by lentiviral gene addition. However, definitive hematologic improvement remains to be shown, potentially owing to oligoclonal reconstitution with corrected stem cells.34 Studies in metabolic disorders suggest that in vivo delivery of modified mRNA using a non-viral approach may be an alternative treatment approach that relies on repeat dosing. Our studies lay out basic observations on key variables of LNP delivery, durability, and immunogenicity toward that goal and demonstrate promise for LNP treatment in FA.
To this end, we delivered LNPs IF and IV to determine the efficiency of delivery to HSPCs in the BM. Consistent with other reports employing systemic LNP delivery,15,20 we observe dose-dependent transfection efficiency, with high rates of transfection in a Cre-sensitive tdTomato reporter mouse model. Side-by-side comparisons of systemic and direct delivery modes highlight the decreased access to BM after systemic delivery, which is increased to levels found in the spleen by directly injecting LNP into the medullary space. These data suggest that low transfection efficiency by systemic delivery in the BM is primarily the product of decreased physical access to BM cells through normal blood circulation. This decrease is likely due to systemic dissemination of LNPs to organs of high blood perfusion such as the spleen, liver, and lungs, decreasing the effective concentration of LNPs reaching the BM and resulting in diminished transfection. Therefore, direct delivery to the BM might increase transfection efficiency through bypassing LNP loss in circulation prior to arrival in the BM. Interestingly, our data demonstrate that LNPs delivered IF achieve similar levels of expression in typically high-expressing tissues in systemic delivery models, suggesting that elevated initial concentrations are enough to significantly improve local medullary transfection efficiency, despite subsequent systemic dissemination. While direct access to the BM compartment is less common in the clinic than other routes for administration of drug product, the means do exist, offering the potential for reduced hepatotoxicity. In fact, several US Food and Drug Administration-approved devices to simplify access to the BM already exist (e.g., EZ-IO system) and are typically employed to allow vascular access for infusions in the absence of a viable IV route. Recently, mRNA BM transfection efficiency was shown to be increased by targeting LNPs through surface modifications to specifically bind the c-kit receptor (CD117) on HSPCs.15 A combination of methods may be required to improve access to the HSC niche for maximum impact and to minimize any potentially negative off-target effects.
While HSC defects are a near-uniform feature in FA, dysfunction within BM mesenchymal stroma has also been reported.35,36,37 FA BM MSCs demonstrate compromised osteoblast differentiation and decreased bone formation.35 Additionally, murine BM MSCs display impaired hematopoietic support, as evidenced by decreased homing and engraftment by transplanted HSCs.37 While it is currently unknown what consequences FA gene correction may have in cells other than BM HSCs, it is possible that mRNA therapies could improve other functional aspects of the BM niche. Using the Cre-reporter system, our data show consistent uptake and expression of mRNA cargo in both hematopoietic and non-hematopoietic immunophenotyped cell populations, including stromal populations such as osteogenic MSCs (CD45−,CD31−,CD51+,Sca1+). This consistent uptake across BM populations suggests that transfection efficiency in our models relies primarily on local LNP concentrations rather than cell-specific interactions or tissue tropism. While the effects of FA complementation in non-hematopoietic cells is unknown, future work will elucidate whether compartment-wide transfection by Fancc LNPs in the BM could offer additional benefits.
While viral gene addition provides permanent HSPC correction, mRNA delivery will have transient effects that may require redosing for any durable clinical benefit. In addition, unlike common viral approaches, dosing LNP-mediated mRNA delivery is not known to be limited by adaptive immunoreactivity.38 Nevertheless, reduced dosing frequency will reduce logistical burdens of treatment. To that end, one existing approach to extend mRNA expression lifetime is by circularization of mRNA constructs. Our results using circularized variants of our FLuc and Fancc LNP demonstrated 2–3 days of additional expression by functional assays. Additionally, work in FA and other congenital disorders has demonstrated that select patient mutations may be amenable to LNP-mediated CRISPR-based editing approaches for permanent repair.15,39,40 Another potential therapeutic modality that we did not explore is the use of self-replicating RNA (srRNA) vectors.41 These vectors enable longer-term expression of protein products up to 7 weeks.42 Unfortunately, currently available srRNA backbones are virally derived, and their RNA products tend to elicit immune responses.43 For this reason, we have not yet explored their use. While the reporter systems employed here are less logistically complex than existing editing approaches, the potential efficiency of such an approach is illustrated by our Cre-based LNP reporter system. We find that an LNPCre dose of roughly 25% of the magnitude of the LNPmCherry mRNA mass employed in the mCherry experiments (3 vs. 12.5 μg) yields recombination in over 40% of cells, while an equivalent dose of LNPCre (12.5 μg) yields greater than 90% recombination in LSK BM populations (Figure S7). The apparent differences in reporter positivity as a function of mRNA dosing between the mCherry and Cre experiments highlight that the observed LNP mRNA delivery efficiency to cells is subject to the functional output of the mRNA gene product and likely needs be determined empirically. In the case of fluorescent reporters, for example, observed positivity rates are limited by factors such as mRNA translation rates, reporter lifetime, and detector sensitivity.
In the context of our Fancc LNP, one limitation of the Fancc mouse model used in these studies is that mutant mice do not naturally present with BM failure. This limitation complicates the faithful modeling of endogenous HSPC rescue by our Fancc LNP in restoring function in the BM, compounded by the transient nature of the protective effects we show here. A more robust model of BM failure, along with repeat dosing schemes, will likely be necessary to achieve time-resolved metrics concerning long-term therapeutic effects of LNP-mediated management of FA.
While safety has been demonstrated in ex vivo lentiviral modification of HSCs, immune recognition is a natural barrier to using lentiviral vectors in vivo.38 Indeed, lentiviral vectors promote low-level inflammation and can elicit adaptive innate immune responses, but this has not been explored in FA.44,45 LNPs such as those we present here have also demonstrated innate inflammatory activity; however, our results demonstrate no significant inflammatory responses in HSPCs after exposure to LNPs. Despite this, one common approach for immunosuppression in the context of drug delivery is pre-treatment with broadly immunomodulatory glucocorticoids, such as dexamethasone. Dexamethasone typically suppresses adaptive T cell response signals in the context of LNPs.46 This approach does not seem to blunt the beneficial LNPFancc response in vitro (Figure S8) and may optimize repeat dosing regimens to extend expression durability beyond that of a single mRNA dose. This or other more specific immunosuppression approaches may prove vital if adaptive immunity is eventually realized as a complication of LNP delivery for protein replacement therapies. Additionally, FA HSCs are known to exist in an elevated inflammatory state, which has been hypothesized to assist in the proliferation of FA HSCs despite cell-cycle blockades by p53/p21 activity. Several of the previously reported inflammatory signals caused by LNPs are known to act upstream of MYC, a strong promoter of proliferative capacity through the activity of downstream effector targets. Our limited studies suggest that LNP treatment of FA HSPC may provoke only a limited innate immune response that is largely similar to what is observed in WT cells. Interestingly, our data suggest a possible trend of decreased immune cytokine expression in FA HSCs based on transcript levels measured by quantitative real-time PCR (Figure 6), although additional studies are required to further qualify this observation.
In aggregate, our results demonstrate that LNP access to the HSC niche can deliver mRNA for protein replacement to restore FA HSPC proliferative capacity and alkylator resistance. This work serves as a first step toward clinical translation and illustrates the successful adaptation of an LNP platform for a congenital blood disorder.
Materials and methods
Materials
A general list of materials and their sources is provided in Table S1.
Animal husbandry
All animal studies were approved by the Children’s Hospital of Philadelphia institutional animal care and use committee and performed according to their guidelines. Fancc−/− and WT mice were bred from C57BL/6 FANCC chimeric mice genetically modified to present a single deletion of exon 8 of the FANCC gene, resulting in a downstream frameshift and, therefore, deletion of the C-terminal half of Fancc.47 Mice were housed with up to four littermates with unlimited access to chow. For BM harvests, mice were sacrificed via CO2 asphyxiation, and the pelvis, femurs, and tibias were isolated from tissue and flushed with Hank’s balanced salt solution using a 26G needle. Transplantation of ex vivo expanded HSCs was performed on lethally irradiated (two doses of 5.2 Gy 4 h apart) mice by tail vein injection of 100 μL PBS containing 420,000 ex vivo expanded cells and 250,000 whole BM cells from healthy C57BL/6 donors. Blood samples were obtained through retro-orbital access in anesthetized mice. Ai14 mice were purchased from The Jackson Laboratory and are a Cre reporter strain presenting a loxP-flanked STOP cassette upstream a tdTomato reporter gene located at the Gt(ROSA)26Sor locus.
LNPs
Linear and circular synthetic mRNA constructs were produced with either 5′ CleanCap or IRES sequence, a 101-repeat poly(A) tail, and N1-methylpseudouridine modifications. Circularization was achieved using a Tetrahymena thermophila ribozyme-based RNA circularization system. RNA products were purified by size-exclusion chromatography and precipitated before resuspension at 1 mg/mL for use. LNPs (polydispersity index 0.01–0.1, diameter 80 ± 5 nm) were obtained from Acuitas Therapeutics (Vancouver, BC, Canada) and contained a proprietary ionizable cationic lipid/phosphatidylcholine/cholesterol/polyethylene glycol (PEG)-lipid as described in patent application WO2020146805A1, or were produced using the commercially available ionizable lipid SM-102 (Broadpharm), along with helper lipid distearoylphosphatidylcholine (DSPC; Avanti Polar Lipids, AVP), cholesterol (Sigma), and either ALC-0159 (AVP) or DMG-PEG 2000 (AVP). SM-102 LNP used molar ratios of 50:10:38.5:1.5 for SM-102:DSPC:cholesterol:DMG-PEG 2000. After formulation, LNPs were concentrated by spin column and sterile filtered before use. Final RNA content and concentration were determined by Ribogreen assay. The respective formulation used for each experiment is highlighted in Table S2.
LNP injections
LNPs were delivered to mice either IV or IF. IV delivery was performed by diluting stock LNP to a 100-μL volume dose containing up to 12.5 μg mRNA in PBS and administering the dose via tail vein injection using a 28G insulin syringe. IF delivery was achieved using a 28G insulin syringe to access the medullary space of the left femur axially through the distal end. A custom plastic spacer was used to control injection depth, and up to 20 μL LNP solution was administered manually.
In vivo luciferase expression
C57BL/6 WT mice between 7 and 9 weeks of age were treated by IV or IF injection of 10 μg LNPLFluc. After 24 h, mice were administered 200 μL 16.7 mg/mL d-luciferin substrate solution (Regis Technologies). After a 10-min rest time, mice were sacrificed, and femurs, tibias, liver, spleen, lungs, heart, and kidneys were excised. For each mouse, all organs were imaged simultaneously except for liver and spleen due to their relatively elevated radiance. Mice were processed sequentially to ensure equal luciferin rest times and organ extraction delays prior to imaging by IVIS Spectrum (PerkinElmer) live animal imager. All images were captured within 15 min of sacrificing. Images were processed by custom MATLAB scripts using calibrated values of intensity to radiance relationships.
Cell culture
All ex vivo LSK or Lin− populations were cultured in STEMSPAN media (STEMCELL Technologies) with 10 ng/mL interleukin-3 (IL-3), 10 ng/mL IL-6, and 50 ng/mL stem cell factor (SCF). HSC (KSL,CD48−, CD150+) populations were expanded in ex vivo cultures based on previously reported methods for efficient ex vivo expansion.48 Briefly, sorted HSCs were seeded on human plasma fibronectin (Sigma-Aldrich)-coated 24-well tissue culture plates in F12 medium supplemented with 1 mg/mL polyvinyl alcohol, 0.01 M HEPES, 1% (v/v) 100× penicillin-streptomycin-glutamine (Gibco), 1% (v/v) 100× insulin-transferrin-selenium-ethanolamine (Gibco), 100 ng/mL murine thrombopoietin, and 10 ng/mL murine SCF. Cells were expanded up to 14 days with initial media changes 5–6 days post-seeding and in 3-day intervals afterward. Mouse embryonic fibroblasts were cultured in DMEM with 10% v/v fetal bovine serum and 1% v/v penicillin-streptomycin.
Flow cytometry analysis
For flow cytometric analysis, whole BM was flushed from the femurs using sterile PBS with 1% v/v penicillin/streptomycin. Flushed BM was strained through a 70-μm filter, centrifuged at 400 rcf for 5 min, and resuspended in RBC lysis buffer (BioLegend) diluted to working concentration in deionized water. Cells were washed with PBS, resuspended in immunophenotyping buffer for 20 min (BM: FITC Mouse Lineage Antibody Cocktail, ckit, Sca-1, CD150, CD48; BM niche cells: Ter119, CD45, CD51, CD31, Sca-1), and rinsed again with PBS prior to FACS analysis. All samples were analyzed using a Cytek Aurora spectral analyzer, and spectral data were processed using custom MATLAB scripts. For reporter analyses, positivity thresholds for the mCherry reporter were defined as the intensity value yielding 0.5% positivity in unstained cell populations within each cohort. MFI values are reported as the mean intensity of cells above the positivity threshold.
quantitative real-time PCR
To analyze relative concentrations of exogenous mCherry and Fancc mRNA, total RNA was extracted from whole BM or spleen cells (RNeasy Micro Plus Kit, Qiagen), and reverse transcribed (SuperScript IV VILO, Invitrogen). cDNA was either analyzed immediately using real-time PCR (PowerUp SYBR Green Master Mix, Applied Biosystems; ViiA 7 Real-Time PCR System, Thermo Fisher), or stored at −80°C until analysis. Primers were obtained from Harvard PrimerBank unless otherwise specified. Primers for synthetic mRNA constructs mCherry and cotFANCC were custom designed, ordered from Integrated DNA Technologies, and validated before use. RT2 Profiler arrays used for screening immune gene expression were loaded with 4 ng ex vivo expanded HSCs per well in SYBR Green Master Mix. Relative mRNA concentrations are approximated by normalization to glyceraldehyde 3-phosphate dehydrogenase within biological replicates, followed by normalization to a common sample and expressed as the binary log of the relative fold change.
Immunofluorescence imaging and foci counting
Mouse embryonic fibroblasts derived from fetal WT and mutant FANCC embryos harvested between embryonic day (E)12 and E14 were seeded at 20,000 cells/well in optically thin F-bottom 96-well plates for 2 days before treatment with LNP, MMC, or both. Four hours after MMC treatment, cells were fixed in 4% paraformaldehyde, permeabilized in PBS with 0.1% v/v Triton X-100 for 15 min, blocked for 1 h in a blocking buffer (5% BSA in PBS-Tween 20 [PBS-T]), and then stained for 1 h with Alexa Fluor 488 anti-γH2AX and Alexa Fluor 647 anti-Fancd2 in PBS-T. Wells were then rinsed three times with PBS-T before incubation in 1 μg/mL DAPI in PBS-T for 5 min before a final PBS-T rinse. Wells were imaged on a Zeiss Axio Observer 7 widefield microscope using a 40× 0.95 numerical aperture air objective operated through the Zen Blue software. Tiled images were taken at the center of each well, with triplicate wells per condition. Foci and nuclei were analyzed in MATLAB using a custom script.
Colony-forming assays
For Lin− CFUs, whole BM from WT or mutant FANCC mice was flushed and enriched for hematopoietic progenitor cells using EasySep Mouse Hematopoietic Progenitor Cell Isolation Kit (STEMCELL Technologies). Lin− cells were counted and seeded at 60,000 cells/well in a 48-well non-treated tissue culture plate in STEMSPAN media with growth factors. Cells were then treated with 0.5 μg LNP. At 24 h after LNP treatment, cells were exposed to 20 nM MMC for 24 h before media change and additional 0.5 μg LNP treatment. At 6 h after the second treatment, cells were plated in methylcellulose media at 5,000 cells/mL and cultured for 8 days in a 6-well plate. Colonies were imaged using a STEMvision imager (STEMCELL Technologies) and analyzed in MATLAB using a custom script.
Proliferation assays
Liquid culture assays to probe Fancc−/−) survival after MMC treatment in ex vivo expanded HSCs were prepared by treating 13-day expanded LT-HSCs with LNP for 24 h, followed by MMC treatment for 24, 48, 72, 96, or 120 h. Cells remaining in wells 6 days post-LNP treatment were counted via FACS (Cytek Aurora) in 96-well v-bottom plates. Survival was approximated by comparing the number of cells in MMC-treated replicates to the number of cells in MMC-free wells and normalized to the performance of WT cells in the same conditions. For dexamethasone studies, whole BM from WT or mutant FANCC mice was flushed and enriched for hematopoietic progenitor cells using EasySep Mouse Hematopoietic Progenitor Cell Isolation Kit (STEMCELL Technologies). Cells were counted and seeded at 30,000 cells/well in a 48-well non-treated tissue culture plate. For in vitro assays, cells were then treated with 0.25 μg LNP. At 24 h after LNP treatment, cells were exposed to 20 nM MMC for 24 h before media change and additional 0.25 μg LNP treatment. Cells were cultured for 5 days and were counted using a DeNovix CellDrop FL automated counter.
Statistical analysis
All statistical analyses and graphs were generated by either GraphPad Prism 9 or MATLAB (R2021a) (p > 0.05 for all analyses). In cases where one-way ANOVA was performed and multiple comparisons were performed post-hoc, the Tukey method was employed when comparing all means, and Bonferroni-Dunn was employed when comparing pairs of means. All data used in statistical comparisons were assumed to be homoscedastic and normally distributed.
Data and code availability
All flow cytometry, quantitative real-time PCR, and imaging raw data are maintained on institutionally managed servers and are available upon request.
Acknowledgments
This work was funded in part by the Cell & Gene Therapy Collaborative at the Children's Hospital of Philadelphia. L.O. was supported by 5-T32-HL-007150-47 (Poncz/Chou, principal investigators). We thank Dr. Florin Tuluc and Jennifer Murray of the Children’s Hospital of Philadelphia Flow Cytometry core facility and Dr. Benjamin Davis and Dr. Houping Ni from the Engineered mRNA and Targeted Nanomedicine core for technical assistance. We thank Danielle Kobulsky for editorial review. Graphical representations were produced using BioRender.
Author contributions
O.B. and P.K. conceptualized the study and experimental design and edited the manuscript. O.B. performed experiments, analyzed data, and wrote the manuscript. S.A. performed experiments, analyzed data, and edited the manuscript. L.O., S.A., S.J., and T.P. assisted in performing experiments. M.-G.A., H.S., and D.W. designed and produced mRNA and formulated and provided mRNA-LNP. Y.T. formulated and provided LNPs. S.R. and M.-G.A. provided insights into the experimental design and findings. All authors read and approved the manuscript.
Declaration of interests
Y.T., D.W., and M.-G.A. are named on patents that describe LNPs for the delivery of nucleic acid therapeutics, including mRNA, and the use of modified mRNA in LNPs as a vaccine platform.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102423.
Supplemental information
References
- 1.Kelaidi C., Makis A., Petrikkos L., Antoniadi K., Selenti N., Tzotzola V., Ioannidou E.D., Tsitsikas K., Kitra V., Kalpini-Mavrou A., et al. Bone Marrow Failure in Fanconi Anemia: Clinical and Genetic Spectrum in a Cohort of 20 Pediatric Patients. J. Pediatr. Hematol. Oncol. 2019;41:612–617. doi: 10.1097/mph.0000000000001549. [DOI] [PubMed] [Google Scholar]
- 2.Bagby G. Recent advances in understanding hematopoiesis in Fanconi Anemia. F1000Res. 2018;7:105. doi: 10.12688/f1000research.13213.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milletti G., Strocchio L., Pagliara D., Girardi K., Carta R., Mastronuzzi A., Locatelli F., Nazio F. Canonical and Noncanonical Roles of Fanconi Anemia Proteins: Implications in Cancer Predisposition. Cancers. 2020;12 doi: 10.3390/cancers12092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amenábar J.M., Torres-Pereira C.C., Tang K.D., Punyadeera C. Two enemies, one fight: An update of oral cancer in patients with Fanconi anemia. Cancer. 2019;125:3936–3946. doi: 10.1002/cncr.32435. [DOI] [PubMed] [Google Scholar]
- 5.Hughes A.D., Kurre P. The impact of clonal diversity and mosaicism on haematopoietic function in Fanconi anaemia. Br. J. Haematol. 2022;196:274–287. doi: 10.1111/bjh.17653. [DOI] [PubMed] [Google Scholar]
- 6.Baxter-Lowe L.A., Maiers M., Spellman S.R., Haagenson M.D., Wang T., Fernandez-Vina M., Marsh S.G.E., Horowitz M., Hurley C.K. HLA-A disparities illustrate challenges for ranking the impact of HLA mismatches on bone marrow transplant outcomes in the United States. Biol. Blood Marrow Transplant. 2009;15:971–981. doi: 10.1016/j.bbmt.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Río P., Navarro S., Guenechea G., Sánchez-Domínguez R., Lamana M.L., Yañez R., Casado J.A., Mehta P.A., Pujol M.R., Surrallés J., et al. Engraftment and in vivo proliferation advantage of gene-corrected mobilized CD34(+) cells from Fanconi anemia patients. Blood. 2017;130:1535–1542. doi: 10.1182/blood-2017-03-774174. [DOI] [PubMed] [Google Scholar]
- 8.Río P., Navarro S., Wang W., Sánchez-Domínguez R., Pujol R.M., Segovia J.C., Bogliolo M., Merino E., Wu N., Salgado R., et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019;25:1396–1401. doi: 10.1038/s41591-019-0550-z. [DOI] [PubMed] [Google Scholar]
- 9.Cappelli E., Degan P., Bruno S., Pierri F., Miano M., Raggi F., Farruggia P., Mecucci C., Crescenzi B., Naim V., et al. The passage from bone marrow niche to bloodstream triggers the metabolic impairment in Fanconi Anemia mononuclear cells. Redox Biol. 2020;36 doi: 10.1016/j.redox.2020.101618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mombled M., Rodriguez L., Avalon M., Duchez P., Vlaski-Lafarge M., Debeissat C., Pérard B., Sawai K.M., Pasquet J.M., Bijou F., et al. Characteristics of cells with engraftment capacity within CD34+ cell population upon G-CSF and Plerixafor mobilization. Leukemia. 2020;34:3370–3381. doi: 10.1038/s41375-020-0982-y. [DOI] [PubMed] [Google Scholar]
- 11.Morgan R.A., Gray D., Lomova A., Kohn D.B. Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned. Cell Stem Cell. 2017;21:574–590. doi: 10.1016/j.stem.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zonari E., Desantis G., Petrillo C., Boccalatte F.E., Lidonnici M.R., Kajaste-Rudnitski A., Aiuti A., Ferrari G., Naldini L., Gentner B. Efficient Ex Vivo Engineering and Expansion of Highly Purified Human Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy. Stem Cell Rep. 2017;8:977–990. doi: 10.1016/j.stemcr.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cannon P., Asokan A., Czechowicz A., Hammond P., Kohn D.B., Lieber A., Malik P., Marks P., Porteus M., Verhoeyen E., et al. Safe and Effective In Vivo Targeting and Gene Editing in Hematopoietic Stem Cells: Strategies for Accelerating Development. Hum. Gene Ther. 2021;32:31–42. doi: 10.1089/hum.2020.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berraondo P., Martini P.G.V., Avila M.A., Fontanellas A. Messenger RNA therapy for rare genetic metabolic diseases. Gut. 2019;68:1323–1330. doi: 10.1136/gutjnl-2019-318269. [DOI] [PubMed] [Google Scholar]
- 15.Breda L., Papp T.E., Triebwasser M.P., Yadegari A., Fedorky M.T., Tanaka N., Abdulmalik O., Pavani G., Wang Y., Grupp S.A., et al. In vivo hematopoietic stem cell modification by mRNA delivery. Science. 2023;381:436–443. doi: 10.1126/science.ade6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X., Xu T., Shi H., Hong J., Jin X., Cao L., Wang J., Lin Y., Pan Z., Wang S., et al. Cartilage-targeting mRNA-lipid nanoparticles rescue perifocal apoptotic chondrocytes for integrative cartilage repair. Chem. Eng. J. 2023;465 doi: 10.1016/j.cej.2023.142841. [DOI] [Google Scholar]
- 17.Herrera-Barrera M., Ryals R.C., Gautam M., Jozic A., Landry M., Korzun T., Gupta M., Acosta C., Stoddard J., Reynaga R., et al. Peptide-guided lipid nanoparticles deliver mRNA to the neural retina of rodents and nonhuman primates. Sci. Adv. 2023;9 doi: 10.1126/sciadv.add4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B., Manan R.S., Liang S.Q., Gordon A., Jiang A., Varley A., Gao G., Langer R., Xue W., Anderson D. Combinatorial design of nanoparticles for pulmonary mRNA delivery and genome editing. Nat. Biotechnol. 2023;41:1410–1415. doi: 10.1038/s41587-023-01679-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huayamares S.G., Lokugamage M.P., Rab R., Da Silva Sanchez A.J., Kim H., Radmand A., Loughrey D., Lian L., Hou Y., Achyut B.R., et al. High-throughput screens identify a lipid nanoparticle that preferentially delivers mRNA to human tumors in vivo. J. Contr. Release. 2023;357:394–403. doi: 10.1016/j.jconrel.2023.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarez D., Masse-Ranson G., Sedimbi S.K., Wisti P., Rodriguez L., Santana J., Manning T., Towner T., Geilich B., Mihai C., et al. Functional mRNA delivery to hematopoietic stem and progenitor cells in vivo. bioRxiv. 2022 doi: 10.1101/2022.12.15.520650. Preprint at. [DOI] [Google Scholar]
- 21.Garaycoechea J.I., Patel K.J. Why does the bone marrow fail in Fanconi anemia? Blood. 2014;123:26–34. doi: 10.1182/blood-2013-09-427740. [DOI] [PubMed] [Google Scholar]
- 22.Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R., et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melamed J.R., Hajj K.A., Chaudhary N., Strelkova D., Arral M.L., Pardi N., Alameh M.-G., Miller J.B., Farbiak L., Siegwart D.J., et al. Lipid nanoparticle chemistry determines how nucleoside base modifications alter mRNA delivery. J. Contr. Release. 2022;341:206–214. doi: 10.1016/j.jconrel.2021.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J., Xu Y., Zhou M., Xu S., Varley A.J., Golubovic A., Lu R.X.Z., Wang K.C., Yeganeh M., Vosoughi D., Li B. Combinatorial design of ionizable lipid nanoparticles for muscle-selective mRNA delivery with minimized off-target effects. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2309472120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dilliard S.A., Siegwart D.J. Passive, active and endogenous organ-targeted lipid and polymer nanoparticles for delivery of genetic drugs. Nat. Rev. Mater. 2023;8:282–300. doi: 10.1038/s41578-022-00529-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki H., Zuo Y., Wang J., Zhang M.Q., Malhotra A., Mayeda A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006;34 doi: 10.1093/nar/gkl151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C., Liu H. Factors influencing degradation kinetics of mRNAs and half-lives of microRNAs, circRNAs, lncRNAs in blood in vitro using quantitative PCR. Sci. Rep. 2022;12:7259. doi: 10.1038/s41598-022-11339-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wesselhoeft R.A., Kowalski P.S., Anderson D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 2018;9:2629. doi: 10.1038/s41467-018-05096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walter D., Lier A., Geiselhart A., Thalheimer F.B., Huntscha S., Sobotta M.C., Moehrle B., Brocks D., Bayindir I., Kaschutnig P., et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- 30.Ceccaldi R., Parmar K., Mouly E., Delord M., Kim J.M., Regairaz M., Pla M., Vasquez N., Zhang Q.S., Pondarre C., et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11:36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodríguez A., Zhang K., Färkkilä A., Filiatrault J., Yang C., Velázquez M., Furutani E., Goldman D.C., García de Teresa B., Garza-Mayén G., et al. MYC Promotes Bone Marrow Stem Cell Dysfunction in Fanconi Anemia. Cell Stem Cell. 2021;28:33–47.e38. doi: 10.1016/j.stem.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alameh M.G., Tombácz I., Bettini E., Lederer K., Sittplangkoon C., Wilmore J.R., Gaudette B.T., Soliman O.Y., Pine M., Hicks P., et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity. 2021;54:2877–2892.e2877. doi: 10.1016/j.immuni.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ndeupen S., Qin Z., Jacobsen S., Bouteau A., Estanbouli H., Igyártó B.Z. The mRNA-LNP platform's lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. iScience. 2021;24 doi: 10.1016/j.isci.2021.103479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gentner B., Naldini L. In Vivo Selection for Gene-Corrected HSPCs Advances Gene Therapy for a Rare Stem Cell Disease. Cell Stem Cell. 2019;25:592–593. doi: 10.1016/j.stem.2019.10.004. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Y., He Y., Xing W., Zhang P., Shi H., Chen S., Shi J., Bai J., Rhodes S.D., Zhang F., et al. An abnormal bone marrow microenvironment contributes to hematopoietic dysfunction in Fanconi anemia. Haematologica. 2017;102:1017–1027. doi: 10.3324/haematol.2016.158717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lecourt S., Vanneaux V., Leblanc T., Leroux G., Ternaux B., Benbunan M., Chomienne C., Baruchel A., Marolleau J.P., Gluckman E., et al. Bone marrow microenvironment in fanconi anemia: a prospective functional study in a cohort of fanconi anemia patients. Stem Cell. Dev. 2010;19:203–208. doi: 10.1089/scd.2009.0062. [DOI] [PubMed] [Google Scholar]
- 37.Li Y., Chen S., Yuan J., Yang Y., Li J., Ma J., Wu X., Freund M., Pollok K., Hanenberg H., et al. Mesenchymal stem/progenitor cells promote the reconstitution of exogenous hematopoietic stem cells in Fancg-/- mice in vivo. Blood. 2009;113:2342–2351. doi: 10.1182/blood-2008-07-168138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shirley J.L., de Jong Y.P., Terhorst C., Herzog R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020;28:709–722. doi: 10.1016/j.ymthe.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siegner S.M., Ugalde L., Clemens A., Garcia-Garcia L., Bueren J.A., Rio P., Karasu M.E., Corn J.E. Adenine base editing efficiently restores the function of Fanconi anemia hematopoietic stem and progenitor cells. Nat. Commun. 2022;13:6900. doi: 10.1038/s41467-022-34479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C., Georgakopoulou A., Mishra A., Gil S., Hawkins R.D., Yannaki E., Lieber A. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal γ-globin in β-YAC mice. Blood Adv. 2021;5:1122–1135. doi: 10.1182/bloodadvances.2020003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aliahmad P., Miyake-Stoner S.J., Geall A.J., Wang N.S. Next generation self-replicating RNA vectors for vaccines and immunotherapies. Cancer Gene Ther. 2023;30:785–793. doi: 10.1038/s41417-022-00435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geall A.J., Verma A., Otten G.R., Shaw C.A., Hekele A., Banerjee K., Cu Y., Beard C.W., Brito L.A., Krucker T., et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA. 2012;109:14604–14609. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pepini T., Pulichino A.M., Carsillo T., Carlson A.L., Sari-Sarraf F., Ramsauer K., Debasitis J.C., Maruggi G., Otten G.R., Geall A.J., et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017;198:4012–4024. doi: 10.4049/jimmunol.1601877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maertens G.N., Engelman A.N., Cherepanov P. Structure and function of retroviral integrase. Nat. Rev. Microbiol. 2022;20:20–34. doi: 10.1038/s41579-021-00586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Annoni A., Gregori S., Naldini L., Cantore A. Modulation of immune responses in lentiviral vector-mediated gene transfer. Cell. Immunol. 2019;342 doi: 10.1016/j.cellimm.2018.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen S., Zaifman J., Kulkarni J.A., Zhigaltsev I.V., Tam Y.K., Ciufolini M.A., Tam Y.Y.C., Cullis P.R. Dexamethasone prodrugs as potent suppressors of the immunostimulatory effects of lipid nanoparticle formulations of nucleic acids. J. Contr. Release. 2018;286:46–54. doi: 10.1016/j.jconrel.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 47.Chen M., Tomkins D.J., Auerbach W., McKerlie C., Youssoufian H., Liu L., Gan O., Carreau M., Auerbach A., Groves T., et al. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nat. Genet. 1996;12:448–451. doi: 10.1038/ng0496-448. [DOI] [PubMed] [Google Scholar]
- 48.Wilkinson A.C., Ishida R., Kikuchi M., Sudo K., Morita M., Crisostomo R.V., Yamamoto R., Loh K.M., Nakamura Y., Watanabe M., et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature. 2019;571:117–121. doi: 10.1038/s41586-019-1244-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All flow cytometry, quantitative real-time PCR, and imaging raw data are maintained on institutionally managed servers and are available upon request.