

Abstract
Receptor-stimulated generation of intracellular reactive oxygen species (ROS) modulates signal transduction, although the mechanism(s) is unclear. One potential basis is the reversible oxidation of the active site cysteine of protein tyrosine phosphatases (PTPs). Here, we show that activation of the antigen receptor of T cells (TCR), which induces production of ROS, induces transient inactivation of the SH2 domain-containing PTP, SHP-2, but not the homologous SHP-1. SHP-2 is recruited to the LAT–Gads–SLP-76 complex and directly regulates the phosphorylation of key signaling proteins Vav1 and ADAP. Furthermore, the association of ADAP with the adapter SLP-76 is regulated by SHP-2 in a redox-dependent manner. The data indicate that TCR-mediated ROS generation leads to SHP-2 oxidation, which promotes T-cell adhesion through effects on an SLP-76-dependent signaling pathway to integrin activation.
Keywords: adhesion, reactive oxygen species, protein tyrosine phosphatase, signal transduction, T lymphocyte
Introduction
Regulation of cellular functions in response to exogenous stress, for example, oxidative stress, irradiation, etc., has been characterized in evolutionarily conserved systems from microbial organisms to plants and mammals. It is now clear that cell surface receptors also utilize changes in redox balance to regulate signal transduction. Receptor stimulation with ligands as diverse as PDGF (Meng et al, 2002), insulin (Mahadev et al, 2001) or angiotensin II (Ushio-Fukai et al, 1998) induces the intracellular production of reactive oxygen species (ROS). In these studies, ROS function as requisite second messengers, regulating protein kinase activation, gene expression and/or proliferative responses. The mechanism(s) for this redox-dependent regulation of biologic responses, however, remains unclear.
One redox-sensitive target that regulates signaling is the family of protein tyrosine phosphatases (PTPs), which have an oxidation-sensitive, active site cysteine (Rhee et al, 2000). Insulin-induced ROS generation leads to oxidative inactivation of the PTPs, PTP1B and TC45 (Mahadev et al, 2001; Meng et al, 2004). Both phosphatases control phosphorylation of the insulin receptor or associated proteins, and insulin-induced PTP oxidation regulates downstream signaling. Oxidative inactivation of PTPs was also induced by ROS generation associated with stimulation with EGF (PTP1B) (Lee et al, 1998) or PDGF (LM-PTP, SHP-2, PTEN) (Chiarugi et al, 2001; Meng et al, 2002; Kwon et al, 2004) and this is a pivotal step in the biological effects of these ligands.
Previous studies have shown that signal transduction through the antigen receptor of T cells (TCR) can also be regulated by receptor-mediated production of ROS (Jackson et al, 2004). In mature T cells, TCR crosslinking induces rapid (within 2–4 min) ROS generation and the data indicate that TCR-stimulated ROS production regulates activation of MAPK, cytokine secretion and gene expression (Devadas et al, 2002; Kwon et al, 2003). In particular, TCR-induced production of hydrogen peroxide selectively inhibits activation of the MEK/Erk pathway (Kwon et al, 2003). However, the mechanisms by which ROS regulate TCR signaling are still poorly understood. Exposure to exogenous oxidants leads to oxidative inactivation of PTPs in T cells (O'Shea et al, 1992), but there is no evidence to date on the effects of TCR-induced ROS production on PTP function.
TCR signaling depends upon coordinated interactions of multiple signaling pathways, including PTPs, protein kinases and adapter proteins (e.g., LAT, SLP-76, Grb2, Gads) that ‘nucleate' key proteins (Jordan et al, 2003; Mustelin et al, 2003). The membrane-localized adapter LAT (linker for activation of T cells), upon tyrosine phosphorylation, recruits a number of proteins via their SH2 domains (e.g., Grb2, Gads, PLCγ1) (Wange, 2000). Gads (Grb2-like adapter downstream of Shc) is an adapter protein that, via one of its SH3 domains, interacts with the proline-rich domain of SLP-76 and recruits it to LAT and the membrane (Liu et al, 2001). In addition to the proline-rich domain, SLP-76 (SH2-containing leukocyte-specific protein of 76 kDa) has multiple functional interaction domains, including phosphorylated tyrosines and an SH2 domain (Myung et al, 2001) that bind proteins, including Vav1, Nck, Itk and ADAP (Myung et al, 2001). ADAP (adhesion- and degranulation-promoting adaptor protein), also known as Fyn SH2-binding protein (Fyb) or SLAP-130, binds the SH2 domain of SLP-76 upon ADAP phosphorylation (Peterson, 2003). ADAP is necessary for optimal T-cell responses and the inside-out activation of integrins (Griffiths et al, 2001; Peterson et al, 2001). Thus, the adapters Gads, SLP-76 and ADAP are critical regulators of T-cell development and TCR signaling pathways.
The goal of the current report was to investigate the molecular basis for redox control of TCR signal transduction by intracellular production of ROS. In the current study, we show that SHP-2 is oxidized by ROS produced upon TCR stimulation and that the active site cysteine is the primary site of oxidation. We further identify a site of action for SHP-2 in dephosphorylation of Vav1 and ADAP associated with SLP-76, although it does not dephosphorylate SLP-76 itself. The data support the hypothesis that SHP-2 exerts a negative role in dephosphorylation of ADAP and Vav1, which limits TCR-induced adhesion and LFA-1 clustering. TCR-induced ROS production selectively inhibits SHP-2-mediated dephosphorylation of Vav1 and ADAP associated with SLP-76, leading to redox-dependent changes in TCR-stimulated adhesion and integrin clustering.
Results
TCR-induced oxidation of SHP-2
Our previous data have demonstrated that TCR-induced production of hydrogen peroxide selectively inhibited activation of the MEK/Erk kinase pathway (Kwon et al, 2003; Jackson et al, 2004). SHP-2 has been shown to promote ERK activation in T cells (Frearson and Alexander, 1998). Therefore, we analyzed TCR-induced oxidation of SHP-2.
A biotinylated thiol reactive probe was used to label reduced/nonoxidized thiols in whole-cell lysates under anaerobic conditions. The labeling conditions lead to selective alkylation of reactive thiols, such as active site cysteines of PTPs (Kim et al, 2000). Loss of biotin incorporation into immunoprecipitated SHP-2 indicates oxidation of reactive thiols (Figure 1A). Decreased biotin labeling in SHP-2 was detected upon TCR crosslinking in Jurkat T cells (Figure 1B) and primary murine T-cell blasts (Figure 1D), which occurs coincident with the kinetics of TCR-stimulated ROS generation (Kwon et al, 2003; Jackson et al, 2004). The loss was transient, however, indicating that TCR-stimulated thiol oxidation was reversible.
Figure 1.
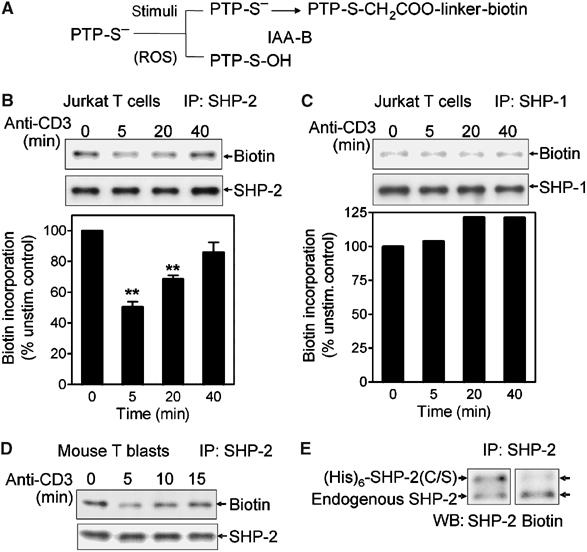
TCR stimulation induces oxidative modification of SHP-2. (A) Reduced thiols (PTP-S−) are labeled with PEO-iodoacetyl biotin, but oxidized thiols (PTP-S-OH; sulfenic acid) are not. (B, C) After anti-CD3 stimulation for the indicated times, Jurkat T cells were lysed and labeled with PEO-iodoacetyl biotin as described in Materials and methods. SHP-2 (B) or SHP-1 (C) was immunoprecipitated and biotinylation detected with HRP-conjugated streptavidin and ECL. Equal application of protein was confirmed by immunoblot analysis. The level of biotin incorporation (by densitometry) was corrected for total protein levels and the signal at time=0 was normalized to 100%. The graphs represent the average of three separate experiments±s.e.m. **Significantly different from unstimulated controls; P<0.01. (D) Mouse T-cell blasts were stimulated for the indicated times by anti-CD3 crosslinking. SHP-2 oxidation was measured as in (B). (E) Jurkat T cells were transiently transfected with an expression vector encoding (His)6-SHP-2(C/S) and cells were left unstimulated. Incorporation of biotin into endogenous or tagged SHP-2 was measured as in (B).
To test the selectivity of TCR-induced ROS generation in PTP oxidation, oxidation of SHP-1 was measured. SHP-1 also has tandem SH2 domains, exhibits similar substrate specificity as SHP-2 and has been proposed to affect TCR signal transduction (reviewed in Neel et al, 2003). Under conditions in which SHP-2 oxidation was observed, little or no oxidation of SHP-1 was detected (Figure 1C).
Although the labeling procedure is designed to detect oxidation of the active site cysteine, iodoacetamide could react with other free thiols in SHP-2. Therefore, Jurkat T cells were transfected to express (His)6-tagged SHP-2 in which the active site cysteine was mutated to serine (SHP-2(C/S)). Due to the presence of the (His)6 tag, the ectopically expressed SHP-2 has a slower mobility in gels and can be distinguished from endogenous SHP-2. In unstimulated cells, biotin labeling of endogenous SHP-2 was readily detected, but there was little incorporation of the label into the SHP-2(C/S) protein (Figure 1E). The data suggest that the majority of the iodoacetamide labeling of SHP-2, under these conditions, was at the active site. Thus, the transient loss of iodoacetamide labeling in SHP-2 upon TCR stimulation indicates reversible oxidation of the active site cysteine of SHP-2. SHP-2 phosphatase activity was also inhibited under conditions in which thiol oxidation was detected. Using a colorimetric assay (Dechert et al, 1994), phosphatase activity in SHP-2 immunoprecipitates was decreased upon TCR stimulation in both Jurkat T cells (16.72 to 11.1 pmol/min/106 cells) and mouse T-cell blasts (14.35 to 10.72 pmol/min/106 cells).
The labeling procedure above measures a loss of reactivity in protein thiols under nondenaturing conditions. The results could be affected by accessibility of thiols, and interpretation of the data relies upon the ability to detect decreased biotin incorporation. Therefore, SHP-2 oxidation was assayed using a method in which reduced thiols were blocked under denaturing conditions and the reversibly oxidized thiols were re-reduced and labeled with iodoacetamide-biotin (Kwon et al, 2004). This ‘positive labeling' of oxidized thiols will show a gain of signal upon thiol oxidation induced by TCR-stimulated ROS generation. Consistent with the results in Figure 1, TCR crosslinking of human T blasts induced a transient oxidation of SHP-2 (Figure 2A). The level of biotin incorporation was similar to that observed upon treatment of cells with 100 μM exogenous hydrogen peroxide. Under the same conditions, neither TCR stimulation nor treatment with 100 μM hydrogen peroxide induced SHP-1 oxidation. Exposure to 1 mM H2O2, however, did lead to detectable oxidation of SHP-1 (Figure 2B). TCR stimulation in Jurkat T cells also led to marked oxidation of SHP-2 (Figure 2C), but not SHP-1 (Figure 2D). Thus, TCR-stimulated production of ROS selectively induces oxidation of SHP-2 in T cells.
Figure 2.
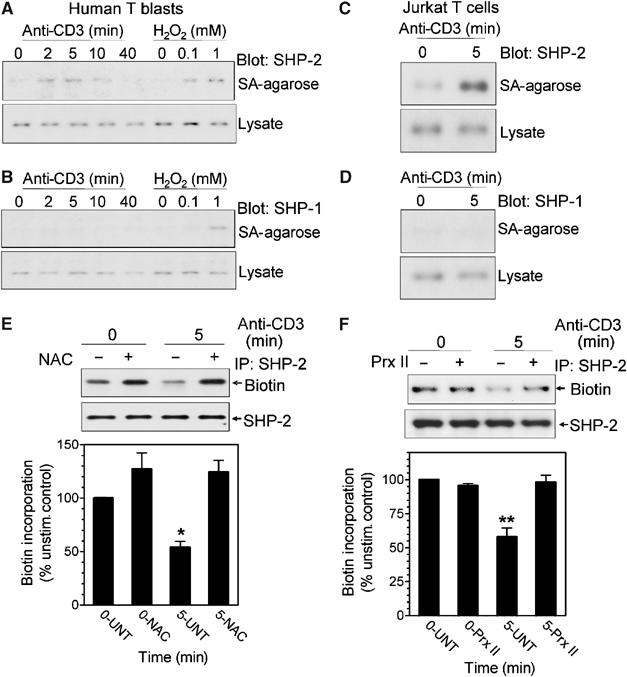
SHP-2 oxidation measured by ‘positive labeling'. Human T blasts (A, B) or Jurkat T cells (C, D) were stimulated by anti-CD3 crosslinking for the indicated times. Oxidized thiols were labeled with PEO-iodoacetyl biotin in a ‘positive' labeling method as described in Materials and methods. Biotinylated proteins were isolated on Neutravidin-Sepharose, separated by SDS–PAGE and immunoblotted for SHP-2 (A, C) or SHP-1 (B, D). An aliquot of the lysates was reserved to blot for total SHP-2 or SHP-1. (E, F) Antioxidants inhibit SHP-2 oxidation. (E) Mouse T-cell blasts were preincubated in the absence (−) or presence (+) of 20 mM NAC for 60 min. (F) Jurkat T cells were transfected with an empty vector (−) or one encoding Prx II (+) and prepared as described in Materials and methods. Cells were stimulated by anti-CD3 crosslinking for the indicated times and SHP-2 oxidation was detected as in Figure 1. The level of biotin incorporation (by densitometry) was corrected for total protein levels and the signal at time=0 was normalized to 100%. The graphs represent the average of three separate experiments±s.e.m. **Significantly different from unstimulated controls; P<0.01.
The two labeling approaches suggested that SHP-2 was more sensitive to intracellular ROS than SHP-1. This was directly tested by exposing Jurkat cells to graded concentrations of hydrogen peroxide and measuring oxidation of both SHP-1 and SHP-2 by the loss of biotin-iodoacetamide incorporation as in Figure 1 (Supplementary Figure 1). Oxidation of both SHP-1 and SHP-2 is detected, but at least two- to three-fold more hydrogen peroxide is required to detect oxidation of SHP-1.
Antioxidants and antioxidant enzymes were used to indicate a causal role for TCR-induced ROS generation in SHP-2 oxidation. Coincubation of mouse T cells with the antioxidant N-acetyl-cysteine (NAC) inhibited the loss of SHP-2 labeling, suggesting that ROS produced upon TCR stimulation were responsible for the loss of labeling (Figure 2E). The presence of NAC also produced a modest increase in biotin labeling of SHP-2 in unstimulated cells, suggesting a basal level of SHP-2 oxidation in cells. To further establish a role for hydrogen peroxide, the antioxidant enzyme peroxiredoxin II (Prx II) was overexpressed in Jurkat T cells. Previous studies have used overexpression of Prx II to eliminate hydrogen peroxide produced upon TCR stimulation (Devadas et al, 2002). As opposed to that observed in vector-transfected cells, TCR stimulation of cells overexpressing Prx II did not show decreased incorporation of biotin into SHP-2 (Figure 2F). The data indicate that TCR-induced production of hydrogen peroxide leads to transient oxidation of SHP-2.
Expression of inactive SHP-2 in T cells
The active site mutant of SHP-2 (SHP-2(C/S)) was expressed in cells to mimic the conditions in which SHP-2 was oxidatively inactivated. Expression of such mutated PTPs has also been used to identify potential phosphatase substrates (Neel and Tonks, 1997). Overexpression of wild-type SHP-2 (SHP-2 WT) was used to control for the effects of increased SHP-2 protein and the possible effects of increased SHP-2 activity in cells. Anti-HA immunoprecipitation from anti-CD3-stimulated cells expressing HA-tagged SHP-2(C/S) showed increased association of multiple phosphoproteins (Figure 3A). The literature suggests that some of these SHP-2-associated proteins may be LAT (Frearson and Alexander, 1998), Vav1 (Wakino et al, 2004) and/or Gab2 (Yamasaki et al, 2001). The presence of LAT and the adapter protein ADAP in the anti-HA immunoprecipitates was confirmed by Western blot (Figure 3A). The current results and previous reports support an interaction of SHP-2 with LAT and the multiprotein complex formed around LAT. Therefore, to investigate the potential implications of SHP-2 oxidation, the effect of SHP-2(C/S) expression was examined on TCR signaling in and around the complexes dependent upon LAT and SLP-76.
Figure 3.
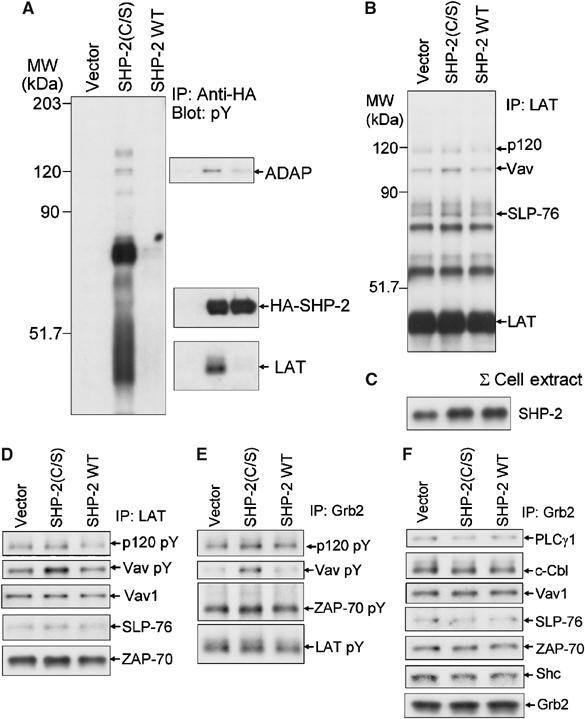
Association of proteins with SHP-2(C/S) upon TCR stimulation. (A) Jurkat T cells were transiently transfected with an empty vector (Vector) or an expression vector encoding HA-SHP-2(C/S) or HA-SHP-2 WT. Cells were stimulated by anti-CD3 crosslinking for 5 min and lysed. Anti-HA immunoprecipitates were blotted with anti-phosphotyrosine and the membrane was stripped and reprobed with antibodies to the indicated proteins. (B–F) Effect of SHP-2(C/S) expression on association of signaling molecules with LAT and Grb2. Jurkat T cells were transfected and stimulated as in (A). (C) Levels of SHP-2 in total cell lysates were determined by immunoblot analysis. (B, D) Anti-LAT or (E, F) anti-Grb2 immunoprecipitates were blotted with (B, E) anti-phosphotyrosine and (D, F) the membranes were stripped and reprobed for the indicated proteins.
Immunoprecipitation of LAT from Jurkat cells transfected to express SHP-2(C/S) and SHP-2 WT showed minor changes in associated phosphoproteins upon stimulation with anti-CD3 (Figure 3B). In these studies, the level of SHP-2(C/S) or ectopically expressed SHP-2 WT was titered to avoid overwhelming the system (Figure 3C). There was a modest increase in signals from phosphoproteins comigrating with SLP-76, Vav1 and ADAP in cells expressing SHP-2(C/S). There were no apparent changes in phosphorylation of LAT or ZAP-70, suggesting a selective effect. Immunoblots suggested that the minor increases in the phosphotyrosine signal in Vav1 were not mirrored by changes in Vav1 protein associated with LAT, but SLP-76 protein levels in the immunoprecipitates did show similar increases as in the phosphotyrosine blot (Figure 3D). In general, expression of wild-type SHP-2 did not markedly change either phosphorylation or levels of proteins co-immunoprecipitating with LAT.
Grb2 and Gads are the major adapter proteins that bind LAT. Grb2 immunoprecipitates showed minor differences in the association of tyrosine phosphorylated proteins in cells expressing either wild-type or mutant SHP-2 (Figure 3E and F). There was an increased intensity in the phosphotyrosine blot for a band that comigrated with Vav1, although there was no change in Vav1 protein in the immunoprecipitates by immunoblot. Thus, expression of the inactive SHP-2(C/S) induced small changes in phosphorylation and association of proteins with LAT, and the effect(s) does not appear to be centered upon proteins known to be associated with Grb2.
Effect of SHP-2(C/S) on Gads- and SLP-76-associated proteins
Analysis of phosphoproteins that co-immunoprecipitated with Gads from TCR-stimulated cells expressing SHP-2(C/S) extended the results observed in LAT precipitates. There was an increase (approximately two-fold by densitometry) in tyrosine phosphorylation of bands corresponding to ADAP and Vav1 as compared to cells expressing SHP-2 WT or vector (Figure 4A and Supplementary Figure 2A). In both cases, overexpression of SHP-2 WT led to small decreases in phosphorylation of the proteins as compared to vector control cells. Immunoblot for total ADAP protein showed commensurate changes in ADAP protein (Figure 4A). The data are consistent with the model of SHP-2 regulating binding of phosphorylated ADAP to SLP-76. The changes in Vav1 phosphorylation, however, were not reflected by increased Vav1 protein associated with Gads (Figure 4A). These results were consistent with the model of the SH2 domain of Vav1 binding to phosphorylated SLP-76, and SHP-2 acting to dephosphorylate Vav1. Identical results were observed in SLP-76 immunoprecipitates (Figure 4B and Supplementary Figure 2B). The effects of SHP-2 were selective in that total protein and tyrosine phosphorylation of both SLP-76 and LAT and association of other proteins including PLCγ1 were largely unchanged by expression of either active site mutant or wild-type SHP-2. These results suggested that SHP-2 dephosphorylated proteins associated with SLP-76, but did not target SLP-76 itself. The data further suggest that Vav1 and ADAP are SHP-2 substrates and SHP-2 regulates their activation and/or association with SLP-76.
Figure 4.
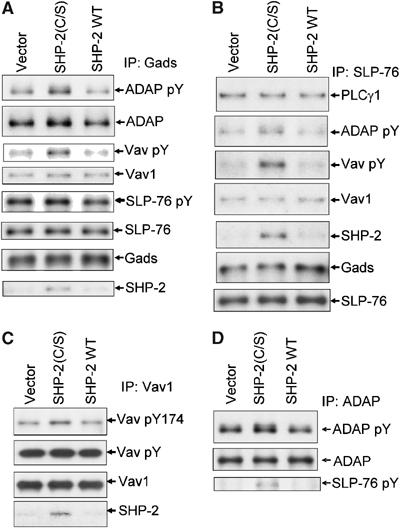
Effect of SHP-2(C/S) on phosphorylation and association of proteins with Gads and SLP-76. Jurkat T cells were transiently transfected with an empty vector (Vector) or an expression vector encoding SHP-2(C/S) or SHP-2 WT. Cells were stimulated for 5 min by anti-CD3 crosslinking. Lysates were immunoprecipitated with (A) anti-Gads, (B) anti-SLP-76, (C) anti-Vav1 or (D) anti-ADAP antibody. Immunoprecipitates were separated by SDS–PAGE, immunoblotted for phosphotyrosine and then stripped and reprobed for the indicated proteins. (C) Anti-Vav1 immunoprecipitates were also blotted with a phosphospecific antibody to Vav1 phosphorylated at Y174.
The consistent effect on Vav1 phosphorylation led us to measure directly changes in Vav1 phosphorylation in Vav1 immunoprecipitates (Figure 4C). Vav1 has multiple possible phosphorylation sites, and SHP-2(C/S) expression induced a modest increase in total Vav1 phosphorylation. When analyzed with a phosphospecific antibody to tyrosine 174 (Vav1 Y174), however, the presence of SHP-2(C/S) showed an increase (greater than 50% over vector control) in Vav1 phosphorylation. Thus, SHP-2 activity seems to have a selective effect on the phosphorylation of Vav1 at tyrosine 174, which is critical in activation of Vav1 GEF function (Turner and Billadeau, 2002).
Direct immunoprecipitation of ADAP supported the observation that SHP-2(C/S) expression led to increased ADAP phosphorylation (Figure 4D). These data further substantiate the model of SHP-2 regulating ADAP–SLP-76 association. Immunoprecipitation of Gads from anti-CD3-stimulated SLP-76-deficient Jurkat cells (J.14) did not show evidence of Vav1 and low levels of ADAP in Gads immunoprecipitates (Supplementary Figure 2C). Co-immunoprecipitation of SHP-2 with Gads, on the other hand, was largely unchanged in J.14 cells.
The SH2 domains of SHP-2 and SHP-1 show selective recognition, and are generally not interchangeable (Neel et al, 2003). Nevertheless, expression of high levels of SHP-2(C/S) might alter SHP-1 function. To test the potential role of SHP-1 in TCR-mediated changes in phosphorylation of proteins in the Gads–SLP-76 complex, SHP-1(C/S) was expressed side by side with SHP-2(C/S) in Jurkat cells (Supplementary Figure 3). In cells expressing SHP-1(C/S), anti-SLP-76 immunoprecipitates did not show the increase in phosphorylated Vav1 or phospho-ADAP that was observed in cells expressing SHP-2(C/S). In contrast, there was decreased phosphorylation of bands corresponding to ADAP, LAT and Vav1.
Therefore, the data support a model in which SHP-2 associates with a Gads–SLP-76 complex but does not target SLP-76 itself as a substrate. The data suggest that SHP-2 controls phosphorylation of Vav1 bound to SLP-76 and regulates the phosphorylation of ADAP and therefore its association with SLP-76.
Role of ROS in protein association with Gads–SLP-76
SHP-2(C/S) was expressed to mimic oxidatively inactivated SHP-2 in cells where ROS generation was occurring. Quenching TCR-induced ROS production with antioxidants, which inhibited SHP-2 oxidation (Figure 2), should show the opposite effect produced by SHP-2(C/S) expression. Thus, the effects of Prx II overexpression in TCR-stimulated Jurkat T cells were analyzed in immunoprecipitates of both Gads and SLP-76 (Figure 5). As was suggested by our previous findings (Kwon et al, 2003), overall anti-CD3-induced tyrosine phosphorylation was not markedly affected by Prx II overexpression as measured in total cell lysates (Supplementary Figure 4A). In Gads (Figure 5A) and SLP-76 (Figure 5B) immunoprecipitates, anti-CD3-induced ADAP phosphorylation and association were decreased nearly two-fold in cells expressing Prx II as compared to vector control. Direct immunoprecipitation of ADAP confirmed these observations, as Prx II overexpression inhibited TCR-stimulated phosphorylation of total ADAP (Figure 5C). As above, these effects were selective since phosphorylation of SLP-76 and association of proteins such as PLCγ1 were not affected by expression of Prx II. In contrast, the results with Vav1 in Prx II-overexpressing cells were not the converse of those with SHP-2(C/S). Vav1 phosphorylation and Vav1 protein levels in Gads immunoprecipitates showed little to no change as compared to vector control (Figure 5A). Thus, redox regulation of Vav1 phosphorylation in Gads–SLP-76 complexes appears to be multifaceted. The data clearly show that tyrosine phosphorylation of ADAP and its association with SLP-76 are influenced by ROS, potentially through oxidation of SHP-2.
Figure 5.
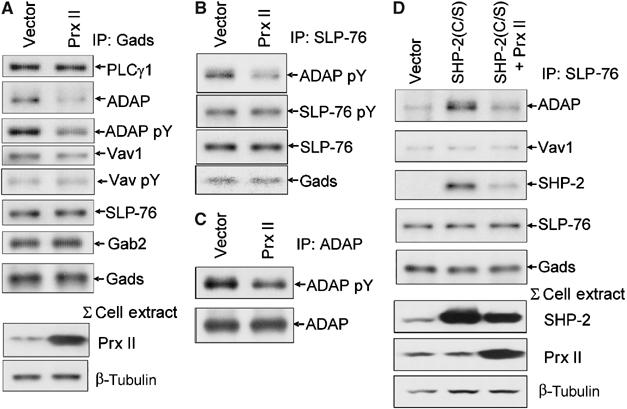
Effect of Prx II on association of proteins with Gads and SLP-76. Jurkat T cells were transfected with an empty vector or an expression vector encoding Prx II, and cells were stimulated by anti-CD3 crosslinking. Lysates were immunoprecipitated with (A) anti-Gads, (B) anti-SLP-76 or (C) anti-ADAP antibody. Immunoprecipitates were separated by SDS–PAGE, immunoblotted for phosphotyrosine and were stripped and reprobed for the indicated proteins. (A) Total cell lysates were probed for Prx II to measure overexpression and β-tubulin to control for loading. (D) Effects of coexpression of SHP-2(C/S) and Prx II. Jurkat T cells were transfected with an empty vector or an expression vector encoding SHP-2(C/S) alone or in combination with one encoding Prx II (SHP-2(C/S)+Prx II) and the cells were stimulated by anti-CD3 crosslinking. Lysates were immunoprecipitated with anti-SLP-76 antibody and probed with antibodies to the indicated proteins. Total lysates were probed for SHP-2 and Prx II to measure expression levels and β-tubulin was probed to control loading.
The effects of Prx II (Figure 5A) or SHP-2(C/S) (Figure 4A) on SLP-76–ADAP association might result from redox regulation of SHP-2 localized with the Gads–SLP-76 complex. Cotransfection to express both SHP-2(C/S) and Prx II showed an intermediate effect as compared to either protein alone. Levels of ADAP protein co-immunoprecipitating with SLP-76 were nearly 2.5-fold less in cells coexpressing Prx II and SHP-2(C/S) as compared to that observed in cells expressing SHP-2(C/S) alone (Figure 5D). The formation of an SLP-76–ADAP complex in cells expressing both proteins was still almost twice as much as in vector control cells as compared to the effects of Prx II alone, which was two-fold less than vector control (Figure 5A). Similarly, levels of SHP-2 in SLP-76 immunoprecipitates from cells expressing both SHP-2(C/S) and Prx II were half that in cells expressing the mutant SHP-2 alone, although the levels were still considerably higher than that observed in vector control. The data suggest that localized changes in redox balance control the availability of active SHP-2 to the Gads–SLP-76 signaling complex.
Effects of ROS and SHP-2 on adhesion
ADAP association with SLP-76 has been proposed to regulate TCR-induced adhesion of T cells through inside-out signaling to β-integrins (Griffiths et al, 2001; Peterson et al, 2001). Using adhesion to fibronectin-coated plates, SHP-2(C/S) expression significantly increased TCR-stimulated adhesion (Figure 6A). In contrast, overexpression of SHP-2 WT or Prx II, both of which lead to increased functional SHP-2 in TCR-stimulated cells, significantly inhibited adhesion.
Figure 6.
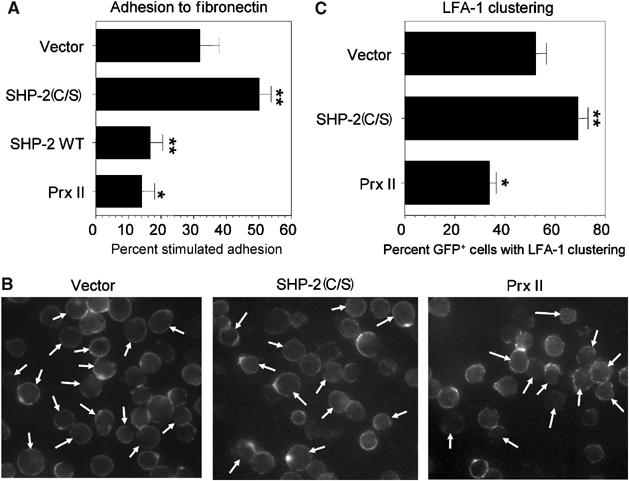
Effect of SHP-2 or Prx II on TCR-stimulated adhesion to fibronectin and LFA-1 clustering. (A) Jurkat T cells were transiently transfected with a β-gal expression plasmid in the presence of an empty vector (Vector) or an expression vector encoding SHP-2(C/S), SHP-2 WT or Prx II. Adhesion of anti-CD3-stimulated cells to fibronectin-coated wells was performed as described in Materials and methods. The data are expressed as the percent anti-CD3-induced increase in β-gal activity in the wells±s.e.m. and represent the average of triplicate samples in at least six separate experiments. (B, C) Jurkat T cells were transfected with an expression vector for GFP with a two-fold excess of empty vector, or expression vectors for SHP-2(C/S) or Prx II. Cells were stimulated for 15 min by anti-CD3 crosslinking and were stained with anti-human LFA-1 (TS2/4) as in Materials and methods and analyzed by fluorescence microscopy. (B) Representative fields showing LFA-1 staining. Arrows indicate the productively transfected GFP+ cells. (C) The data show the percentage of GFP+ T cells with LFA-1 clustering±s.e.m. from at least five separate experiments with at least 50 GFP+ cells counted per sample. For both experiments, *P<0.05 as compared to vector control cells and **P<0.01 as compared to vector control cells.
Inside-out signaling to integrins involves TCR-stimulated clustering of LFA-1 (Peterson, 2003). Fluorescent microscopic analysis of LFA-1 in T cells expressing SHP-2(C/S) or Prx II supported the adhesion data (Figure 6B). TCR crosslinking induced clustering of LFA-1 and this was significantly augmented in cells expressing SHP-2(C/S) and it was significantly inhibited in cells overexpressing Prx II (Figure 6C). Thus, redox regulation of SLP-76–ADAP association is reflected by changes in integrin activation and adhesion of TCR-stimulated cells (Figure 7).
Figure 7.
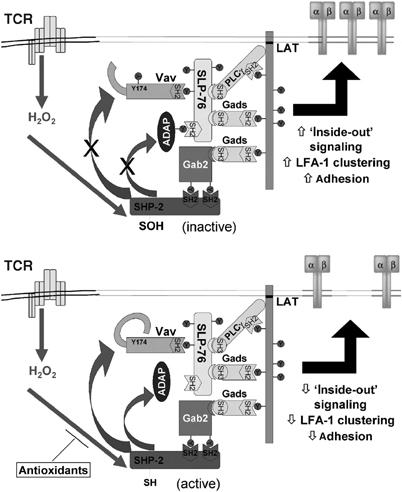
Model showing protein associations in the Gads–SLP-76 complex and the proposed targets for SHP-2. (Top) TCR stimulation induces production of ROS (H2O2), which oxidizes SHP-2 present in the Gads–SLP-76 complex. SHP-2 association is independent of SLP-76 and potentially is recruited by another Gads protein bound to the SHP-2 binding adapter Gab2. Oxidation of SHP-2 promotes phosphorylation of ADAP and Vav1, which is translated to increased LFA-1 clustering and T-cell adhesion. (Bottom) In the presence of antioxidants, for example, the effects of ROS are blunted and SHP-2 in the complex remains more active. This leads to less ADAP–SLP-76 association, inhibits LFA-1 clustering and decreases adhesion.
Discussion
Appropriate signal transduction and optimal cell activation require a balance between the activity of protein tyrosine kinases and PTPs. Recent evidence has shown that receptor-mediated generation of ROS can act to fine-tune this balance (Finkel, 2003). Herein, we show that the PTP SHP-2, but not the highly homologous SHP-1, is transiently oxidized in T cells by TCR-stimulated generation of ROS. Furthermore, we have identified two putative SHP-2 substrates, ADAP and Vav1, in T cells. The data suggest that SHP-2 oxidation promotes TCR-induced phosphorylation of ADAP and Vav1 in the Gads–SLP-76 complex and these changes increase integrin clustering and T-cell adhesion.
Signal transduction by ligands such as EGF, PDGF or insulin leads to generation of intracellular ROS and redox regulation of PTP function (Rhee et al, 2005). TCR-stimulated ROS production induced oxidation of SHP-2, primarily at the active site cysteine. SHP-2 oxidation was transient, returning to baseline within 15–30 min after stimulation. Oxidative inactivation of PTPs by ROS can be reversible or irreversible, depending upon the oxidation state (Claiborne et al, 1999). Enzymatic reduction of the active site thiol by glutaredoxin or thioredoxin can lead to recovery of phosphatase activity (Lee et al, 1998; Barrett et al, 1999). Using positive labeling, SHP-2 was found to have DTT-reducible form(s) of oxidized thiols upon TCR-induced ROS generation. Based upon previous reports of cysteine oxidation in PTPs, these may include sulfenic acids or disulfides formed with nearby intraprotein thiols or glutathione (Rhee et al, 2005). Recently, it has been shown that, upon oxidation of the active site thiol, PTP1B formed a novel intraprotein cyclic sulfenyl amide (Salmeen et al, 2003; van Montfort et al, 2003). These modifications are important because they are reversible and protect the active site thiol from further irreversible oxidation.
It appears that oxidation of SHP-2 is somewhat selective in T cells. SHP-1, the only other known mammalian PTP with tandem SH2 domains (Neel et al, 2003), was not detectably oxidized upon TCR stimulation and required higher amounts of exogenous hydrogen peroxide to demonstrate similar levels of thiol oxidation as SHP-2. Interestingly, PDGF-stimulated ROS generation also induced oxidation of SHP-2, but not SHP-1 (Meng et al, 2002). This may be due to different spatial orientation of the active site cysteines in relationship to positively charged residues in the active site pocket of the two phosphatases (Groen et al, 2005). The results may also reflect selective oxidation of PTP(s) present in a specific subcellular fraction by localized production of ROS. Consistent with this model, PDGF-induced SHP-2 oxidation did not occur in cells expressing a mutated PDGF receptor that was unable to recruit SHP-2 (Meng et al, 2002).
The current data suggest that SHP-2 associates with SLP-76 upon TCR stimulation and oxidized SHP-2 is localized within the Gads–SLP-76 complex. Association of inactive SHP-2 with proteins may be due to prolonged binding to phosphorylated substrates (Frearson and Alexander, 1998; Meng et al, 2002). In cells expressing the inactive SHP-2(C/S), both anti-Gads and anti-SLP-76 immunoprecipitates showed increased SHP-2 association. Similarly, immunoprecipitation of HA-SHP-2 (C/S) also revealed a limited association of SHP-2 with tyrosine phosphorylated proteins primarily involved in a LAT–Gads–SLP-76 complex.
Coexpression of SHP-2(C/S) and Prx II suggested localized redox regulation of SHP-2, since combined expression partially reversed the effect of either one alone. Thus, expressing SHP-2(C/S) to inhibit/compete for the increased levels of active SHP-2 produced by expression of Prx II led to increased ADAP phosphorylation and SLP-76 association when compared to vector controls or cells expressing Prx II alone. Conversely, inhibiting localized oxidation of SHP-2 with Prx II expression increased the amount of active SHP-2 in the Gads–SLP-76 complex. This competed with inactive SHP-2(C/S), dephosphorylated ADAP and decreased ADAP–SLP-76 binding. Therefore, the data suggest that changes in redox balance control the association of SHP-2 with the Gads–SLP-76 complex and its ability to dephosphorylate substrate proteins.
The interactions that lead SHP-2 to be recruited to SLP-76 are, as of yet, unclear. In SLP-76-deficient cells, co-immunoprecipitation of SHP-2 with Gads appeared unaffected, suggesting that SLP-76 itself or proteins recruited to Gads via SLP-76 (Vav1, Nck, Itk) did not recruit SHP-2 to the complex. A major SHP-2-binding protein is the adapter Gab2 (Yamasaki et al, 2001), which binds the SH3 domains of Grb2 and Gads (Yamasaki et al, 2003). In our hands, Gab2 was not detected in Grb2 immunoprecipitates (data not shown), while association with Gads was readily detected. Gab2 and SLP-76 were shown to compete for binding to the C-terminal SH3 domain of Gads (Yamasaki et al, 2003), while it is unclear if any proteins bind the N-terminal SH3 domain of Gads (Liu et al, 2001). Therefore, in our working model (Figure 7), SHP-2 is brought in proximity to substrate proteins in the LAT–Gads–SLP-76 complex via binding of its SH2 domains to phosphorylated tyrosines in Gab2, which is bound to another Gads molecule interacting with the same or neighboring phosphorylated LAT.
Despite its proximity to many proteins in the Gads–SLP-76 complex, the effects of SHP-2 and ROS are selective. Phosphorylation or co-immunoprecipitation of proteins such as SLP-76 itself and PLCγ1 was not affected by expression of SHP-2(C/S) or by removing ROS with Prx II. Because Gads and PLCγ1 promote association of SLP-76 with LAT (Houtman et al, 2004), the data suggest that SHP-2 does not affect formation of the multi-adapter complex (LAT–Gads–SLP-76) but does selectively dephosphorylate proteins bound to that complex. The findings suggest that Vav1 and ADAP are direct substrates of SHP-2. Phosphorylation of total ADAP and total Vav1 was augmented by expression of SHP-2(C/S), and if only the pools of Vav1 or ADAP that were associated with Gads–SLP-76 were analyzed, this effect was magnified. These data support the notion of a localized and selective effect of SHP-2 on proteins in the Gads–SLP-76 complex upon TCR triggering.
Our findings are further supported by a recent report that SHP-2 regulates Vav1 phosphorylation and Rho activation in rat aortic smooth muscle cells (Wakino et al, 2004). Y174 in Vav1 contributes to maintenance of a closed or inactive conformation for Vav1, and mutation of this residue leads to a constitutively active protein with transforming activity (Turner and Billadeau, 2002). Thus, SHP-2 localization to SLP-76 may control the activation of Vav1 bound to phosphorylated SLP-76, inhibiting subsequent activation of Rho family proteins, Rho, Rac1/2 or cdc42. It is not clear why Prx II expression did not further inhibit Vav1 phosphorylation. Vav1 has multiple phosphorylation sites whose phosphorylation may be affected by other redox-sensitive elements.
In contrast to Vav1, tyrosine phosphorylation of ADAP promotes binding to the SH2 domain in SLP-76 (Peterson, 2003). There is still some controversy as to which tyrosine(s) mediates ADAP–SLP-76 association (Raab et al, 1999; Boerth et al, 2000). All the same, decreased SHP-2 activity, either through expression of SHP-2(C/S) or through ROS generation, promotes co-immunoprecipitation of ADAP with Gads–SLP-76. Conversely, augmenting SHP-2 activity through SHP-2 WT expression or quenching ROS with Prx II inhibited ADAP phosphorylation and co-precipitation with Gads–SLP-76. Thus, association of Vav1 with SLP-76 is SHP-2 independent, while ADAP–SLP-76 binding is sensitive to SHP-2 and changes in redox balance.
ADAP appears to be required for TCR-induced clustering of LFA-1 and associated changes in adhesion of antigen-stimulated T cells (Peterson, 2003). Interaction of ADAP with SLP-76 may be required for TCR-induced LFA-1 clustering (Myung et al, 2001), although this remains to be demonstrated. ADAP may regulate T-cell adhesion through the adapter SKAP55 (Wang et al, 2003), or via Ena/VASP family members, which may affect integrin clustering through effects on cytoskeletal elements (Krause et al, 2000). Because association of ADAP with these proteins is independent of ADAP phosphorylation, redox regulation of ADAP–SLP-76 interactions may be a critical regulatory step in TCR-induced adhesion. Alternatively, Fyn association with ADAP is dependent upon ADAP phosphorylation (Peterson, 2003), so Fyn recruitment to a Gads–SLP-76 complex may also be altered by changes in redox balance.
The biological role of SHP-2 in T cells has been difficult to ascertain because of the unavailability of SHP-2-deficient lymphocytes (Qu et al, 2001). Frearson and Alexander (1998) showed that expression of SHP-2(C/S) inhibited TCR-induced ERK activation, suggesting a positive role for SHP-2 in T cells. Conversely, a negative role for SHP-2 in activation of the PI3K pathway was also proposed in T cells (Yamasaki et al, 2001). The current data support a negative role for SHP-2 in regulating ‘inside-out' signals from TCR stimulation leading to LFA-1 clustering and adhesion. By extension, TCR-induced ROS generation serves to promote TCR signaling in this context.
In conclusion, the current study has identified SHP-2 as a biological target of TCR-induced ROS. A recent report showing integrin-induced association of SHP-2 with the antioxidant enzyme catalase (Yano et al, 2004) suggests that redox regulation may be a common element in signaling pathways and that mechanisms to protect SHP-2 from oxidation have evolved. Furthermore, the data suggest that redox regulation of SHP-2 modulates TCR-induced LFA-1 clustering and adhesion. This implies that endogenous ROS or proinflammatory conditions may promote antigen-induced adhesion and arrest prior to extravasation and recruitment to the periphery. In the case of T cells, the effects may be via modulation of phosphorylation and/or association of key effector proteins within the protein complex formed through the adapters Gads–SLP-76. Thus, based upon the data, we hypothesize that localized production of ROS upon TCR triggering selectively shifts the balance between kinases and phosphatases to modulate T-cell function and immune responses.
Materials and methods
Antibodies and reagents
Antibodies to Fyb/SLAP-130, c-Cbl, SHP-2, Grb2, ZAP-70, SHP-1, Vav1 and Shc were from Transduction Labs/BD-Biosciences (San Jose, CA). Antibodies to Vav1, Cbl, SHP-2, Grb2, Vav1 (pY174) and SHP-1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to LAT, SLP 76, Gads, Vav1, Gab2, phosphotyrosine (4G10) and HRP-anti-sheep IgG were from Upstate Biotechnology Inc. (Lake Placid, NY). HA-tagged proteins were immunoprecipitated using the Profound HA-Tag IP/Co-IP Kit (Pierce, Rockford, IL). Anti-human ADAP was provided by Dr G Koretzky (University of Pennsylvania), and polyclonal anti-LAT sera was provided by Dr L Samelson (NICHD, NIH). The mixture of anti-PLCγ1 antibodies for immunoblot was from Dr SG Rhee (NHLBI, NIH) and anti-human LFA-1 (TS2/4) serum was provided by Dr L Zhang (University of Maryland School of Medicine). All other chemicals were obtained from Sigma (St Louis, MO) and all cell culture supplies were from Life Technologies (Carlsbad, CA).
Plasmids
Mammalian expression vectors for green fluorescent protein (GFP; EGFP-N1) and Prx II have been described previously (Devadas et al, 2002). Mammalian expression vectors for wild-type and catalytically inactive SHP-2 (SHP-2 C459S) have been described previously (Yu et al, 2003). The cDNA from these vectors was subcloned into pcDNA3.1/His and pCMV-HA vectors (Clontech, Palo Alto, CA) to make expression vectors for (His)6-tagged- and HA-tagged SHP-2 proteins, respectively. The mammalian expression vector for SHP-1(C/S) has been described previously (Yu et al, 2005).
Cells
The human T leukemic cell line, Jurkat (ATCC, Rockville, MD), was maintained in exponential growth phase in RPMI 1640 medium supplemented with 10% FBS, antibiotics and 50 μM 2-mercaptoethanol (complete medium). SLP-76-deficient Jurkat cells (J.14) were generously provided by Dr A Weiss (University of California, San Francisco). Mouse and human T blasts were prepared essentially as previously described (Jackson et al, 2004).
PEO-iodoacetyl biotin labeling for detection of oxidized thiols
Jurkat T cells or mouse T blasts were serum-starved and subsequently stimulated with anti-CD3 as described previously (Kwon et al, 2003; Jackson et al, 2004). Alternatively, cells were treated with graded concentrations of hydrogen peroxide for 5 min. Cells were lysed for 1 h at 25°C in an anaerobic chamber with 1 ml of O2-free lysis buffer (20 mM HEPES (pH 7.0), 1% Triton X-100, 10% glycerol, 2 mM EGTA, 10 mM β-glycerophosphate, 20 mM NaF, 10 μg/ml aprotinin and leupeptin, and 1 mM AEBSF) containing 0.4 mM PEO-iodoacetyl biotin (Pierce). Biotin incorporation into immunoprecipitated proteins was detected by Western blotting with HRP-conjugated streptavidin and ECL.
‘Positive' labeling of oxidized thiols was performed as described (Kwon et al, 2004). Briefly, T cells were lysed under anaerobic conditions and all free thiols were masked with 10 mM NEM and 10 mM iodoacetamide. Reversibly oxidized thiols were re-reduced with 4 mM DTT in the anaerobic chamber and then were labeled with 1 mM PEO-iodoacetyl biotin. Biotinylated proteins were precipitated with Neutravidin-agarose and the bound proteins were separated by SDS–PAGE.
Cell adhesion assays
Adhesion assays were performed essentially as described with minor modifications (Epler et al, 2000). LumiNunc Maxi-Sorp 96-well plates were coated with 1 μg/well fibronectin (from A Belkin, University of Maryland School of Medicine). Jurkat cells were transfected with 3 μg of β-galactosidase-coding plasmid and 15 μg of an empty vector, or expression plasmids for genes encoding SHP-2(C/S), SHP-2 WT or Prx II. Following incubation for 16 h, viable cells were precoated with anti-CD3 on ice as above. Cells (50 000 cells/well) were added to wells with anti-mouse IgG (2 μg/ml), incubated at 4°C for 1 h and then in a 37°C water bath for 30 min. Plates were washed gently and bound cells were assayed for β-galactosidase using the Gal-Screen System (Applied Biosystems, Bedford, MA).
Fluorescence microscopy analysis
Paraformaldehyde-fixed cells were incubated with anti-human LFA-1 (TS2/4) serum in PBS/0.2% BSA for 1 h on ice. After staining with Texas red-conjugated goat anti-mouse IgG1 (Southern Biotech, Birmingham), cells were plated on poly-L-lysine-coated slides and were visualized by fluorescent microscopy (Nikon Eclipse E400 microscope with a Photometrics CCD camera). Image capture was analyzed using Cool SNAP v.1.1. For each experiment, about 50 randomly selected GFP-positive cells were analyzed for LFA-1 staining pattern. Cells were divided into quadrants and were considered to have clustered LFA-1 if the staining pattern showed LFA-1 polarized to one of the quadrants of the cell.
Immunoprecipitation and Western blotting
Cells were lysed in ice-cold lysis buffer containing 1% Nonidet P-40 (v/v) in 20 mM HEPES (pH 7. 4) and 130 mM NaCl. The lysis buffer contained 10% glycerol, 1 mM Na4VO3, 2 mM EDTA, 10 mM β-glycerophosphate, 20 mM NaF, 10 μg/ml aprotinin and leupeptin, and 1 mM AEBSF. After centrifugation, the cell lysates were precleared with protein G-Sepharose 4B (Amersham) at 4°C and were immunoprecipitated with the indicated antibodies and protein G-Sepharose 4B. The beads were washed three times with lysis buffer. Cell lysates were subjected to immunoblot analysis essentially as described (Jackson et al, 2004). Densitometric analysis was performed using Personal Densitometer SI (Amersham Biosciences, Piscataway, NJ).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
We thank Dr G Koretzky, Dr L Samelson, Dr SG Rhee and Dr A Weiss for generously providing reagents for this study. We also thank Dr D Leitenberg and Dr A Keegan for critical review of the manuscript. This work was supported by a Scientist Development Grant and a Grant-In-Aid to MSW from the American Heart Association.
References
- Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB (1999) Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem 274: 34543–34546 [DOI] [PubMed] [Google Scholar]
- Boerth NJ, Judd BA, Koretzky GA (2000) Functional association between SLAP-130 and SLP-76 in Jurkat T cells. J Biol Chem 275: 5143–5152 [DOI] [PubMed] [Google Scholar]
- Chiarugi P, Fiaschi T, Taddei ML, Talini D, Giannoni E, Raugei G, Ramponi G (2001) Two vicinal cysteines confer a peculiar redox regulation to low molecular weight protein tyrosine phosphatase in response to platelet-derived growth factor receptor stimulation. J Biol Chem 276: 33478–33487 [DOI] [PubMed] [Google Scholar]
- Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ III, Charrier V, Parsonage D (1999) Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 38: 15407–15416 [DOI] [PubMed] [Google Scholar]
- Dechert U, Adam M, Harder KW, Clark-Lewis I, Jirik F (1994) Characterization of protein tyrosine phosphatase SH-PTP2. Study of phosphopeptide substrates and possible regulatory role of SH2 domains. J Biol Chem 269: 5602–5611 [PubMed] [Google Scholar]
- Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS (2002) Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med 195: 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epler JA, Liu R, Chung H, Ottoson NC, Shimizu Y (2000) Regulation of beta 1 integrin-mediated adhesion by T cell receptor signaling involves ZAP-70 but differs from signaling events that regulate transcriptional activity. J Immunol 165: 4941–4949 [DOI] [PubMed] [Google Scholar]
- Finkel T (2003) Oxidant signals and oxidative stress. Curr Opin Cell Biol 15: 247–254 [DOI] [PubMed] [Google Scholar]
- Frearson JA, Alexander DR (1998) The phosphotyrosine phosphatase SHP-2 participates in a multimeric signaling complex and regulates T cell receptor (TCR) coupling to the Ras/mitogen-activated protein kinase (MAPK) pathway in Jurkat T cells. J Exp Med 187: 1417–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EK, Krawczyk C, Kong YY, Raab M, Hyduk SJ, Bouchard D, Chan VS, Kozieradzki I, Oliveira-Dos-Santos AJ, Wakeham A, Ohashi PS, Cybulsky MI, Rudd CE, Penninger JM (2001) Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap. Science 293: 2260–2263 [DOI] [PubMed] [Google Scholar]
- Groen A, Lemeer S, van der Wijk T, Overvoorde J, Heck AJ, Ostman A, Barford D, Slijper M, den Hertog J (2005) Differential oxidation of protein-tyrosine phosphatases. J Biol Chem 280: 10298–10304 [DOI] [PubMed] [Google Scholar]
- Houtman JC, Higashimoto Y, Dimasi N, Cho S, Yamaguchi H, Bowden B, Regan C, Malchiodi EL, Mariuzza R, Schuck P, Appella E, Samelson LE (2004) Binding specificity of multiprotein signaling complexes is determined by both cooperative interactions and affinity preferences. Biochemistry 43: 4170–4178 [DOI] [PubMed] [Google Scholar]
- Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS (2004) T cells express a phagocyte-type NADPH oxidase which is activated upon TcR stimulation. Nat Immunol 5: 818–827 [DOI] [PubMed] [Google Scholar]
- Jordan MS, Singer AL, Koretzky GA (2003) Adaptors as central mediators of signal transduction in immune cells. Nat Immunol 4: 110–116 [DOI] [PubMed] [Google Scholar]
- Kim JR, Yoon HW, Kwon KS, Lee SR, Rhee SG (2000) Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal Biochem 283: 214–221 [DOI] [PubMed] [Google Scholar]
- Krause M, Sechi AS, Konradt M, Monner D, Gertler FB, Wehland J (2000) Fyn-binding protein (Fyb)/SLP-76-associated protein (SLAP), Ena/vasodilator-stimulated phosphoprotein (VASP) proteins and the Arp2/3 complex link T cell receptor (TCR) signaling to the actin cytoskeleton. J Cell Biol 149: 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J, Devadas S, Williams MS (2003) T cell receptor-stimulated generation of hydrogen peroxide inhibits MEK–ERK activation and lck serine phosphorylation. Free Radic Biol Med 35: 406–417 [DOI] [PubMed] [Google Scholar]
- Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG (2004) Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci USA 101: 16419–16424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Kwon KS, Kim SR, Rhee SG (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem 273: 15366–15372 [DOI] [PubMed] [Google Scholar]
- Liu SK, Berry DM, McGlade CJ (2001) The role of Gads in hematopoietic cell signalling. Oncogene 20: 6284–6290 [DOI] [PubMed] [Google Scholar]
- Mahadev K, Zilbering A, Zhu L, Goldstein BJ (2001) Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem 276: 21938–21942 [DOI] [PubMed] [Google Scholar]
- Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK (2004) Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem 279: 37716–37725 [DOI] [PubMed] [Google Scholar]
- Meng TC, Fukada T, Tonks NK (2002) Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell 9: 387–399 [DOI] [PubMed] [Google Scholar]
- Mustelin T, Rahmouni S, Bottini N, Alonso A (2003) Role of protein tyrosine phosphatases in T cell activation. Immunol Rev 191: 139–147 [DOI] [PubMed] [Google Scholar]
- Myung PS, Derimanov GS, Jordan MS, Punt JA, Liu QH, Judd BA, Meyers EE, Sigmund CD, Freedman BD, Koretzky GA (2001) Differential requirement for SLP-76 domains in T cell development and function. Immunity 15: 1011–1026 [DOI] [PubMed] [Google Scholar]
- Neel BG, Gu H, Pao L (2003) The ‘Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28: 284–293 [DOI] [PubMed] [Google Scholar]
- Neel BG, Tonks NK (1997) Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol 9: 193–204 [DOI] [PubMed] [Google Scholar]
- O'Shea JJ, McVicar DW, Bailey TL, Burns C, Smyth MJ (1992) Activation of human peripheral blood T lymphocytes by pharmacological induction of protein-tyrosine phosphorylation. Proc Natl Acad Sci USA 89: 10306–10310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson EJ (2003) The TCR ADAPts to integrin-mediated cell adhesion. Immunol Rev 192: 113–121 [DOI] [PubMed] [Google Scholar]
- Peterson EJ, Woods ML, Dmowski SA, Derimanov G, Jordan MS, Wu JN, Myung PS, Liu QH, Pribila JT, Freedman BD, Shimizu Y, Koretzky GA (2001) Coupling of the TCR to integrin activation by Slap-130/Fyb. Science 293: 2263–2265 [DOI] [PubMed] [Google Scholar]
- Qu CK, Nguyen S, Chen J, Feng GS (2001) Requirement of Shp-2 tyrosine phosphatase in lymphoid and hematopoietic cell development. Blood 97: 911–914 [DOI] [PubMed] [Google Scholar]
- Raab M, Kang H, da Silva A, Zhu X, Rudd CE (1999) FYN–T-FYB–SLP-76 interactions define a T-cell receptor zeta/CD3-mediated tyrosine phosphorylation pathway that up-regulates interleukin 2 transcription in T-cells. J Biol Chem 274: 21170–21179 [DOI] [PubMed] [Google Scholar]
- Rhee SG, Bae YS, Lee SR, Kwon J (2000) Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE 2000: PE1. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA (2005) Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol 17: 183–189 [DOI] [PubMed] [Google Scholar]
- Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D (2003) Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423: 769–773 [DOI] [PubMed] [Google Scholar]
- Turner M, Billadeau DD (2002) VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol 2: 476–486 [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Alexander RW, Akers M, Griendling KK (1998) p38 mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem 273: 15022–15029 [DOI] [PubMed] [Google Scholar]
- van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H (2003) Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423: 773–777 [DOI] [PubMed] [Google Scholar]
- Wakino S, Hayashi K, Kanda T, Tatematsu S, Homma K, Yoshioka K, Takamatsu I, Saruta T (2004) Peroxisome proliferator-activated receptor gamma ligands inhibit Rho/Rho kinase pathway by inducing protein tyrosine phosphatase SHP-2. Circ Res 95: e45–e55 [DOI] [PubMed] [Google Scholar]
- Wang H, Moon EY, Azouz A, Wu X, Smith A, Schneider H, Hogg N, Rudd CE (2003) SKAP-55 regulates integrin adhesion and formation of T cell-APC conjugates. Nat Immunol 4: 366–374 [DOI] [PubMed] [Google Scholar]
- Wange RL (2000) LAT, the linker for activation of T cells: a bridge between T cell-specific and general signaling pathways. Sci STKE 2000: RE1. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Nishida K, Hibi M, Sakuma M, Shiina R, Takeuchi A, Ohnishi H, Hirano T, Saito T (2001) Docking protein Gab2 is phosphorylated by ZAP-70 and negatively regulates T cell receptor signaling by recruitment of inhibitory molecules. J Biol Chem 276: 45175–45183 [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Nishida K, Sakuma M, Berry D, McGlade CJ, Hirano T, Saito T (2003) Gads/Grb2-mediated association with LAT is critical for the inhibitory function of Gab2 in T cells. Mol Cell Biol 23: 2515–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S, Arroyo N, Yano N (2004) SHP2 binds catalase and acquires a hydrogen peroxide-resistant phosphatase activity via integrin-signaling. FEBS Lett 577: 327–332 [DOI] [PubMed] [Google Scholar]
- Yu WM, Hawley TS, Hawley RG, Qu CK (2003) Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling. Oncogene 22: 5995–6004 [DOI] [PubMed] [Google Scholar]
- Yu WM, Wang S, Keegan AD, Williams MS, Qu CK (2005) Abnormal Th1 cell differentiation and IFN-gamma production in T lymphocytes from motheaten viable mice mutant for Src homology 2 domain-containing protein tyrosine phosphatase-1. J Immunol 174: 1013–1019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4