

Abstract
TP63, an important epithelial developmental gene, has significant homology to p53. Unlike p53, the expression of p63 is regulated by two different promoters resulting in proteins with opposite functions: the full-length transcriptionally active TAp63 and the dominant-negative ΔNp63. We investigated the downstream mechanisms by which TAp63α elicits apoptosis. TAp63α directly transactivates the CD95 gene via the p53 binding site in the first intron resulting in upregulation of a functional CD95 death receptor. Stimulation and blocking experiments of the CD95, TNF-R and TRAIL-R death receptor systems revealed that TAp63α can trigger expression of each of these death receptors. Furthermore, our findings demonstrate a link between TAp63α and the mitochondrial apoptosis pathway. TAp63α upregulates expression of proapoptotic Bcl-2 family members like Bax and BCL2L11 and the expression of RAD9, DAP3 and APAF1. Of clinical relevance is the fact that TAp63α is induced by many chemotherapeutic drugs and that inhibiting TAp63 function leads to chemoresistance. Thus, beyond its importance in development and differentiation, we describe an important role for TAp63α in the induction of apoptosis and chemosensitivity.
Keywords: apoptosis, CD95, chemosensitivity, TAp63, TRAIL
Introduction
p63 and p73 give rise to proteins that have p53-agonistic as well as p53-antagonistic functions and new functions. One reason for this diversity in p53/p63/p73 function lies in their gene structure. p53 has a single promoter with three conserved domains, namely the transactivation domain (TA), the specific DNA-binding domain and the oligomerization domain. In contrast, p63 and p73 have two promoters, resulting in two different types of proteins with opposing functions: p53-like proteins containing the TA domain (TAp63 and TAp73), and inhibitory proteins lacking TA, called ΔNp63 and ΔNp73. These inhibitory proteins retain their DNA binding and tetramerization competence and, thus, can act as dominant-negative inhibitors of p53 and of themselves (Yang et al, 1999; Melino et al, 2002, 2003; Zaika et al, 2002; Moll and Slade, 2004). In addition, both of these genes undergo alternative splicing at the COOH-terminus producing three and nine different species of TAp63 and TAp73, respectively, named α, β, γ, δ, ɛ, etc., with α being the full length.
Hence, p63 and p73 share some p53 functions, such as induction of cell cycle arrest and apoptosis (Osada et al, 1998; Vousden, 2000; Melino et al, 2003; Moll and Slade, 2004). However, there are many functional differences between p53, p63 and p73. Studies of p53-, p63- and p73-deficient mice established that the expression of p63 and p73 is more important for mouse development than the expression of p53. Knockout p63 mice are not viable and show severe structural deficiencies, such as the complete absence of skin, lack of limbs as well as other epithelial structures (Mills et al, 1999; Yang et al, 1999, 2002) and severe craniofacial dysplasia (Celli et al, 1999; Mills et al, 1999; Yang et al, 1999). The reason for these deficiencies lies in the lack of stem cells that are required for the development and differentiation of such complex epithelial structures (Yang et al, 1999, 2002). p63 is the only gene known to be of essential relevance for the survival of epithelial stem cells (Pellegrini et al, 2001), which is diametrically opposed to the function of p53, which again is strongly linked to cell cycle arrest and cell death.
There is mounting evidence that p63 and p73 play an important role in human cancer (Casciano et al, 2002; Flores et al, 2002; Melino et al, 2003, 2004; Moll and Slade, 2004; Westfall and Pietenpol, 2004; Wu et al, 2005), although their precise roles in tumorigenesis remain to be clarified.
There is also intense debate on whether and how p63 and p73 interact with p53 in apoptosis and tumor suppression (Benchimol, 2004). An example of cooperativity among the three p53 family members has been reported in E1A-expressing mouse embryo fibroblasts and in primary neuronal cultures (Flores et al, 2002). However, results of a recent study indicate that at least in thymocytes, p53-dependent apoptosis occurs independently of p63 and p73 (Senoo et al, 2004).
To further define the interactions between the three p53 family members in human cancer, it is essential to investigate common/distinct targets in the apoptosis pathways triggered by p63, p73 and p53.
The aim of our study has been to provide insight into the molecular mechanisms accounting for the role of TAp63 in cancer and its involvement in cell death induced by chemotherapeutic drugs. We have analyzed the downstream mechanisms of TAp63α-induced apoptosis in different cellular systems. Here we report that TAp63α, like p53, activates major apoptosis pathways by triggering signaling via death receptors and mitochondria and thus sensitizes cancer cells toward chemotherapy. Of note, we found that endogenous TAp63α is induced by many chemotherapeutic agents and that blocking TAp63α function confers chemoresistance.
Results
TAp63α-mediated apoptosis involves activation of caspases
Adenoviral transfer of the TAp63α gene into Hep3B cells induced apoptosis in a dose- and time-dependent manner (Figure 1).
Figure 1.
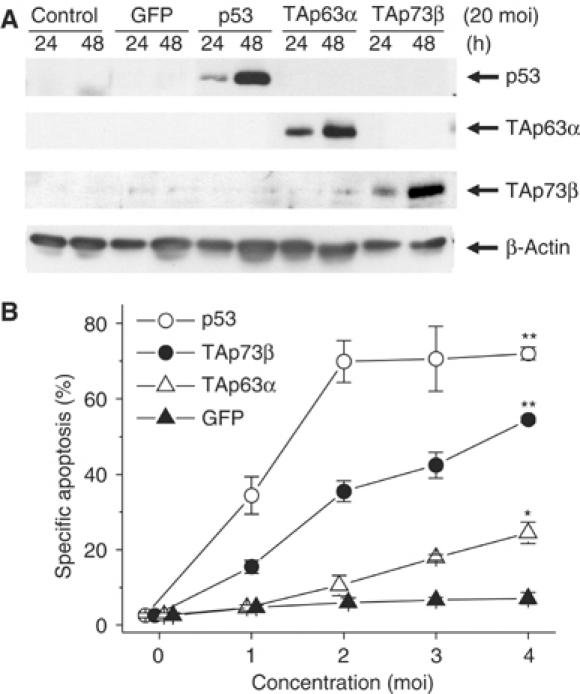
TAp63α induces apoptosis of hepatoma cells. (A) Comparison of protein expression following rAd-p53, rAd-TAp63α or rAd-TAp73β expression in Hep3B cells. (B) Like rAd-p53 and rAd-TAp73β, rAd-TAp63α induces dose-dependent apoptosis of Hep3B cells within 72 h. Propidium iodide staining (Nicoletti et al, 1991) and FACScan® analysis were performed. Three independent experiments were performed, and a representative result is shown, mean±s.d., n=3. *P<0.01, **P<0.001, multivariate analysis of variance (MANOVA), between-subject effect compared to rAd-GFP.
TAp63α-mediated apoptosis was strongly inhibited by the caspase inhibitors ZVAD-FMK, DEVD-FMK, Z-IETD-FMK and Z-LEHD-FMK (Figure 2). This confirms the involvement of caspases in TAp63α-mediated apoptosis.
Figure 2.
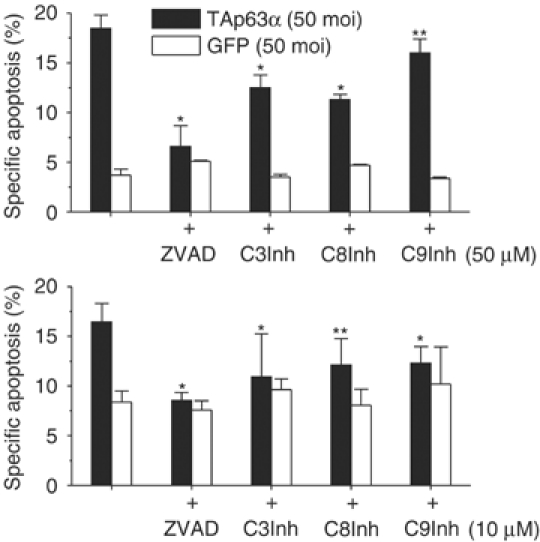
Apoptosis induced by TAp63α involves activation of caspases. FACScan® analysis of propidium iodide-stained nuclei (Nicoletti et al, 1991) of Hep3B cells, which were either transduced by rAd-GFP or rAd-TAp63α for 72 h. TAp63α-dependent apoptosis was blocked by the broad-spectrum caspase inhibitor ZVAD-FMK and by DEVD-FMK (C3Inh), Z-IETD-FMK (C8Inh) and Z-LEHD-FMK (C9Inh). Data obtained in two separate experiments were averaged. Presented is mean±s.d., n=6. *P<0.001, **P<0.05; Wilcoxon's test compared to rAd-TAp63α.
Microarray analysis of TAp63α-mediated apoptosis
Following adenoviral TAp63α expression in Hep3B cells, the genes encoding for the death receptors CD95, TNF-R1, TRAIL-R1 and TRAIL-R2 were found to be upregulated (Table I). Induction of TNF, TRAF (TNF receptor-associated factor)-interacting protein (TRIP) and DAP3 (death-associated protein-3) provides further evidence for the involvement of receptor-mediated signaling.
Table 1.
List of selected TAp63α-regulated genes in Hep3B cells, according to the MIAME criteria (Brazma et al, 2001; The Tumor Analysis Best Practices Working Group, 2004)
| Feature |
Reporter |
Composite sequence |
Fold activation |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coordinates on arrays |
Reporter ID (RZPD defined) | Biosequence type | Clone ID | GenBank access. no. | Reporter usage | Control type | Comp. ID | Designation | Related gene symbol | Database entry | 24 h | 48 h | 72 h | ||||
| X-Pos | Y-Pos | Spot plate | Row | Column | |||||||||||||
| Death receptor-mediated apoptosis | |||||||||||||||||
| 34 | 27 | 1 | A | 13 | 1048421 | cDNA clone | IMAGp998E031157 | X65019 | Exp | — | 005-B03 | Caspase-1 | CASP1 | LocusID834 | +4.08 | ||
| 43 | 12 | 1 | M | 10 | 49729 | cDNA clone | IMAGp998G22281 | U13737 | Exp | — | 002-G05 | Caspase-3 | CASP3 | LocusID836 | +5.33 | ||
| 1 | 15 | 1 | L | 24 | 302539 | cDNA clone | IMAGp998C20678 | U28014 | Exp | — | 004-F12 | Caspase-4 | CASP4 | LocusID837 | +1.61 | +1.46 | |
| 14 | 18 | 3 | K | 20 | 341763 | cDNA clone | IMAGp998F04780 | U28015 | Exp | — | 006-F10 | Caspase-5 | CASP5 | LocusID838 | +1.37 | ||
| 47 | 15 | 3 | L | 9 | 338776 | cDNA clone | IMAGp998I17772 | X98175 | Exp | — | 009-D12 | Caspase-8 | CASP8 | LocusID841 | +1.12 | ||
| 37 | 12 | 1 | M | 12 | 121693 | cDNA clone | IMAGp998D14114 | U60521 | Exp | — | 002-G06 | Caspase-9 | CASP9 | LocusID842 | +3.80 | +3.04 | |
| 42 | 20 | 2 | J | 11 | 1693595 | cDNA clone | IMAGp998L124300 | M10988 | Exp | — | 007-E06 | Tumor necro- sis factor | TNF | LocusID7124 | +3.00 | +0.38 | |
| 6 | 38 | 2 | D | 23 | 2521744 | cDNA clone | IMAGp998F176279 | M33294 | Exp | — | 007-B12 | Tumor necro- sis factor receptor superfamily- member 1A | TNFRSF1A/TNF-R1 | LocusID7132 | +2.07 | +2.25 | |
| 18 | 26 | 2 | H | 19 | 309671 | cDNA clone | IMAGp998L24696 | M67454 | Exp | — | 007-D10 | Tumor necro- sis factor receptor superfamily-member 6 | TNFRSF6/CD95 | LocusID355 | +2.43 | +1.83 | +1.54 |
| 60 | 26 | 2 | H | 5 | 526788 | cDNA clone | IMAGp998C131262 | U90875 | Exp | — | 007-D03 | Tumor necro- sis factor receptor superfamily- member 10A | TNFRSF10A/TRAIL-R1 | LocusID8797 | +2.81 | +0.25 | |
| 72 | 14 | 2 | L | 1 | 3948060 | cDNA clone | IMAGp958L13810 | AF016268 | Exp | — | 007-F01 | Tumor necro- sis factor receptor superfamily-member 10b | TNFRSF10B/TRAIL-R2 | LocusID8795 | +3.16 | ||
| 19 | 12 | 1 | M | 18 | 27476 | cDNA clone | IMAGp998M21142 | U77845 | Exp | — | 002-G09 | TRAF (TNF receptor- associated factor) interacting protein | TRIP | LocusID10293 | +4.65 | ||
| Mitochondrial genes | |||||||||||||||||
| 37 | 18 | 1 | K | 12 | 293401 | cDNA clone | IMAGp998G02654 | AF013263 | Exp | — | 002-F06 | Apoptotic protease- activating factor | APAF1 | LocusID317 | +1.64 | +3.13 | |
| 43 | 30 | 1 | G | 10 | 300194 | cDNA clone | IMAGp998B03672 | AF032458 | Exp | — | 002-D05 | BCL2-like protein 11 | BCL2L11 | LocusID10018 | +1.71 | +10.41 | |
| 54 | 2 | 2 | P | 7 | 812290 | cDNA clone | IMAGp998K112005 | U18321 | Exp | — | 007-H04 | Death- associated protein-3 | DAP3 | LocusID7818 | +2.80 | ||
| 35 | 3 | 3 | p | 13 | 713617 | cDNA clone | IMAGp998L021748 | U53174 | Exp | — | 011-H07 | RAD9 (S. pombe) homolog (nuclear- and mito- chondrial- localized apoptosis inducer) | RAD9 | LocusID5883 | +1.71 | ||
| Control clones | |||||||||||||||||
| 32 | 9 | 3 | N | 14 | 148920 | cDNA clone | IMAGpC01228 | H13168 | Control | Pos | — | Hemoglobin, alpha 1 | HBA1 | LocusID3039 | Nan | Nan | Nan |
| 20 | 33 | 3 | F | 18 | 203166 | cDNA clone | IMAGpG07392 | R44290 | Control | Pos | — | Actin, beta | actin1 | LocusID51164 | Nan | Nan | Nan |
| 44 | 9 | 3 | N | 10 | 201154 | cDNA clone | IMAGpC11387 | R17745 | Control | Pos | — | Transferrin | TF | LocusID7018 | Nan | Nan | Nan |
| Shown is the fold activation of TAp63α-dependent genes relative to GFP (=1) as evaluated by SAM (six independent hybridizations). Nan=not a number. http://www.ebi.ac.uk/arrayexpress/; accession number E-MEXP-199. | |||||||||||||||||
Caspase-1, -3, -4, -5, -8 and -9 were induced by rAd-TAp63α (Table I and Figures 2, 4B and C). Furthermore, we identified the genes encoding the proapoptotic Bcl-2 family member BCL2L11 and the genes encoding RAD9 and APAF1 as targets for transcriptional upregulation by TAp63α (Table I). Thus, microarray analyses provide evidence that TAp63α stimulates both, genes that regulate the extrinsic apoptosis pathways initiated by ligation of death receptors and genes that regulate the intrinsic/mitochondrial apoptosis pathway.
Figure 4.
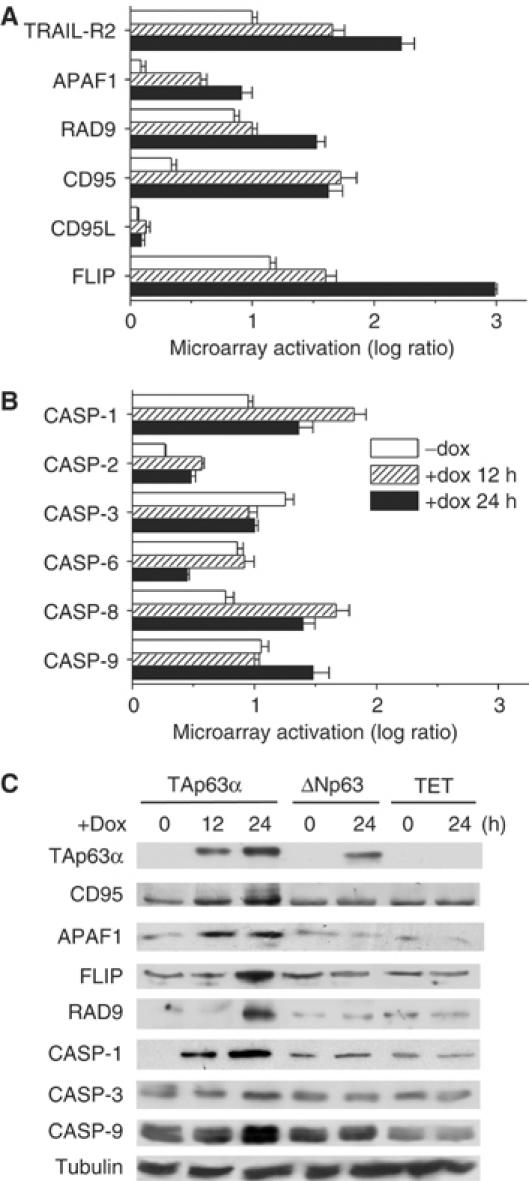
TAp63α induces the expression of proapoptotic genes in Tet-On Saos2 cells. (A, B) TAp63α induces upregulation of death receptors and caspases. Cells were analyzed by microarray technique. The data presented are mean±s.d., n=3. http://www.ebi.ac.uk/arrayexpress/; accession number E-MEXP-199. (C) Validation of targets identified in panels A and B by Western blot. Cells overexpressing ΔNp63α and dox-treated control cells (TET) were included to underline the TAp63α-specific upregulation of the target genes. Panel C shows representative results of two independent experiments.
Induction of apoptosis in TAp63α-inducible p53-negative cells
In order to generalize these results, we performed similar experiments using a completely different cellular system. We generated Tet-On-inducible osteosarcoma cells overexpressing TAp63α. Saos2 cells are p53 negative, and show no detectable levels of p63 and p73 at either mRNA or protein level. The expression of TAp63α protein was induced in a time-dependent manner following treatment with 2.5 μg/ml doxycycline (dox; Figure 3A). The expression of TAp63α was functional, as shown by the ability to induce expression of p21. Following the induction of p21, the cells showed a G1 cell cycle arrest, with a significant reduction in S and G2/M phases, as indicated in Figure 3B. Consistent with the induction of TAp63α, Saos2 cells underwent apoptosis, as measured by flow cytometric analysis of sub-G1 events (Figure 3C).
Figure 3.

TAp63α induces apoptosis in Tet-On Saos2 cells. (A) Different clones were selected showing the ability to induce TAp63α under the control of exogenous dox. At 24 h, both p63 (HA tag) and p21 were upregulated in all Saos2 clones. (B) G1 cell cycle arrest and reduction of the relative portion of S and G2/M phases induced by TAp63α overexpression (dead cells were gated out). (C) Induction of apoptosis (sub-G1 hypodiploid peak by flow cytometry in propidium iodide-stained cells) by TAp63α. (D) Subcellular localization of TAp63α (HA tag) and p21. Both proteins colocalize in the nucleus. The expression of p21 is related to that of p63, both in dox-induced cells (middle row) and in noninduced cells (lower row), showing some degree of leakiness of the promoter used. (E) Subcellular localization of TAp63α (HA tag) and Ki67. The data presented in panel C are mean±s.d., n=3. Panels show representative results of four to six independent experiments.
TAp63α is localized only in the nucleus, and is strictly correlated to the expression of p21 both in induced and noninduced leaky cells (Figure 3D), and it is independent of the expression of Ki67 (Figure 3E).
We performed microarray analyses in this second model of apoptosis. Similarly to the results obtained in Hep3B cells, TAp63α was able to induce expression of several proapoptotic genes (Figure 4A and B) and their corresponding proteins: CD95, APAF1, RAD9, caspase-1, caspase-3 and caspase-9 (Figure 4C).
TAp63α is a transcriptional activator of the CD95 gene
We have previously shown that the CD95 gene is a transcriptional target of wild-type (wt) p53, whose expression is induced through binding of wt p53 to a regulatory region within its first intron (Müller et al, 1997, 1998). Based on these observations and on our microarray data, we investigated the possibility that TAp63α transactivates the CD95 gene (Figure 5A). This was carried out by transient transfection assays, employing a plasmid in which the expression of a luciferase reporter gene is driven by regulatory DNA elements from the CD95 gene. These regulatory sequences include the physiological sequence of the CD95 gene, the CD95 promoter, exon 1 and a region from intron 1 encompassing the p53-responsive element (p1142CD95-luc). Figure 5C shows that cotransfection of TAp63α, like cotransfection of p53 (Müller et al, 1997, 1998), significantly increased p1142CD95-luc activity. The TAp63α-dependent transactivation strictly depends on the intronic p53 binding site of the CD95 gene, as it is totally abrogated when using a CD95 luciferase construct with a mutated intronic p53 binding site (Figure 5B and C). This strongly argues in favor of the conclusion that the CD95 gene is a direct transcriptional target for TAp63α. A direct evidence has been found by chromatin immunoprecipitation (ChIP). Figure 5D shows the ability of p63 protein to bind directly the p53/p63 binding site in the first intron of the CD95 gene.
Figure 5.
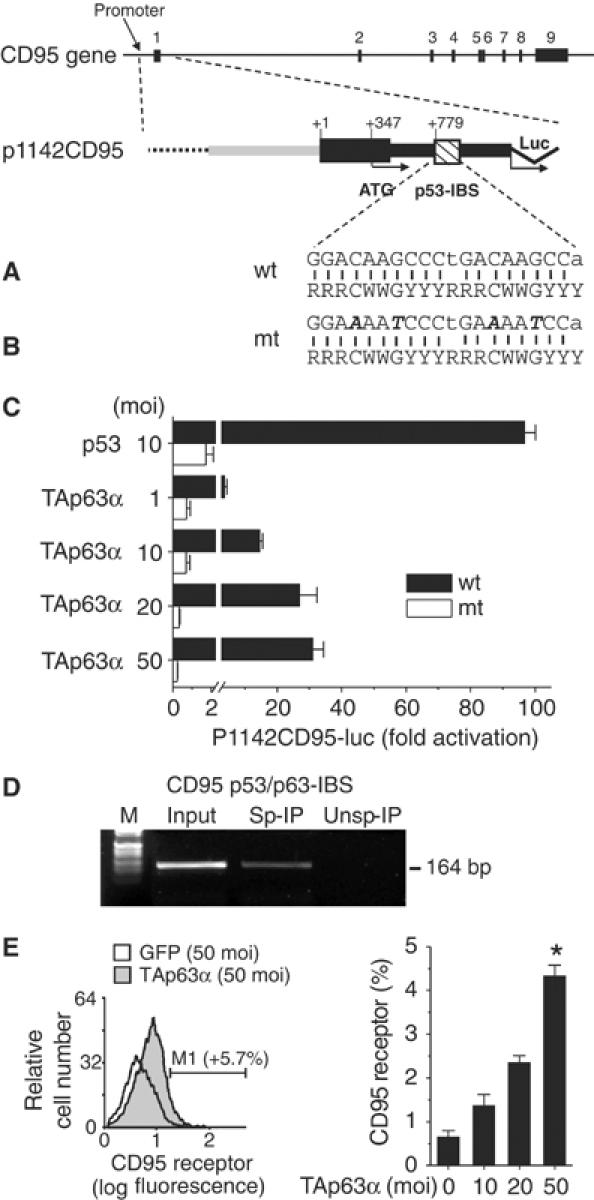
TAp63α directly transactivates the CD95 gene via binding to the intronic p53 binding site and induces upregulation of the CD95 receptor. Map of the human CD95 gene. Exons 1–9 are numbered according to Wada et al (1995). The striped box indicates the intronic p53 binding site (p53-IBS) in the first intron of the CD95 gene (Müller et al, 1998). (A) Wild-type (wt) sequence of the CD95 p53-IBS (Müller et al, 1998). This sequence (top line) is compared with the consensus p53 binding site (bottom line) (El-Deiry et al, 1992). Missing vertical bars indicate deviations in the wt sequence from the consensus. R=purine, Y=pyrimidine, W=A or T. (B) In plasmid mt p1142CD95-luc, essential nucleotides for the binding of wt p53 protein have been mutated in the p53-IBS. (C) TAp63α, like p53, transactivated the CD95 gene. Mutation of the p53-IBS (mt p1142CD95-luc) completely abrogated transactivation by TAp63α. Hep3B cells were transfected with 1 μg of plasmid p1142CD95-luc together with 10 moi rAd-p53 or of rAd-p63 (1, 10, 20 or 50 moi). Presented is the fold p53- or TAp63α-dependent activation of the p1142CD95-luc reporter plasmid, calculated relative to the value obtained with the same adenovirus in the absence of p53 or TAp63α. Four independent experiments were performed, and a representative result is shown (mean±s.d., n=3). (D) ChIP of p63 on the CD95 gene. Crosslinked chromatin was extracted from dox-induced Saos2 cells and subjected to immunoprecipitation with specific (Sp-IP) and nonspecific (Unsp-IP) antibodies. DNA recovered from immunocomplexes and input material (Input) was PCR amplified using primers designed across the p53/p63 responsive element in intron 1 of the CD95 gene. A representative result of two independent experiments is shown (M: molecular weight marker). (E) Transfer of rAd-TAp63α (72 h) restores the ability of Hep3B cells to increase the CD95 receptor. Assays were performed in triplicate, and six independent experiments were performed; a representative result is shown (mean±s.d., n=3). *P<0.0001, ANOVA for effect of TAp63α-dependent upregulation of the CD95 receptor.
TAp63α induces upregulation of the CD95 receptor and sensitizes toward CD95-mediated apoptosis
Importantly, FACS analysis revealed that overexpression of TAp63α also led to an increase in the amount of CD95 death receptors displayed on the cell surface (dose-dependent, P<0.001; Figure 5E). Next we tested whether the induction of CD95 resulted in a sensitization toward CD95-mediated apoptosis. In fact, the agonistic antibody anti-APO-1 triggered cell death in TAp63α-overexpressing Hep3B cells (Figure 6). These data indicate that the CD95 death receptor induced by TAp63α is functional.
Figure 6.
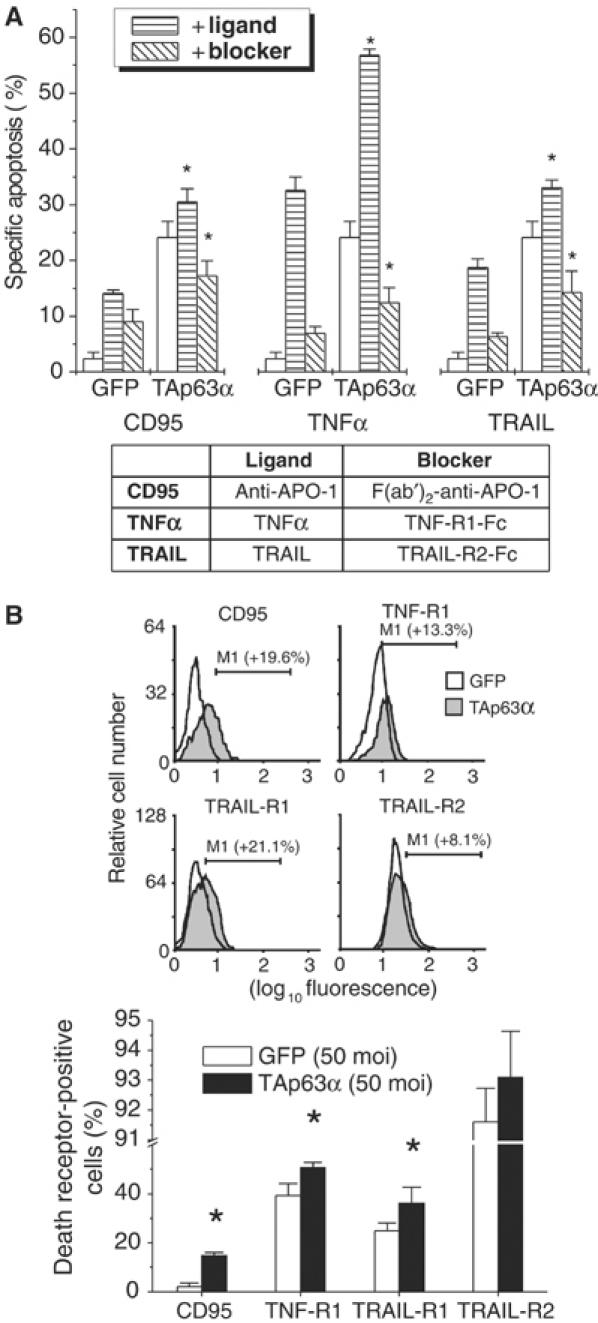
TAp63α sensitizes hepatoma cells toward death receptor-mediated apoptosis. (A) Addition of the specific ligands following adenoviral expression of TAp63α sensitized Hep3B cells toward CD95-, TNF-R-, and TRAIL-R-mediated apoptosis. Following adenoviral transfer of TAp63α (48 h), specific ligands were added for further 24 h. This led to a significant increase of apoptosis mediated by the CD95, TNF and TRAIL death receptor systems. Addition of the specific blockers of these death receptors significantly reduced apoptosis triggered by TAp63α. Shown is one representative out of three experiments performed. Presented is mean±s.d., n=3. *P<0.05, Wilcoxon's test compared to TAp63α. (B) Transfer of rAd-TAp63α (72 h) increases cell surface expression of CD95, TNF-R1, TRAIL-R1 and TRAIL-R2. Assays were performed in triplicate, and three independent experiments were performed; a representative result is shown (mean±s.d., n=3). *P<0.05, Wilcoxon's test compared to GFP.
TAp63α sensitizes hepatoma cells toward TNF-R- and TRAIL-R-mediated apoptosis
To further dissect the death receptor pathways involved in the mediation of TAp63α-induced apoptosis, we performed stimulation and blocking experiments of CD95, TNF-R and TRAIL-R. As shown for the CD95 death receptor system, addition of the specific ligands (TNFα or human LZ-TRAIL) led to a further increase of TAp63α-mediated apoptosis.
Addition of the specific blockers of these death receptors, F(ab′)2-anti-APO-1, human TNF-R1-Fc and TRAIL-R2-Fc, significantly reduced apoptosis triggered by rAd-TAp63α but not by rAd-GFP (green fluorescent protein) transfer (Figure 6A). Flow cytometry analysis confirmed upregulation of CD95, TNF-R1, TRAIL-R1 and TRAIL-R2 following rAd-TAp63α transfer (Figure 6B). Thus, it is evident that TAp63α-induced apoptosis is not solely mediated by the CD95 system, but by a set of death receptors including the TNF and TRAIL receptor system.
TAp63α induces the mitochondrial apoptosis pathway
In order to further characterize the molecular mechanisms of TAp63α-mediated apoptosis, we investigated the influence of TAp63α on mitochondrial apoptosis. FACScan® analysis revealed an alteration of the mitochondrial membrane potential following adenoviral TAp63α transfer in Hep3B cells (Figure 7A). To investigate the possible involvement of Bax in TAp63α-induced apoptosis, we performed transient transfection assays using a reporter plasmid (Figure 7B), containing the full-length Bax promoter placed upstream of a luciferase cDNA. Figure 7C shows that cotransfection of TAp63α significantly increased Bax promoter activity. Western blot analysis confirmed induction of endogenous Bax protein following rAd-TAp63α transfer (Figure 7D). In addition, as shown above by microarray and immunoblot analyses, BCL2L11, APAF1, caspase-9, RAD9 and DAP3 were induced. Thus, TAp63α contributes to apoptosis by inducing the expression of several proapoptotic proteins acting on mitochondria.
Figure 7.
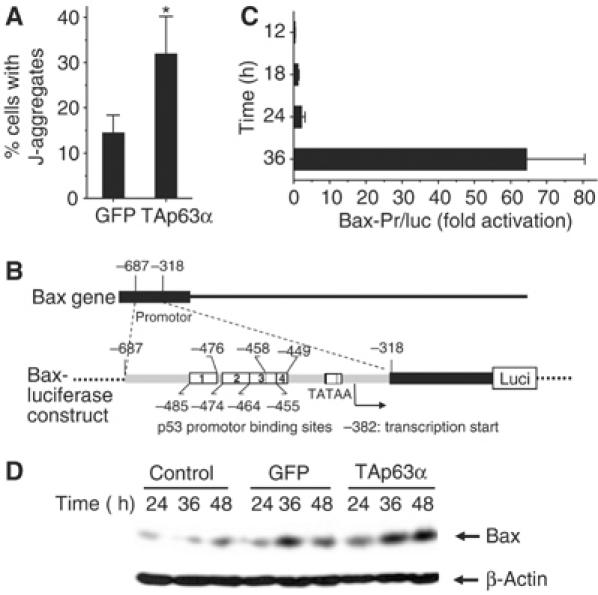
TAp63α engages mitochondrial apoptosis pathways. (A) FACScan® analysis of Hep3B cells following JC-1 staining (Zuliani et al, 2003) showed alteration of the mitochondrial membrane potential and a significant increase in J-aggregates due to TAp63α (72 h). Data obtained in two separate experiments, each performed in triplicate, were averaged. Presented is mean±s.d., n=6. *P<0.005, Wilcoxon's test. (B) Overview of the Bax-luciferase gene construct used in panel C, containing a luciferase construct of the Bax promoter with its four p53 promoter binding sites (white boxes) (Miyashita and Reed, 1995). (C) TAp63α transactivates the Bax gene. Hep3B cells were transfected with 1 μg of the reporter plasmid presented in panel B together with 100 ng of a TAp63α plasmid. Shown is the fold TAp63α-dependent activation of the Bax reporter plasmid, calculated relative to the value obtained with the same reporter in the absence of TAp63α. Shown is one representative out of three experiments performed. Presented is mean±s.d., n=3. (D) Immunoblot of endogenous Bax expression following rAd-TAp63α transfer.
TAp63α sensitizes hepatoma cells toward chemotherapy
p53 is frequently mutated in hepatocellular carcinoma. This has been implicated in resistance toward anticancer therapy. We investigated whether TAp63α could restore response of hepatoma cells toward chemotherapeutic drugs. Figure 8A shows that rAd-TAp63α enhances cell killing by bleomycin. Transfection assays revealed that the additive action of anticancer drugs and TAp63α is partially due to a cooperative effect on the transactivation of the CD95 gene (Figure 8B).
Figure 8.
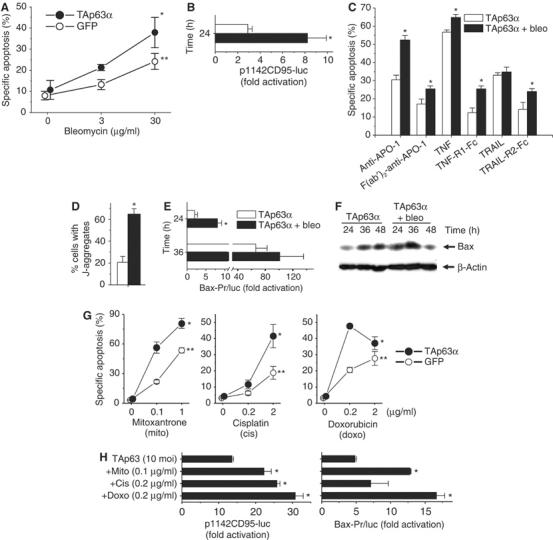
TAp63α sensitizes hepatoma cells toward chemotherapy. (A) TAp63α and chemotherapeutic agents synergize in the induction of apoptosis in Hep3B cells. Cells were treated either with bleomycin alone or in combination with rAd-TAp63α (10 moi) or rAd-GFP (10 moi) for 72 h. One out of four experiments performed in triplicate is shown. Mean±s.d., n=3. *P<0.01, ANOVA for between-subject effect (TAp63α versus GFP); **P<0.001, ANOVA for dose dependence of the effect of bleomycin on specific apoptosis. (B) The synergistic action of rAd-TAp63α and bleomycin is in part due to a cooperative effect on the transactivation of the CD95 gene. Presented is the fold rAd-TAp63α (10 moi)-dependent activation of the p1142CD95-luc reporter plasmid (1 μg), calculated relative to the value obtained with the same adenovirus in the absence of TAp63α or bleomycin. Assays were performed in triplicate in three independent experiments. One representative experiment is shown (mean±s.d., n=3). *P<0.05, Wilcoxon's test. (C) A set of death receptors is involved in the augmentation of TAp63α-mediated apoptosis by chemotherapeutic drugs. Blocking of the CD95, TNF or TRAIL receptor system cannot prevent the further enhancement of TAp63α-mediated apoptosis by chemotherapeutic drugs. Presented is one out of three independent experiments performed. Shown is mean±s.d., n=3. *P<0.05, Wilcoxon's test compared to the corresponding left column without bleomycin. (D) Following rAd-TAp63α transfer, mitochondrial membrane potential was significantly increased by addition of bleomycin (JC-1 staining, Hep3B cells, 72 h). Data obtained in two separate experiments, each performed in triplicate, were averaged. Presented is mean±s.d., n=6. *P<0.005, Wilcoxon's test. (E) The cooperative action of TAp63α transfer and chemotherapy leads to enhancement of Bax gene transactivation. Combined treatment with 3 μg/ml bleomycin led to a further increase of the transactivation of the Bax promoter. Treatment conditions are as detailed in Figure 7C. Shown is one representative out of three experiments performed, mean±s.d., n=3. *P<0.005, ANOVA, for time=24 h. (F) Western blot analysis confirms the cooperative induction of endogenous Bax protein by rAd-TAp63α and bleomycin. (G) TAp63α synergizes with different chemotherapeutic drugs in the induction of apoptosis. Cells were treated either with DNA-damaging agents alone or in combination with rAd-TAp63α (10 moi) or rAd-GFP (10 moi) for 72 h. One out of three experiments performed in triplicate is shown. Mean±s.d., n=3. *P<0.01, ANOVA for between-subject effect (TAp63α versus GFP), **P<0.001, ANOVA for dose -dependence of the effect of chemotherapy on specific apoptosis. (H) TAp63α cooperates with a variety of chemotherapeutic agents in the transactivation of CD95 and Bax genes. Treatment conditions are as detailed in Figures 5C and 7C. Shown is one representative out of three experiments performed, mean±s.d., n=3. *P<0.05, Wilcoxon's test.
As TAp63α in fact triggers a set of different death receptors, we investigated if blocking of the CD95, TNF or TRAIL receptor system can abolish the further enhancement of TAp63α-mediated apoptosis by chemotherapeutic drugs. Blocking of a single death receptor system does not prevent the augmentation of TAp63α-induced apoptosis by anticancer drugs (Figure 8C). Thus, the additive effect of chemotherapeutic drugs on TAp63α-induced apoptosis is due, at least in part, to a concomitant stimulation of a set of different death receptors.
FACScan® analysis showed that the change of the mitochondrial membrane potential caused by TAp63α was significantly increased by addition of bleomycin (Figure 8D). The cooperative action of TAp63α and bleomycin on mitochondrial apoptosis is further evidenced by the fact that combined treatment led to a significant increase of the transactivation of the Bax gene (Figure 8E). This was validated on protein level; combined treatment led to a further increase of endogenous Bax protein (Figure 8F).
Mitoxantrone, cisplatin and doxorubicin also showed cooperativity with TAp63α in induction of apoptosis (Figure 8G) and transactivation of CD95 and Bax genes (Figure 8H). Thus, the findings with bleomycin could be generalized using different chemotherapeutic drugs.
TAp63α is a determinant of chemotherapeutic efficacy
Of note, endogenous TAp63α is induced by many chemotherapeutic drugs (Figure 9A). We next asked if physiological levels of TAp63α contribute to chemotherapy-induced apoptosis. To define more clearly the role of TAp63α for chemotherapeutic efficacy, an siRNA approach was used to inhibit the accumulation of TAp63α in tumor cells following DNA damage (Figure 9B). p63 siRNA conferred protection against a variety of chemotherapeutic drugs. This is due to inhibition of specific apoptosis (Figure 9C) and due to inhibition of caspase-3, -8 and -9 activation (Figure 9D). Furthermore, blocking endogenous TAp63 impaired mitochondrial activity (Figure 9E). These findings indicate that downregulation of endogenous TAp63α leads to chemoresistance of tumor cells.
Figure 9.
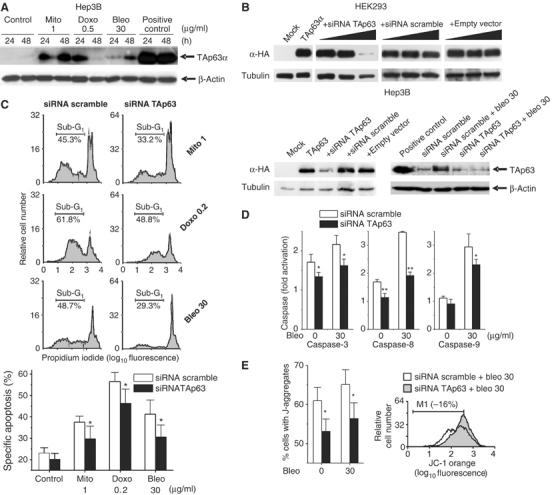
Blocking TAp63 function confers chemoresistance. (A) Endogenous TAp63α is induced by chemotherapy. Anti-p63α and anti-β-actin immunoblot of Hep3B cells treated with different chemotherapeutic drugs. Saos2 cells transduced by rAd-TAp63α were used as positive control. (B) Anti-p63 and anti-tubulin immunoblots of HEK293 and Hep3B cells cotransfected with pcDNA-TAp63α and siRNA directed against TAp63 at 1:1, 1:2 and 1:4 ratios. In parallel, cells were cotransfected with annealing buffer only (mock), scrambled TAp63 siRNA or vector alone. Transfection of siRNA TAp63 but not siRNA scramble led to decreased TAp63α in response to DNA damage induced by bleomycin treatment (lower right panel). Positive control is as detailed in panel A. (C) Blocking TAp63 function with siRNA inhibits chemotherapy-induced apoptosis. Hep3B cells transfected with the indicated siRNAs were treated with different anticancer drugs for 72 h. FACScan® analysis of propidium iodide-stained nuclei was performed. Data obtained in two separate experiments were averaged. Shown is mean±s.d., n=6. *P<0.05, Wilcoxon's test compared to the corresponding left column (siRNA scramble). (D) Hep3B cells transfected with the indicated siRNAs were treated with bleomycin, and caspase-3, -8 or -9 activation assays were performed 48 h later. Fold activation represents the caspase-3, -8 or -9 activity measured in cells transfected with siRNA scramble or siRNA TAp63 compared to untreated cells. Data obtained in two separate experiments were averaged. Shown is mean±s.d., n=6. *P<0.05, **P<0.005, Wilcoxon's test. (E) Blocking TAp63 diminishes alteration of the mitochondrial membrane potential. Hep3B cells transfected with the indicated siRNAs were treated with bleomycin and mitochondrial membrane potential was assessed by flow cytometry (JC-1 staining, 72 h). Data obtained in two separate experiments were averaged. Shown is mean±s.d., n=6. *P<0.05, Wilcoxon's test. (C–E) Results similar to transfection with scrambled siRNA were seen with mock transfection or transfection with the empty pSuperRetro vector alone (data not shown).
Discussion
Recent studies indicate that the differential expression of TAp63 and TAp73 isoforms, together with the interaction between TAp63 and TAp73 isoforms themselves, and with p53, might be crucial in controlling p53 function and programmed cell death (Flores et al, 2002; Zaika et al, 2002; Melino et al, 2004; Westfall and Pietenpol, 2004).
Data obtained in the present study allow us to propose a model for the regulation of apoptosis and chemosensitivity of cancer cells by TAp63α. We found that p63 can regulate genes with diverse roles in apoptosis in distinct cellular models (Figure 10 and Table I).
Figure 10.
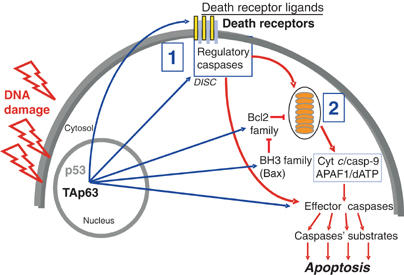
Model of TAp63α-induced apoptosis. Upon DNA damage, TAp63α activates multiple apoptosis pathways. In particular, both the death receptor (pathway 1) and the mitochondrial (pathway 2) cascades are induced by TAp63α.
We show that TAp63α is involved in the activation of both, the extrinsic/death receptor-mediated apoptosis pathway and the intrinsic/mitochondria-mediated apoptosis pathway (Figure 10).
Microarray, immunoblot and FACS analyses, stimulation and blocking experiments of the CD95, TNF-R and TRAIL-R death receptor systems revealed that TAp63α can trigger each of these death receptors and consequently sensitize tumor cells toward apoptosis. Additional evidence for the involvement of receptor-mediated signaling in TAp63α-induced apoptosis was provided by the fact that TRIP and DAP3 were found to be upregulated. TRIP acts as a receptor-proximal regulator that influences signals responsible for cell activation/proliferation and cell death induced by members of the TNF-R superfamily (Lee and Choi, 1997). DAP3 has been shown to mediate TNFα-, CD95L- and TRAIL-induced apoptosis. DAP3 also associates with the pro-caspase-8-binding adapter protein Fas-associated death domain (FADD) and links FADD to the TRAIL receptors –TRAIL-R1 and –TRAIL-R2 (Kissil et al, 1999; Miyazaki and Reed, 2001).
Recently, DAP3 was reported to localize to mitochondria (Mukamel and Kimchi, 2004). In addition to its effect on death receptor-mediated apoptosis, TAp63α contributes to apoptosis by inducing the expression of several proapoptotic proteins acting on mitochondria. We identified Bax, BCL2L11, APAF1, caspase-9, RAD9 and DAP3 to be upregulated in tumor cells expressing TAp63α.
We have previously shown that the CD95 gene is a transcriptional target of wt p53. To determine if TAp63α directly binds the CD95 endogenous regulatory region in vivo, we performed ChIP assays. We found that p63 protein binds the intron 1 p53/p63 site, the key p63-responsive element in the CD95 regulatory region. Taken together, our results implicate CD95 as a direct p63 target gene. In addition, similarly to p53, TAp63α was able to transactivate the Bax promoter in hepatoma cells.
Presumably, the genes, which we have shown to be induced by TAp63α (Table I), are shared transcriptional targets of both p53 and p63. Thus, we performed microarray analyses to examine if p53 also induces these genes (Supplementary Table II). Indeed, the genes encoding for CD95, TRAIL-R1 and -R2, TRIP, TNF, caspase-1, -3, -4, -5 and -8, BCL2L11 and DAP3 were found to be upregulated by rAd-p53 in Hep3B cells. Among these, the genes encoding for CD95 (Müller et al, 1998), TRAIL-R1 (Liu et al, 2004), TRAIL-R2 (Wu et al, 1997), caspase-1 (Gupta et al, 2001) and caspase-8 (Liedtke et al, 2003) have previously been described to contain p53 response elements. To identify formerly unrecognized p53 response elements, we performed data base analyses, which identified at least one putative p53 response element in each of the genes (Table I) shown to be upregulated by TAp63α (Supplementary Tables III and IV; Supplementary Figure 11). To our knowledge, none of these genes has been described to be upregulated by TAp63α, so far.
In conclusion, TAp63α, like p53 (Vousden, 2000), engages multiple distinct apoptosis pathways in the cell stimulating death receptor signaling, activation of caspases and apoptosis emanating from mitochondria.
Furthermore, our results show a relevant role for TAp63α in chemosensitivity of hepatoma cells. Combination of TAp63α gene transfer and chemotherapeutic treatment led to a synergistic effect regarding the induction of apoptosis. TAp63α activates both, death receptor- and mitochondria–mediated apoptosis pathways, and both mechanisms are clearly reinforced by concomitant treatment with chemotherapeutic drugs. Of clinical importance, we found that endogenous TAp63α is induced by a variety of chemotherapeutic agents and that blocking TAp63α function leads to enhanced chemoresistance. These data are consistent with recent observations that p63 participates in p53-mediated DNA damage responses (Flores et al, 2002; Petitjean et al, 2005). There have been several reports, which have demonstrated that p73 is essential for apoptosis induced by many cytotoxic agents and that inactivation of p73 by a dominant-negative mutation or RNA interference leads to resistance of cells to apoptosis induced by genotoxic agents (Agami et al, 1999; Gong et al, 1999; White and Prives, 1999; Yuan et al, 1999; Costanzo et al, 2002; Ben Yehoyada et al, 2003; Gonzalez et al, 2003; Irwin et al, 2003). To our knowledge, this is the first report that links chemosensitivity to TAp63α function. Thus, chemosensitivity is determined by p53, p73 and p63 function. Synergistic effects of p63 and chemotherapeutic drugs should be taken into consideration for the development of future gene therapy strategies.
In summary, TAp63α activates genes exerting roles in different steps of the apoptosis program. Like p53, p63 may simultaneously recruit several genes within the same cell, probably acting additively or synergistically, whereas others may be more cell type-restricted with regard to their requirement for p63-mediated apoptosis. This explains why p53 status is not a universal predictor of treatment response, but instead the status of a network of interactions between the p53 family members in cancer cells.
Materials and methods
Cell lines and culture
Hep3B were cultured in minimum essential medium (MEM; Gibco BRL, Eggenstein, Germany) enriched with 10 mM HEPES buffer pH 7.3 (Gibco BRL), 100 μg/ml gentamycin (Invitrogen, Karlsruhe, Germany), 1 × nonessential amino acids (Invitrogen) and 2 mM L-glutamine (Invitrogen).
Saos2 cells with dox-inducible expression of TAp63α were cultured in a 1:1 mixture of Ham's F-12/DMEM supplemented with 10% FCS (Biochrom, Berlin, Germany). TAp63α-inducible cell lines were HA-tagged in order to monitor the steady-state levels of protein induction by Western blotting and laser densitometry. To induce protein expression, cell lines were treated with dox at 2.5 μg/ml.
HEK293 cells were grown in MEM supplemented with 10% FCS (Biochrom).
Treatment with cytostatic drugs
Hep3B cells were treated with bleomycin (3 and 30 μg/ml), doxorubicin (0.02, 0.2 and 2 μg/ml), mitoxantrone (0.1 and 1 μg/ml) or cisplatin (0.2 and 2 μg/ml). The concentrations relevant for therapy are 1.5–3 μg/ml for bleomycin, 0.001–0.02 μg/ml for doxorubicin, 0.03–0.5 μg/ml for mitoxantrone and 0.4–1.6 μg/ml for cisplatin in patients' sera (Müller et al, 1997).
Adenoviral constructs and transduction
Replication-deficient adenoviral vectors were generated (He et al, 1998) encoding the complete human wt p53 cDNA (rAd-p53) or TAp63α cDNA (rAd-TAp63α) together with GFP, or GFP alone (rAd-GFP), each under the control of the cytomegalovirus immediate/early gene (CMV) promoter. Cells were seeded in 6/12/24-well plates at a density of 1.5 × 105/5.5 × 104/3 × 104 cells/cm2 24 h prior to infection. Then, adenoviruses (rAd-GFP, rAd-p53, rAd-TAp63α) were added to the culture medium and cells were incubated with the virus for 4 h. At a multiplicity of infection (moi) of 10 IU/cell, an infection rate of 80–90% of the cells was obtained.
Plasmids
A construct was generated containing 3.2 kb of the CD95 gene, that is, the 3′ part of the promoter, the complete exon 1 and the 5′ part of intron 1. This plasmid is denoted p1142CD95-luc.
Mutants of the intronic p53 binding site of p1142CD95-luc were established using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, Heidelberg, Germany).
The Bax-luciferase reporter plasmid used has been established by Miyashita and Reed (1995).
Transfections
Hep3B cells were transfected by the use of calcium phosphate. After 18 h, the medium was changed; 2 h later, cells were infected with the adenovirus at an moi of 10; and 24 h later, cells were harvested and assayed for luciferase activity (Promega, Heidelberg, Germany).
HEK293 cells were transiently transfected using Lipofectamine 2000 (Invitrogen). Cells were analyzed 2–3 days post-transfection.
Western blot analysis
Saos2 subclones with dox-inducible expression of TAp63α were treated with dox at 2.5 μg/ml and lysed in buffer A (50 mM Tris, pH 8, 150 mM NaCl, 0.5% Nonidet P-40, 0.5 mg/ml leupeptin, 1 mg/ml aprotinin, 0.5 mM phenylmethylsulfonyl fluoride) for 1 h on ice. Cell extracts (20 μg) were resolved by electrophoresis in a 12% SDS–polyacrylamide gel, transferred to a polyvinylidene difluoride membrane and probed with antibodies against HA (TAp63), TAp63, p21, Ki67, CD95, actin, β-tubulin, FLIP, Bax, caspase-1, caspase-3 or caspase-9 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), p73β (Upstate, Lake Placid, NY, USA), RAD9 (Calbiochem, Schwalbach, Germany), APAF1 (Axxora, Grünberg, Germany) or p53 (BD Pharmingen, Heidelberg, Germany).
Subcellular localization
Saos2 cells were grown overnight on glass coverslips and, after the indicated treatment, fixed in 4% paraformaldehyde in phosphate-buffered saline (pH 7.4) for 15 min. Immunofluorescence was carried out using an anti-HA, anti-p21 or anti-Ki67 antibody. Confocal images were acquired by using a four-laser C1 confocal microscope (Nikon) excited with a 488 nm argon-ion laser line or a 542 nm helium–neon laser. Detection was performed with the appropriate filter set (515/30 green filter and 595/70 red filter).
Detection of apoptosis
Quantification of DNA fragmentation was performed by FACS analysis of propidium iodide-stained nuclei (Nicoletti et al, 1991; Müller et al, 1998), carried out in a FACScan® flow cytometer (Becton Dickinson, Heidelberg, Germany) using the CELLQuest® software system.
The broad-spectrum caspase inhibitor ZVAD-FMK (z-Val-Ala-DL-Asp-fluoromethylketone; Bachem, Bubendorf, Germany), DEVD-FMK (Z-Asp(OCH3)-Glu(OCH3)-Val-Asp(OCH3)-FMK; inhibitor of caspase-3 as well as caspase-6, -7, -8 and -10), Z-IETD-FMK (z-Ile-Glu(OMe)-Thr-Asp(OMe)-CH2F; inhibitor of caspase-8), Z-LEHD-FMK (z-Leu-Glu(OMe)-His-Asp(OMe)-CH2F; inhibitor of caspase-9 as well as caspase-4 and -5) were applied (all from Calbiochem, Schwalbach, Germany).
For caspase activation assays, siRNA-transfected cells were harvested 36 and 48 h after bleomycin treatment (caspase-3, -8 and -9 fluorometric assay, R&D systems, Minneapolis, MN, USA).
Apoptosis was inhibited using CD95 receptor-blocking F(ab′)2-anti-APO-1 fragments at 1 μg/ml (Müller et al, 1997), human TNF-R1-Fc (10 μg/ml; Apogenix, Heidelberg) or TRAIL-R2-Fc (10 μg/ml; Apogenix, Heidelberg). To induce CD95 receptor-mediated apoptosis, we used the monoclonal antibody anti-APO-1 IgG3, κ, at a concentration of 1 μg/ml (Trauth et al, 1989; Dhein et al, 1992; Müller et al, 1997). TNFα (Sigma) was added at a concentration of 100 ng/ml, together with 10 μg/ml cycloheximide (Sigma) 12 h prior to harvesting. TRAIL (human leucinzipper (LZ)-TRAIL) was applied at a concentration of 1 μg/ml 24 h before harvesting.
Detection of the death receptors
Cell surface expression of the CD95, TNF-R1, TRAIL-R1 and TRAIL-R2 receptor was assessed by FACScan® as described previously (Müller et al, 1998; Ganten et al, 2004).
Determination of mitochondrial membrane potential
Hep3B cells were incubated with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1, 5 μg/ml; Molecular Probes, Eugene, OR, USA) for 20 min at room temperature (RT) in the dark, washed and analyzed by FACScan® (Zuliani et al, 2003).
Microarray analysis in Hep3B cells
We have developed high-density cDNA arrays in cooperation with the Department of Molecular Genome Analysis, German Cancer Research Center, Heidelberg, Germany. These arrays are now commercially available at www.rzpd.de (Immunofilter, RZPD) and contain PCR products of 1066 specific cDNA clones representing genes that are involved in apoptosis and immunological signaling pathways. For further experimental details, see Supplementary data.
Microarray analysis in Tet-On Saos2 cells
Saos2 cells with dox-inducible expression of TAp63α (7–8 × 106 cells) from pTRE2-Hyg/TAp63α clone 1 were collected after several induction times (0, 12 and 24 h). Total RNA was purified by guanidinium isothiocyanate method (Trizol, Invitrogen). Double-stranded cDNA was generated by using the primer sequence 5′-GGCCAGTGAATTGTAATAATACGACTCACTAT AGGGAGGCGG-(dT)24-3′ and the Superscript Double-Stranded cDNA Synthesis Kit (Invitrogen). Then, biotin-labeled cRNAs were synthesized with the BioArray HighYield RNA Transcript Labeling Kit (Enzo Diagnostics Inc., Farmingdale, NY, USA) and hybridized to the Genechip HuGene FL array (Affymetrix, Santa Clara, USA), which contains probes for about 11 000 mRNA species. One chip was hybridized to cRNAs derived from each time point. Scanned output files were inspected for hybridization artifacts and further analyzed using Genechip 3.3 software (Affymetrix). Ratios were obtained by dividing the average difference of pTRE2-Hyg/TAp63α for each time point with those of the 0 h time point.
Chromatin immunoprecipitation
Cells (1.5 × 106) were crosslinked using 1% formaldehyde in PBS buffer for 10 min at 37°C, stopped by incubation in 0.125 M glycine for 5 min at RT and washed in ice-cold PBS. The cells were harvested in 200 μl SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl, pH 8.1). Cell lysates were sonicated to obtain chromatin fragments of ∼700 bp. After centrifugation at 13 000 r.p.m. for 20 min, 1.8 ml ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 0.0167 M Tris–HCl, 0.167 M NaCl) was added to the reaction. A 100 μg portion of total protein was precleared with 100 μl of protein A-agarose/salmon sperm DNA (Upstate, Chicago, USA) plus 2 μg isotypic IgG for 2 h at 4°C. The precleared extracts were incubated either with 2 μg anti-HA (Babco) or nonspecific cytokeratin 5 (SantaCruz) antibodies overnight at 4°C, followed by incubation with protein A-agarose/salmon sperm DNA (60 μl) for 1 h 30 min at 4°C. The immunocomplexes were washed twice with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, 0.15 M NaCl), five times with high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, 0.5 M NaCl), once with LiCl salt-wash buffer (1 mM EDTA, 10 mM Tris–HCl, 0.25 M LiCl, 1% NP-40, 1% deoxycholate) and twice with TE buffer. The precipitates were extracted twice using 250 μl of IP elution buffer (1% SDS, 0.1 M NaHCO3). Total eluates (500 μl) were pooled by adding 20 μl of 5 M NaCl and incubated at 65°C for at least 6 h to reverse the formaldehyde crosslinking. DNA fragments were purified by phenol–chloroform extraction and ethanol precipitation, and dissolved in 30 μl of sterile water.
DNA samples were then PCR analyzed for the presence of CD95 sequences. The following primers were used to amplify the p53/p63 binding site in the first intron of the CD95 gene: 5′-TCTGGGAAGCTTTAGGGTCG-3′ CD95 intronic p53 binding site F and 5′-TCTGTTCTGAAGGCTGCAGG-3′ CD95 intronic p53 binding site R.
TAp63 siRNA
Silencing of the TAp63 gene was performed using the pSuperRetro plasmid (Oligoengine, Seattle, WA, USA). The following oligonucleotides were subcloned into pSuperRetro that had been digested with BglII and HindIII: 5′-gatccccgaactttgtggatgaaccattcaag agatggttcatccacaaagttctttttggaaa-3′ and 5′-agcttttccaaaaagaactttgtggatgaacc atctcttgaatggttcatccacaaagttcggg-3′. As a control, the following scrambled duplex oligonucleotide was subcloned into pSuperRetro: 5′-gatccccttctccgaacgtgtcacgtttcaag agaacgtgacacgttcggagaatttttggaaa-3′ (sense) and 5′-agcttttccaaaaattctccgaacgtgtcacg ttctcttgaaacgtgacacgttcggagaaggg-3′ (antisense). Hep3B cells were transiently transfected with FuGENE6 (Roche, Basel, Switzerland). Cells were analyzed 2–3 days post-transfection. Transfection efficiency was 30–40%, as assessed by FACS analysis following cotransfection with plasmid DNA coding for GFP.
Statistical analysis
MANOVA or Wilcoxon's analysis was used to test for statistical significance. The statistical software system used was SAS software system (SAS Institute Inc., Cary, NC, USA).
Supplementary Material
Online supplementary information to Gressner et al.
Supplementary Figure
Acknowledgments
We thank Daniela Willen (German Cancer Research Center, Heidelberg) for helpful discussions and Petra Hill for expert technical assistance. This work was supported by grants from the Medizinische Forschungsförderung Heidelberg, the Sonderforschungsbereich 601 and the Tumorzentrum Heidelberg/Mannheim to MM and PHK. This work was in part performed thanks to grants from AIRC, EU (QLG1-1999-00739 and YLK-CT-2002-01956), MIUR and MinSan to GM, EU (QLK3-CT-2002-01956) to GM and MO and EU grant QLG1-1999-00739 to PHK.
References
- Agami R, Blandino G, Oren M, Shaul Y (1999) Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature 399: 809. [DOI] [PubMed] [Google Scholar]
- Ben Yehoyada M, Ben Dor I, Shaul Y (2003) c-Abl tyrosine kinase selectively regulates p73 nuclear matrix association. J Biol Chem 278: 34475–34482 [DOI] [PubMed] [Google Scholar]
- Benchimol S (2004) p53—an examination of sibling support in apoptosis control. Cancer Cell 6: 3–4 [DOI] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J, Vingron M (2001) Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat Genet 29: 365–371 [DOI] [PubMed] [Google Scholar]
- Casciano I, Mazzocco K, Boni L, Pagnan G, Banelli B, Allemanni G, Ponzoni M, Tonini GP, Romani M (2002) Expression of DeltaNp73 is a molecular marker for adverse outcome in neuroblastoma patients. Cell Death Differ 9: 246–251 [DOI] [PubMed] [Google Scholar]
- Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, Newbury-Ecob R, Hennekam RC, Van Buggenhout G, van Haeringen A, Woods CG, van Essen AJ, de Waal R, Vriend G, Haber DA, Yang A, McKeon F, Brunner HG, van Bokhoven H (1999) Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 99: 143–153 [DOI] [PubMed] [Google Scholar]
- Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, Fontemaggi G, Fanciulli M, Schiltz L, Blandino G, Balsano C, Levrero M (2002) DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol Cell 9: 175–186 [DOI] [PubMed] [Google Scholar]
- Dhein J, Daniel PT, Trauth BC, Oehm A, Möller P, Krammer PH (1992) Induction of apoptosis by monoconal antibody anti-APO-1 class switch variants is dependent on cross-linking of APO-1 cell surface antigens. J Immunol 149: 3166–3173 [PubMed] [Google Scholar]
- El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B (1992) Definition of a consensus binding site for p53. Nat Genet 1: 45–49 [DOI] [PubMed] [Google Scholar]
- Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T (2002) p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416: 560–564 [DOI] [PubMed] [Google Scholar]
- Ganten TM, Haas TL, Sykora J, Stahl H, Sprick MR, Fas SC, Krueger A, Weigand MA, Grosse-Wilde A, Stremmel W, Krammer PH, Walczak H (2004) Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ 11 (Suppl 1): S86–S96 [DOI] [PubMed] [Google Scholar]
- Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG Jr, Levrero M, Wang JY (1999) The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 399: 806–809 [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Prives C, Cordon-Cardo C (2003) p73alpha regulation by Chk1 in response to DNA damage. Mol Cell Biol 23: 8161–8171 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gupta S, Radha V, Furukawa Y, Swarup G (2001) Direct transcriptional activation of human caspase-1 by tumor suppressor p53. J Biol Chem 276: 10585–10588 [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG (2003) Chemosensitivity linked to p73 function. Cancer Cell 3: 403–410 [DOI] [PubMed] [Google Scholar]
- Kissil JL, Cohen O, Raveh T, Kimchi A (1999) Structure–function analysis of an evolutionary conserved protein, DAP3, which mediates TNF-alpha- and Fas-induced cell death. EMBO J 18: 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Choi Y (1997) TRAF-interacting protein (TRIP): a novel component of the tumor necrosis factor receptor (TNFR)- and CD30-TRAF signaling complexes that inhibits TRAF2-mediated NF-kappaB activation. J Exp Med 185: 1275–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke C, Groger N, Manns MP, Trautwein C (2003) The human caspase-8 promoter sustains basal activity through SP1 and ETS-like transcription factors and can be up-regulated by a p53-dependent mechanism. J Biol Chem 278: 27593–27604 [DOI] [PubMed] [Google Scholar]
- Liu X, Yue P, Khuri FR, Sun SY (2004) p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res 64: 5078–5083 [DOI] [PubMed] [Google Scholar]
- Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C, Vousden KH (2004) p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem 279: 8076–8083 [DOI] [PubMed] [Google Scholar]
- Melino G, De Laurenzi V, Vousden KH (2002) p73: friend or foe in tumorigenesis. Nat Rev Cancer 2: 605–615 [DOI] [PubMed] [Google Scholar]
- Melino G, Lu X, Gasco M, Crook T, Knight RA (2003) Functional regulation of p73 and p63: development and cancer. Trends Biochem Sci 28: 663–670 [DOI] [PubMed] [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A (1999) p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398: 708–713 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299 [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Reed JC (2001) A GTP-binding adapter protein couples TRAIL receptors to apoptosis-inducing proteins. Nat Immunol 2: 493–500 [DOI] [PubMed] [Google Scholar]
- Moll UM, Slade N (2004) p63 and p73: roles in development and tumor formation. Mol Cancer Res 2: 371–386 [PubMed] [Google Scholar]
- Mukamel Z, Kimchi A (2004) Death-associated protein 3 localizes to the mitochondria and is involved in the process of mitochondrial fragmentation during cell death. J Biol Chem 279: 36732–36738 [DOI] [PubMed] [Google Scholar]
- Müller M, Strand S, Hug H, Heinemann EM, Walczak H, Hofmann WJ, Stremmel W, Krammer PH, Galle PR (1997) Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clin Invest 99: 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 188: 2033–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti I, Migliorati G, Paggliacci MC, Grignani F, Riccardi C (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139: 271–279 [DOI] [PubMed] [Google Scholar]
- Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, Ikawa Y, Nimura Y, Nakagawara A, Obinata M, Ikawa S (1998) Cloning and functional analysis of human p51, which structurally and functionally resembles p53 [see comments] [published erratum appears in Nat Med 1998 Sep;4(9):982]. Nat Med 4: 839–843 [DOI] [PubMed] [Google Scholar]
- Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, Ponzin D, McKeon F, De Luca M (2001) p63 identifies keratinocyte stem cells. Proc Natl Acad Sci USA 98: 3156–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean A, Cavard C, Shi H, Tribollet V, Hainaut P, Caron dF (2005) The expression of TA and DeltaNp63 are regulated by different mechanisms in liver cells. Oncogene 24: 512–519 [DOI] [PubMed] [Google Scholar]
- Senoo M, Manis JP, Alt FW, McKeon F (2004) p63 and p73 are not required for the development and p53-dependent apoptosis of T cells. Cancer Cell 6: 85–89 [DOI] [PubMed] [Google Scholar]
- The Tumor Analysis Best Practices Working Group (2004) Expression profiling—best practices for data generation and interpretation in clinical trials. Nat Rev Genet 5: 229–237 [DOI] [PubMed] [Google Scholar]
- Trauth BC, Klas C, Peters AJM, Matzku S, Möller P, Falk W, Debatin KM, Krammer PH (1989) Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science 245: 301–305 [DOI] [PubMed] [Google Scholar]
- Vousden KH (2000) p53: death star. Cell 103: 691–694 [DOI] [PubMed] [Google Scholar]
- Wada N, Matsumura M, Ohba Y, Kobayashi N, Takizawa T, Nakanishi Y (1995) Transcription stimulation of the Fas-encoding gene by nuclear factor for interleukin-6 expression upon influenza virus infection. J Biol Chem 270: 18007–18012 [DOI] [PubMed] [Google Scholar]
- Westfall MD, Pietenpol JA (2004) p63: molecular complexity in development and cancer. Carcinogenesis 25: 857–864 [DOI] [PubMed] [Google Scholar]
- White E, Prives C (1999) DNA damage enables p73. Nature 399: 734–737 [DOI] [PubMed] [Google Scholar]
- Wu G, Osada M, Guo Z, Fomenkov A, Begum S, Zhao M, Upadhyay S, Xing M, Wu F, Moon C, Westra WH, Koch WM, Mantovani R, Califano JA, Ratovitski E, Sidransky D, Trink B (2005) DeltaNp63alpha up-regulates the Hsp70 gene in human cancer. Cancer Res 65: 758–766 [PubMed] [Google Scholar]
- Wu GS, Burns TF, McDonald ER, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, El-Deiry WS (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene [letter]. Nat Genet 17: 141–143 [DOI] [PubMed] [Google Scholar]
- Yang A, Kaghad M, Caput D, McKeon F (2002) On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet 18: 90–95 [DOI] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F (1999) p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398: 714–718 [DOI] [PubMed] [Google Scholar]
- Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, Lu H, Kharbanda S, Weichselbaum R, Kufe D (1999) p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature 399: 814–817 [DOI] [PubMed] [Google Scholar]
- Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M, Chalas E, Moll UM (2002) DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J Exp Med 196: 765–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuliani T, Duval R, Jayat C, Schnebert S, Andre P, Dumas M, Ratinaud MH (2003) Sensitive and reliable JC-1 and TOTO-3 double staining to assess mitochondrial transmembrane potential and plasma membrane integrity: interest for cell death investigations. Cytometry 54A: 100–108 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online supplementary information to Gressner et al.
Supplementary Figure