

Abstract
The absence of infectivity-associated, protease-resistant prion protein (PrPSc) in the brains of spontaneously sick transgenic (Tg) mice overexpressing PrP linked to Gerstmann–Sträussler Scheinker syndrome, and the failure of gene-targeted mice expressing such PrP to develop disease spontaneously, challenged the concept that mutant PrP expression led to spontaneous prion production. Here, we demonstrate that disease in overexpressor Tg mice is associated with accumulation of protease-sensitive aggregates of mutant PrP that can be immunoprecipitated by the PrPSc-specific monoclonal antibody designated 15B3. Whereas Tg mice expressing multiple transgenes exhibited accelerated disease when inoculated with disease-associated mutant PrP, Tg mice expressing mutant PrP at low levels failed to develop disease either spontaneously or following inoculation. These studies indicate that inoculated mutant PrP from diseased mice promotes the aggregation and accumulation of pre-existing pathological forms of mutant PrP produced as a result of transgene overexpression. Thus, while pathological mutant PrP possesses a subset of PrPSc characteristics, we now show that the attribute of prion transmission suggested by previous studies is more accurately characterized as disease acceleration.
Keywords: Gerstmann–Sträussler Scheinker syndrome, nucleated polymerization, prions, PrPSc-specific antibody
Introduction
The transmissible spongiform encephalopathies (TSEs), which include human Creutzfeldt–Jakob disease (CJD), chronic wasting disease (CWD) of cervids, and bovine spongiform encephalopathy (BSE), are a group of closely related fatal neurodegenerative conditions caused by prions, which are defined as proteinaceous infectious particles that lack informational nucleic acid (Prusiner, 1982). The mechanism of prion replication is unique and features the accumulation of the scrapie isoform of the prion protein (PrP), referred to as PrPSc, which is a conformationally altered, beta-sheet-rich version of the benign host-encoded form of PrP referred to as PrPC. This post-translational structural transition is accompanied by changes in the physicochemical properties of PrP. Most notably, while PrPC is sensitive to proteinase K (PK), enzymatic treatment of full-length PrPSc, referred to as rPrPSc (Tremblay et al, 2004), generates a core molecule consisting predominantly of residues 89–230, referred to as PrP27−30 (Oesch et al, 1985).
Approximately 10–20% of human prion diseases are familial with an autosomal dominant mode of inheritance resulting from mutations in the coding sequence of the human PrP gene (PRNP). Gerstmann–Sträussler Scheinker (GSS) syndrome, which is characterized clinically by ataxia and dementia and neuropathologically by the deposition of PrP amyloid, most commonly results from mutation at codon 102 of PRNP resulting in the substitution of leucine (L) for proline (P) (Hsiao et al, 1989). GSS linked to this mutation is transmissible to non-human primates (Brown et al, 1994), wild-type mice (Tateishi and Kitamoto, 1995), transgenic (Tg) mice expressing a chimeric mouse–human PrP gene expressing the GSS mutation (Telling et al, 1995), and Prnp gene-targeted mice referred to as 101LL (Manson et al, 1999).
To understand how an inherited disease could be infectious, Tg mice expressing mouse PrP (MoPrP) with the equivalent GSS mutation at codon 101, referred to as MoPrP-P101L, were produced. Tg mice overexpressing MoPrP-P101L spontaneously developed central nervous system (CNS) dysfunction and reproduced the neuropathological features of GSS (Hsiao et al, 1990; Telling et al, 1996a). Brain material from such spontaneously sick mice induced disease in inoculated Tg mice expressing lower levels of mutant protein, referred to as Tg196 mice (Hsiao et al, 1994; Telling et al, 1996a). Disease was also induced in Tg196 mice by a mutant synthetic peptide comprising MoPrP residues 89–103 refolded into a beta-sheet conformation (Kaneko K, 2000) and this disease was subsequently propagated to additional Tg196 mice (Tremblay et al, 2004).
Such studies lent strong support to the prion hypothesis since they suggested that pathogenic PrP gene mutations resulted in the spontaneous formation of PrPSc and de novo production of prions (Cohen et al, 1994). However, this explanation met with controversy for several reasons (Chesebro, 1998). Although PrPSc produced by different prion strains has been shown to vary considerably in its resistance to proteases (Bessen and Marsh, 1992), and protease-sensitive forms of PrPSc (sPrPSc) have subsequently been identified using biochemical and immunological methods (Safar et al, 1998; Tzaban et al, 2002), the lack of rPrPSc in the brains of spontaneously sick or recipient mice eliminated a property that, to some, was synonymous with prion infectivity. Moreover, Prnp gene-targeted 101LL mice expressing MoPrP-P101L failed to develop neurodegenerative disease spontaneously (Manson et al, 1999). Also, disease transmission from spontaneously sick mice to wild-type mice did not occur. Finally, spontaneous disease was eventually registered in aged Tg196 mice (Kaneko, 2000; Tremblay et al, 2004), complicating the interpretation of the original transmission experiments.
To address the apparent dissociation of prion infectivity and PrPSc in this well-established Tg model, we sought means other than differential resistance to PK treatment to detect disease-associated forms of PrP in spontaneously sick Tg mice expressing MoPrP-P101L. In recent years, a diverse array of reagents capable of distinguishing between native PrPC and PrPSc has been developed. These include plasminogen (Fischer et al, 2000), anti-DNA antibodies and a DNA binding protein (Zou et al, 2004), RNA aptamers (Rhie et al, 2003; Sayer et al, 2004), PrPSc-specific monoclonal antibodies (Mabs) (Korth et al, 1997; Paramithiotis et al, 2003), and motif-grafted antibodies containing the replicative interface of PrPC (Moroncini et al, 2004). Although the possibility has been raised that reagents such as scrapie-specific Mabs may interact with PrPSc aggregates through nonspecific paratope-independent interactions (Morel et al, 2004), the binding specificity of such interactions may be influenced by factors such as detergent extraction conditions (Shaked et al, 2002).
Here we demonstrate, using the prototypic PrPSc-specific Mab 15B3, that disease in Tg mice overexpressing MoPrP-P101L results from the spontaneous conversion of mutant PrPC to protease-sensitive, 15B3-reactive MoPrP-P101L that accumulates as aggregates in the brains of sick Tg mice. We also show that MoPrP-P101L is capable of adopting at least two pathogenic, 15B3-reactive conformations that differ not only in their degree of resistance to proteolysis but also the extent to which they are endoproteolytically processed in diseased brains. To understand the influence of mutant PrP expression levels on the transmissibility of spontaneously generated pathogenic MoPrP-P101L, we produced mice in which transgene copy numbers and levels of MoPrP-P101L expression were carefully defined. While inoculation of disease-associated MoPrP-P101L accelerated disease in Tg mice expressing MoPrP-P101L from multiple transgenes, disease transmission neither occurred to wild-type nor Tg mice expressing MoPrP-P101L from two transgene copies that do not develop disease spontaneously in their natural lifespan. While these observations are not inconsistent with our current understanding of the mechanisms of nucleation-mediated prion replication, we propose that the term ‘transmissibility' for this disease acceleration phenomenon is inappropriate.
Results
Detection of pathological mutant PrP in the brains of spontaneously sick Tg(GSS) mice using PrPSc-specific Mab 15B3
We re-established Tg mice expressing MoPrP-P101L, referred to here as Tg(GSS) mice, with a range of transgene copy numbers and MoPrP-P101L expression. As previously shown (Hsiao et al, 1994; Telling et al, 1996a), the age of onset of spontaneous neurodegenerative disease was proportional to transgene copy number and level of transgene expression (Table I), and spongiform degeneration, plaque deposits, and reactive astrocytic gliosis were present in the brains of mice at the time they exhibited clinical signs of neurological disease (data not shown).
Table 1.
Neurologic disease in Tg(GSS) mice
| Tg micea | PrP expression | Age at disease onset (mean (days)±s.e.m. (n/n0))b |
|||
|---|---|---|---|---|---|
| Uninoculated | (GSS)22—brain 1 | (GSS)22—brain 2 | RML | ||
| Tg(GSS)22 | 12-fold | 165±3 (28/28) | 134±1 d (8/8) | ND | ND |
| Tg(GSS)12 | Six-fold | 430±2 (5/5) | 220±2 (8/8) | 195±2 (6/6) | 238±2 (8/8) |
| Tg(GSS)6 | Three-fold | 598±1 (9/9) | ND | ND | ND |
| Tg(GSS)2 | 0.5- to 1-fold | >630 (0/9) | >619 (0/5) | >584 (0/8) | 427±1 (8/8) |
| aTg(GSS) mice were distinguished on the basis of transgene copy number, determined by real-time PCR. Thus, Tg(GSS)22, Tg(GSS)12, Tg(GSS)6, and Tg(GSS)2 mice contain 22, 12, six, and two copies of the transgene, respectively. | |||||
| bThe number of mice developing clinical signs of scrapie divided by the original number of mice is shown in parentheses. | |||||
| ND: not determined. | |||||
Western blot analysis confirmed the results of previous studies (Hsiao et al, 1994; Telling et al, 1996a) that brain homogenates from spontaneously sick Tg(GSS) mice did not contain rPrPSc (Supplementary Figure 1A, lanes 6 and 8). Titration of PK concentrations ranging from 20 to 0.005 μg/ml demonstrated that mutant and wild-type MoPrP were equally sensitive to PK at concentrations greater than 0.04 μg/ml (data not shown). In addition, there was no difference in the solubility in nondenaturing detergents of MoPrP-P101L or its cleavage products, the majority of mutant PrP in the brains of both sick and healthy Tg(GSS)22 mice being soluble (Supplementary Figure 1B).
Immunoprecipitation of PrPSc from brain extracts of sick wild-type FVB mice inoculated with the mouse-adapted Rocky Mountain Laboratories (RML) isolate of scrapie prions but not from brains of uninfected FVB mice confirmed the previously demonstrated specificity of Mab 15B3 for PrPSc (Figure 1A, lanes 1 and 5) (Korth et al, 1997). Mab 15B3 also specifically immunoprecipitated mutant MoPrP-P101L from brain homogenates of spontaneously sick Tg(GSS)22 mice (Figure 1A, lane 11) but not from brain homogenates of healthy Tg(GSS)22 mice killed at younger ages (Figure 1A, lane 13) or from asymptomatic Tg(GSS)2 mice killed at ∼570 days of age (Figure 1A, lane 9). Mab 15B3 also failed to immunoprecipitate PrP in brain homogenates from uninfected Tga20 mice, which overexpress wild-type MoPrP (Figure 1A, lane 7) (Fischer et al, 1996). In additional control experiments, an unrelated isotype-matched Mab coupled to anti-mouse IgM-coupled magnetic beads failed to immunoprecipitate PrP from the same brain extracts (data not shown).
Figure 1.
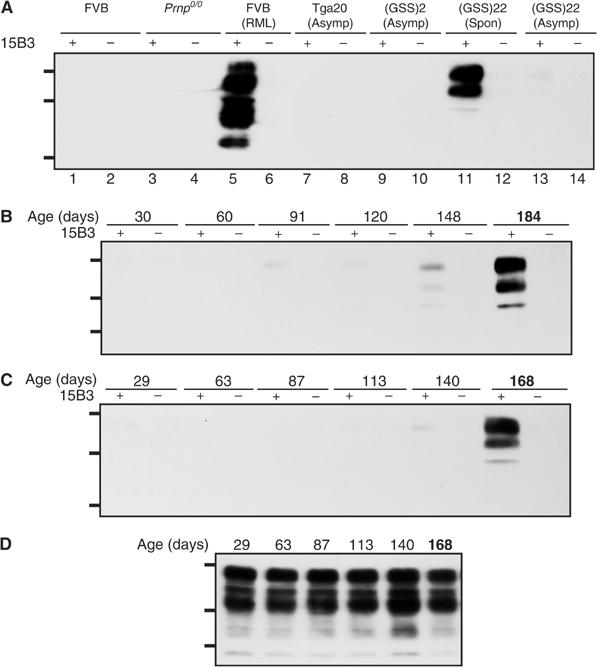
Spontaneous disease in Tg(GSS) mice correlates with the accumulation of 15B3-reactive MoPrP-P101L. (A) Brain extracts were treated with anti-mouse IgM-coupled magnetic beads in the presence or absence of Mab 15B3 as indicated and immunoprecipitates were analyzed by SDS–PAGE and Western blotting as described. Lanes 1 and 2, uninoculated FVB mouse; lanes 3 and 4, Prnp0/0 mouse; lanes 5 and 6, FVB mouse inoculated with mouse-adapted RML scrapie prions; lanes 7 and 8, asymptomatic Tga20 mouse; lanes 9 and 10, asymptomatic Tg(GSS)2 mouse killed at 572 days of age; lanes 11 and 12, spontaneously sick Tg(GSS)22 mouse killed at 188 days of age; lanes 13 and 14, asymptomatic Tg(GSS)22 mouse killed at 30 days of age. (B, C) Kinetics of disease-associated MoPrP-P101L accumulation in Tg(GSS)22 mice. Brain tissues were collected from two independent birth cohorts of Tg(GSS)22 mice killed at various ages, as indicated. The second cohort of Tg(GSS)22 mice was established approximately 1 year after the first. Brain extracts were treated with anti-mouse IgM-coupled magnetic beads in the presence or absence of Mab 15B3 as indicated and immunoprecipitates were analyzed by SDS–PAGE and Western blotting using HRP-conjugated anti-PrP antibody 6H4. (D) Equal amounts of protein in the brains of Tg(GSS)22 mice killed at different ages, as indicated, were analyzed by SDS–PAGE and Western blotting. In (B–D), the ages of asymptomatic Tg(GSS)22 mice are shown in plain text, while mice in which clinical symptoms of disease were manifest at the time of killing are indicated by bold type. The positions of protein molecular weight markers of 35.5, 28.8, and 22.0 kDa from top to bottom are shown.
Longitudinal analyses of two independent birth cohorts of Tg(GSS)22 mice showed that the kinetics of pathological MoPrP-P101L accumulation in brain extracts of Tg(GSS)22 mice killed at different ages was remarkably reproducible (Figure 1B and C). While Mab 15B3-reactive MoPrP-P101L could be detected in the brains of Tg(GSS)22 mice just prior to the onset of clinical symptoms, substantial quantities did not accumulate until mice were clinically sick. In contrast, Western blotting using Mab 6H4 showed that expression of total MoPrP-P101L remained constant over the same time period (Figure 1D).
Characterization of MoPrP-P101L aggregates in the brains of spontaneously sick Tg(GSS) mice
To address whether the protease sensitivity of disease-associated MoPrP-P101L in the brains of spontaneously sick Tg(GSS) mice reflected a difference in aggregation state compared to rPrPSc, we fractionated brain extracts from spontaneously sick Tg(GSS)22 mice and RML-infected FVB mice by density gradient sedimentation followed by immunoprecipitation. Mab 15B3 immunoprecipitated PK-sensitive MoPrP-P101L in fractions 5–12 derived from the brains of spontaneously sick Tg(GSS)22 mice with peak levels occurring in fractions 7–9 (Figure 2E); as expected, 15B3-immunoprecipitable MoPrP-P101L was absent from the corresponding fractions derived from brain extracts of healthy Tg(GSS)22 mice (Figure 2F). Mab 15B3-reactive material was also not detected in fractions from uninfected healthy Tga20 mice (Figure 2G). The sedimentation profile of Mab 15B3-reactive rPrPSc in gradients prepared from RML-infected FVB mice coincided with the distribution of 15B3-reactive MoPrP-P101L in fractions isolated from the brains of spontaneously sick Tg(GSS)22 mice (Figure 2H), demonstrating similar aggregation states of these protease-sensitive and -resistant forms of disease-associated PrP.
Figure 2.
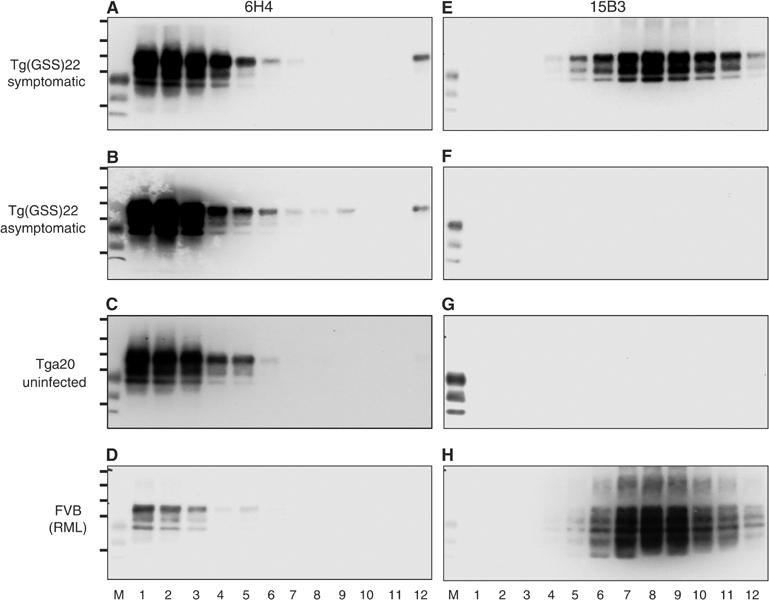
Characterization of MoPrP-P101L aggregates in spontaneously sick Tg(GSS) mice. Sucrose gradient fractionation of brain homogenates followed by immunoprecipitation with Mab 6H4 (A–D) or 15B3 (E–H). (A, E) Spontaneously sick Tg(GSS)22 mouse killed at 181 days of age; (B, F) healthy Tg(GSS)22 mouse killed at 30 days of age; (C, G) uninoculated Tga20 mouse; (D, H) FVB mouse inoculated with mouse-adapted RML scrapie prions. Immunoprecipitated samples were analyzed by SDS–PAGE and Western blotting using HRP-conjugated Mab 6H4. M=PrP27−30 from BSE-infected cow. In each case, lanes 1–12 refer to 1 ml fractions isolated from the top to the bottom of sucrose density gradients. The positions of protein molecular weight markers of 72, 55, 40, 33, and 24 kDa from top to bottom are shown.
Different populations of immunoprecipitable PrP were recognized by Mabs 6H4 and 15B3. The majority of 6H4-immunoprecipitable PrP was confined to fractions 1–3 and, although the relative levels of Mab 6H4-reactive PrP differed between the overexpressing Tg and wild-type FVB mice, this distribution remained constant in spontaneously sick as well as healthy Tg(GSS)22 mice (Figure 2A and B), uninfected Tga20 mice (Figure 2C), and sick RML-infected FVB mice (Figure 2D), demonstrating that the 6H4-immunoprecipitable material corresponded to PrPC.
Inoculation of disease-associated MoPrP-P101L accelerates the age of onset of disease in Tg(GSS) mice
The lack of transmission of brain homogenates from spontaneously sick Tg(GSS)22 mice to wild-type mice (Supplementary Table I) confirmed previous observations (Hsiao et al, 1994; Telling et al, 1996a). Since gene-targeted 101LL mice expressing MoPrP-P101L at physiological levels failed to develop disease spontaneously (Manson et al, 1999), we produced Tg(GSS)2 mice that express equivalent MoPrP-P101L levels from two transgene copies. Since Tg(GSS)2 mice remained free of spontaneous neurologic disease in old age (Table I), we reasoned that they would serve as ideal alternative recipients to wild-type mice for assessing transmission of infectivity from spontaneously sick Tg(GSS)22 mice. However, Tg(GSS)2 mice failed to develop neurologic disease following inoculation with brain homogenates from spontaneously sick Tg(GSS)22 mice killed at 166 and 188 days of age (Table I), and 15B3-immunoprecitable material was not detected in brain homogenates of asymptomatic Tg(GSS)2 mice killed 586 days after inoculation (data not shown).
Although Tg(GSS)12 mice, which overexpress MoPrP-P101L from 12 transgene copies at levels six-fold higher than wild type, eventually develop a spontaneous neurologic disorder, we asked if illness might appear more rapidly if these animals were inoculated. The average age at which Tg(GSS)12 mice inoculated with brain extracts from spontaneously sick Tg(GSS)22 mice developed illness was between 210 and 235 days earlier than the age at which uninoculated Tg(GSS)12 mice developed signs of CNS dysfunction, representing an ∼45–50% decrease in lifespan (Table I). Inoculation of Tg(GSS)22 mice at ∼35 days of age with a brain extract from a spontaneously sick Tg(GSS)22 mouse resulted in an ∼30 days (∼20%) reduction in the age at which mice developed neurologic disease (Table I). Mab 15B3-immunoprecipitable MoPrP-P101L was present in the brains of Tg(GSS)12 mice inoculated with brain material from Tg(GSS)22 mice when they exhibited disease at ∼220 days of age (Figure 3A, lane 1). In contrast, 15B3-reactive MoPrP-P101L was barely detectable in brain extracts of preclinical uninoculated Tg(GSS)12 mice killed as late as 384 days of age (Figure 3A, lane 5).
Figure 3.
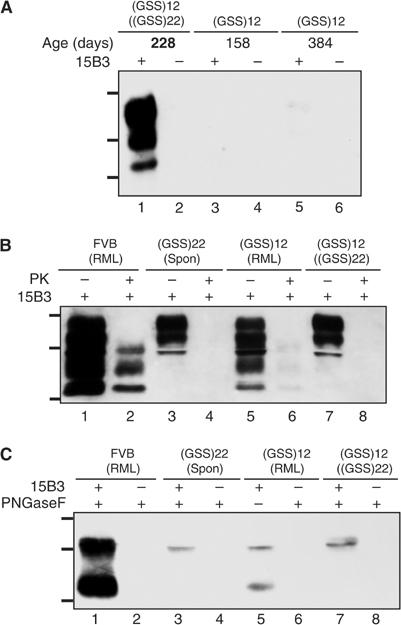
Disease-associated MoPrP-P101L in sick Tg(GSS) mice inoculated with extracts from spontaneously sick Tg(GSS) mice and RML prions. (A) Brain extracts were treated with anti-mouse IgM-coupled magnetic beads in the presence or absence of Mab 15B3 as indicated and immunoprecipitates were analyzed by SDS–PAGE and Western blotting using HRP-conjugated Mab 6H4. Lanes 1 and 2, sick Tg(GSS)12 mouse killed at 228 days of age inoculated at 35 days of age with brain extract from spontaneously sick Tg(GSS)22 mouse, brain 1; lanes 3 and 4, asymptomatic uninoculated Tg(GSS)12 mouse, killed at 158 days of age; lanes 5 and 6, asymptomatic uninoculated Tg(GSS)12 mouse, killed at 384 days of age. The ages of asymptomatic Tg(GSS)12 mice are shown in plain text, while mice in which clinical symptoms of disease were manifest at the time of killing are indicated by bold type. (B) Following immunoprecipitation with Mab 15B3, immune complexes were treated with 20 μg/ml PK for 1 h at 37°C as indicated and analyzed by SDS–PAGE and Western blotting. (C) Following immunoprecipitation with Mab 15B3, immune complexes were treated with PNGase F as indicated and analyzed by SDS–PAGE and Western blotting. Samples in (B, C) are equivalent and are as follows: lanes 1 and 2, sick FVB mouse inoculated with mouse-adapted RML scrapie prions; lanes 3 and 4, spontaneously sick Tg(GSS)22 mouse killed at 159 days of age; lanes 5 and 6, sick Tg(GSS)12 mouse killed at 211 days following inoculation with mouse-adapted RML scrapie prions; lanes 7 and 8, sick Tg(GSS)12 mouse killed at 193 days following inoculation with brain extract from a spontaneously sick Tg(GSS)22 mouse. The positions of protein molecular weight markers of 35.5, 28.8, and 22 kDa from top to bottom are shown.
To confirm that 15B3-precipitable MoPrP-P101L mediated accelerated disease in inoculated Tg(GSS)12 mice, Mab 15B3 immune complexes isolated from the brains of spontaneously sick Tg(GSS)22 mice were inoculated into Tg(GSS)12 mice at ∼35 days of age. All such inoculated Tg(GSS)12 mice developed disease ∼210 days following inoculation. In contrast, Tg(GSS)12 mice inoculated with uncoated beads that were incubated with diseased Tg(GSS)22 mouse brain extract or Mab 15B3-coated beads incubated with diseased Tg(GSS)22 mouse brain extract that was treated with 20 μg/ml PK following immunoprecipitation remained free of neurological disease for >300 days. All Tg(GSS)12 mice inoculated with identically treated samples using a brain extract from an asymptomatic Tg(GSS)22 mouse killed at 29 days of age remained free of CNS disease for >300 days.
Endoproteolytic processing of different pathological MoPrP-P101L conformers in the brains of Tg(GSS) mice
PK treatment of Mab 15B3 immune complexes confirmed the protease sensitivity of disease-associated MoPrP-P101L produced in the brains of spontaneously sick Tg(GSS) mice (Figure 3B, lanes 4 and 8). We asked whether MoPrP-P101L was also capable of adopting a conformation that imparted resistance to PK, by inoculating Tg(GSS) mice with mouse-adapted RML scrapie prions. Tg(GSS)2 and Tg(GSS)12 mice inoculated with mouse-adapted RML scrapie prions at ∼35 days of age developed disease at ∼430 and ∼240 days of age (Table I). While rPrPSc was produced in the brains of sick RML-inoculated Tg(GSS)12 mice (Supplementary Figure 1A, lane 10), the amount was less than rPrPSc produced in the brains of sick RML-infected FVB mice (Supplementary Figure 1A, lane 4). Transmission experiments in wild type FVB mice indicated that the strain properties of RML prions remained unchanged following passage in Tg(GSS)12 mice (Supplementary Information, Supplementary Table I and Supplementary Figure 2).
Interestingly, the electrophoretic profile of 15B3-reactive MoPrP-P101L produced in sick Tg(GSS)12 mice was dependent on whether disease originated from spontaneously sick Tg(GSS)22 mice or RML prions (Figure 3B). While the molecular weight of MoPrP-P101L glycoforms in the brains of sick Tg(GSS)12 mice inoculated with brain material from spontaneously sick Tg(GSS)22 mice ranged from ∼29 to 35 kDa, additional lower molecular weight species extending to ∼21 kDa were present in the brains of Tg(GSS)12 mice inoculated with RML prions. Moreover, the profile of 15B3-reactive MoPrP-P101L in recipient Tg(GSS)12 mice was equivalent to that of disease-associated PrP in the original inocula. Removal of Asn-linked glycans from Mab 15B3 immunoprecipitates with PNGase F demonstrated that these molecular weight differences were related to variations in the endoproteolytic processing of MoPrP-P101L. Previous studies have shown that the endoproteolytic cleavage product of full-length PrPSc appears, following deglycosylation, as an ∼21 kDa C-terminal fragment, referred to as C2 (Chen et al, 1995). While full-length PrPSc and C2 were immuonoprecipitated from brain extracts of sick RML-inoculated wild-type FVB mice or sick RML-inoculated Tg(GSS)12 mice (Figure 3C, lanes 1 and 5). Pathological MoPrP-P101L was not endoproteolytically cleaved in the brains of spontaneously sick Tg(GSS)22 mice (Figure 3C, lanes 3 and 7).
Tremblay et al (2004) established that an ∼22–24 kDa ‘cold PK'-resistant, disease-specific form of PrP could be precipitated from brain extracts of sick Tg(GSS) mice using sodium phosphotungstate (NaPTA). We compared the relative specificities of Mab 15B3 and ‘cold PK'/NaPTA precipitation treatment for detecting disease-associated forms of MoPrP-P101L. While the ‘cold PK'-resistant ∼22–24 kDa fragment was precipitated from the brains of sick Tg(GSS)22 mice by NaPTA (Figure 4, lane 8), additional ‘cold-PK'-resistant 27 and 19 kDa fragments were also precipitated from the brains of sick as well as asymptomatic Tg(GSS)22 mice (Figure 4, lanes 7 and 8). Mab 15B3 immunoprecipitation of ‘cold PK'-treated, NaPTA-precipitated brain extracts demonstrated that only the ∼22–24 kDa fragment was derived from pathological MoPrP-P101L (Figure 4, lanes 9 and 10). Deglycosylated but otherwise untreated brain extracts from RML-infected and uninfected wild-type FVB mice (Figure 4, lanes 1 and 2) indicated that the ∼22–24 kDa fragment corresponded in molecular weight to C2, while the 27 and 19 kDa fragments corresponded to full-length PrP and the PrPC-specific degradation product referred to as C1 (Chen et al, 1995).
Figure 4.
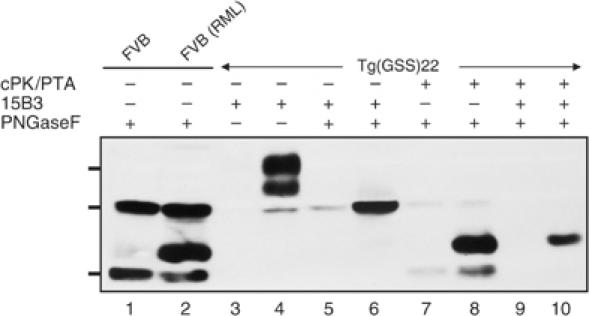
Relative specificities of NaPTA precipitation combined with ‘cold PK' treatment and 15B3 immunoprecipitation for pathological MoPrP-P101L. Lane 1, brain extract from uninfected FVB mouse digested with PNGase F; lane 2, brain extract of sick RML-inoculated FVB mouse digested with PNGase F; lane 3, brain extract from an asymptomatic Tg(GSS)22 mouse immunoprecipitated with Mab 15B3; lane 4, brain extract from a sick Tg(GSS)22 mouse immunoprecipitated with Mab 15B3; lane 5: brain extract from an asymptomatic Tg(GSS)22 mouse immunoprecipitated with Mab 15B3 and digested with PNGase F; lane 6, brain extract from a sick Tg(GSS)22 mouse immunoprecipitated with Mab 15B3 and digested with PNGase F; lane 7, ‘cold PK' digestion followed by NaPTA precipitation, referred to as cPK/PTA, and PNGase digestion of brain extract from an asymptomatic Tg(GSS)22 mouse; lane 8, ‘cold PK' digestion, NaPTA precipitation, and PNGase digestion of brain extract from a sick Tg(GSS)22 mouse; lane 9, ‘cold PK' digestion, NaPTA precipitation, Mab 15B3 immunoprecipitation, and PNGase digestion of brain extract from an asymptomatic Tg(GSS)22 mouse; lane 10, ‘cold PK' digestion, NaPTA precipitation, Mab 15B3 immunoprecipitation, and PNGase digestion of brain extract from a sick Tg(GSS)22 mouse. Samples were analyzed by SDS–PAGE and Western blotting using HRP-conjugated Mab 6H4. The asymptomatic Tg(GSS)22 mouse was killed at 30 days of age while the sick Tg(GSS)22 was killed at 188 days of age. The positions of protein molecular weight markers of 35.5, 28.8, and 22 kDa from top to bottom are shown.
Discussion
Neurodegeneration mediated by protease-sensitive mutant PrP
Despite significant advances, the notion that misfolded PrP is the sole mediator of prion disease still remains contentious (Soto and Castilla, 2004). Since the original Tg(GSS) mice were described, the assertion that de novo production of infectious prions resulted from overexpression of misfolded mutant MoPrP-P101L has met with considerable controversy. Since the prion hypothesis contends that infectivity is associated with PrPSc, which has been defined on the basis of its resistance to protease treatment, the absence of rPrPSc in the brains of sick Tg(GSS) mice as well other examples of disease transmission without rPrPSc (Lasmezas et al, 1997) suggested to some that PrPSc may not be the transmissible component. We reasoned that it might be possible to detect disease-associated forms of mutant PrP in the brains of sick Tg(GSS) mice without resorting to treatment with PK using the PrPSc-specific Mab designated 15B3 (Korth et al, 1997). Here, we demonstrate that Mab 15B3 reacts with a disease-associated, protease-sensitive isoform of MoPrP-P101L, which arises from the spontaneous conversion of mutant PrPC during the course of disease in the brains of overexpressor Tg(GSS) mice.
Characterization of the biochemical properties of pathological MoPrP-P101L
We also show that MoPrP-P101L is capable of producing at least two pathogenic, 15B3-immunoprecipitable protease-sensitive or partially protease-resistant conformations and that the induced conformation depends on the original conformation of PrPSc in the inoculum. These results are consistent with earlier studies showing that the strain-specific properties of prions are enciphered in the tertiary structure of PrPSc (Bessen and Marsh, 1994; Telling et al, 1996b; Mastrianni et al, 1999; Korth et al, 2003). The protease sensitivity of disease-associated MoPrP-P101L produced in spontaneously sick Tg(GSS) mice is not the result of a difference in aggregation state compared to rPrPSc, since the sedimentation profiles of both disease-associated forms are equivalent. These results indicate that Mab 15B3 is capable of recognizing pathological aggregates composed of protease-sensitive and -resistant conformers.
In addition to differences in PK susceptibility, these two MoPrP-P101L conformers were also subject to differences in endoproteolytic processing. PrPSc is cleaved in a process facilitated by calpains (Yadavalli et al, 2004) to form a carboxyl-terminal fragment referred to as C2, which, when deglycosylated, has the same apparent molecular mass as unglycosylated rPrPSc (Chen et al, 1995). While protease-resistant MoPrP-P101L underwent this processing event, protease-sensitive pathological MoPrP-P101L did not (Figure 3C). Thus, the appearance of the C2 fragment appears to correlate with the resistance of PrP to proteolysis by PK. Under conditions of ‘cold PK' digestion in vitro, we confirm that protease-resistant fragments of MoPrP-P101L with molecular weights corresponding to C2 can be detected following precipitation with NaPTA (Tremblay et al, 2004) although Mab 15B3 immunoprecipitation appears to be more selective than NaPTA in precipitating disease-specific forms of PrP (Figure 4).
Effect of mutant transgene expression levels on disease induction by pathological MoPrP-P101L
Another important issue addressed by the current studies relates to the influence of mutant transgene expression levels during disease propagation. The absence of spontaneous disease in Tg(GSS)2 mice is in accordance with the phenotype of 101LL gene-targeted mice (Manson et al, 1999), which have equivalent levels of MoPrP-P101L expression. While accelerated disease was recorded in Tg(GSS)12 mice inoculated either with brain material from spontaneously sick Tg(GSS)22 mice or RML prions, Tg(GSS)2 mice were only susceptible to RML prions with incubation times >530 days. While the lack of disease transmission from spontaneously sick Tg(GSS)22 mice to wild-type mice confirmed previous observations, the lack of transmission to Tg(GSS)2 mice was initially unexpected in light of the uniform susceptibility of Tg196 mice in previous studies (Hsiao et al, 1994; Telling et al, 1996a). However, this apparent discrepancy is readily explained on the basis of transgene copy number and levels of mutant MoPrP-P101L expression in recipient mice. While Tg196 mice did not initially develop spontaneous neurological disease (Hsiao et al, 1994), crossing the transgene array from a wild type to a Prnp0/0 background (Telling et al, 1996a) resulted in spontaneous disease in a subset of mice at ∼550 days of age (Kaneko K, 2000; Tremblay et al, 2004). Tg196 mice were estimated to harbor ∼9 copies of mutant transgene and to express PrP at levels ∼2-fold higher than wild type (Hsiao et al, 1994). The occurrence of spontaneous disease in Tg196 mice is in agreement with the behavior of Tg(GSS)6 mice that harbor ∼6 copies of mutant transgene and develop spontaneous disease at 598±1 days (n=9/9) (Table I).
Our data showing that wild-type and Tg(GSS)2 mice are resistant while Tg(GSS)12 mice are susceptible to the effects of pathological MoPrP-P101L indicate that disease ‘transmission' from spontaneously sick Tg(GSS) mice depends on recipient mice expressing MoPrP-P101L at levels greater than that produced by two transgene copies. Since such levels of overexpression ultimately result in spontaneous disease in older uninoculated recipients, we suggest that the phenomenon of disease ‘transmission' from spontaneously sick Tg(GSS) mice might be more appropriately viewed as disease acceleration whereby inoculation of disease-associated MoPrP-P101L promotes the aggregation of precursors of pathological MoPrP-P101L that result from transgene overexpression. Such a scheme is consistent with a nucleated polymerization mechanism of prion replication originally postulated on the basis of cell-free conversion systems (Kocisko et al, 1994) and subsequently demonstrated to be the basis of prion propagation in lower eukaryotes (Uptain and Lindquist, 2002). According to this model, PrPC is in equilibrium with PrPSc, or its precursor, and the equilibrium normally favors PrPC. Also, PrPSc is stable only in its aggregated form, which can ‘seed' polymerization of additional PrPC, thus converting it into additional PrPSc. However, although 15B3-immunoprecipitable MoPrP-P101L is detected only immediately prior to and during the clinical phase of disease in the brains of spontaneously sick Tg(GSS) mice, we are reluctant to refer to this disease-associated form of mutant PrP as PrPSc in the absence of bona fide disease transmission from sick mice to animals that would not otherwise develop disease.
The role of PrP overexpression in the production of synthetic mammalian prions (SMPs) (Legname et al, 2004) originating from Escherichia coli-derived recombinant MoPrP remains to be determined. While the transmission properties and protease resistance of MoPrP (89–230) SMPs are clearly different from disease-associated MoPrP-P101L, it may be significant that the Tg mice in which these SMPs were initially derived expressed MoPrP(89–231) at levels 16 times higher than normal (Table II).
Table 2.
Accelerated disease in Tg(GSS)12 mice following inoculation of Mab 15B3-immunoprecipitated MoPrP-P101L
| Mab 15B3 on beads | Clinical status of donor Tg(GSS)22 micea | PKb | Incubation time, mean (days)±s.e.m. (n/n0)c |
|---|---|---|---|
| + | Sick | − | 215±4 (8/8) |
| − | Sick | − | >300 (0/8) |
| + | Sick | + | >300 (0/8) |
| + | Healthy | − | >300 (0/8) |
| − | Healthy | − | >300 (0/8) |
| + | Healthy | + | >300 (0/4) |
| aThe sick Tg(GSS)22 mouse was killed at 168 days of age; the healthy Tg(GSS)22 mouse was killed at 28 days of age. | |||
| bTreatment with 20 μg/ml PK for 1 h at 37°C. | |||
| cThe number of inoculated mice developing clinical signs of scrapie divided by the original number of mice is shown in parentheses. | |||
Finally, it is worth comparing the Tg(GSS) mice described in these and previous studies with Tg mice expressing a mouse PrP version of a nine octapeptide insertion associated with familial Creutzfeldt–Jakob disease (CJD), designated Tg(PG14). Tg(PG14) mice exhibit a slowly progressive neurological disorder characterized by apoptotic loss of cerebellar granule cells, gliosis but no spongiosis (Chiesa et al, 1998). Whether the brains of sick Tg(PG14) mice contain 15B3-immunoprecipitable PrP has not yet been reported; however, in both models, mutated PrP adopts different pathologic conformations either spontaneously or following inoculation with authentic prions (Chiesa et al, 2003). Like Tg(GSS) mice, brain homogenates from spontaneously sick Tg(PG14) mice failed to transmit disease to Tg mice that express low levels of mutated PrP and that do not become sick spontaneously. Whether differences in the state of aggregation of PG14spon compared to MoPrP-P101L will affect its ability to accelerate disease progression in overexpressor Tg(PG14) mice remains to be determined.
Materials and methods
Production and characterization of transgenic mice
The substitution of L for P at codon 101 of the MoPrP open reading frame (ORF) was produced by oligonucleotide mismatch, polymerase chain reaction (PCR)-mediated mutagenesis. The resulting ORF cassette was cloned into the cosSHa.Tet cosmid expression vector, which was previously used to produce Tg mice expressing MoPrP-P101L (Telling et al, 1996a). Tg founders were produced by microinjection of fertilized embryos from Prnp0/0 knockout mice on an FVB/N background, referred to as FVB/Prnp0/0. Genomic DNA isolated from mouse tail biopsies was screened by transgene-specific PCR. Transgene copy numbers were determined by real-time PCR using an Applied Biosystems PRISM 7000. Amplification reaction mixtures contained 25 μl of SYBR Green Universal PCR master mix (Applied Biosystems), 5 μl of 18 μM mouse PrP or control mouse actin forward and reverse primers, 5 ng of DNA template, and water to a volume of 50 μl. PCR cycling conditions were 40 cycles of 50°C for 2 min, 95°C for 10 min, and 60°C for 1 min. Nontemplate and Prnp0/0 knockout controls were performed in parallel. All PCR reactions were performed in triplicate. DNA copy numbers were determined using the following equation: gene copy number=2(−ΔΔCt) with

where Ct is the cycle number when the amplified PCR product reaches a fixed threshold, and ΔΔCt is the difference in threshold cycle. Semiquantitative immuno-dot-blotting and Western blotting of brain homogenates from F1 and F2 mice using recombinant PrP-specific Fab D-18 (Peretz et al, 2001) or Mab 6H4 (Korth et al, 1997) were used to estimate PrP expression levels.
Preparation of brain homogenates
Brain homogenates (10% w/v) were prepared by repeated extrusion through an 18-gauge followed by a 21-gauge needle in phosphate-buffered saline (PBS) for transmission experiments, protease digestion, and Western blot analysis and in Prionics Homogenization Buffer for immunoprecipitation experiments. For immunoblot analysis, samples were cleared of cell debris by a brief low-speed centrifugation.
Sources of prion inocula
The mouse-adapted RML scrapie prion isolate (Chandler, 1961) was passaged in Swiss CD-1 mice. For transmission of neurodegeneration from spontaneously sick Tg mice, brain homogenates were prepared as described above. For inoculation studies, mice were anesthetized with a mixture of isoflurane and O2, and inoculated intracerebrally (i.c.) with 30 μl of 1% brain homogenate using a 26-gauge needle inserted into the right parietal lobe.
Determination of clinical symptoms
Groups of uninoculated and inoculated Tg mice were monitored thrice weekly for the development of prion disease. The date of disease was recorded as the age at onset of progressive clinical symptoms of prion disease including truncal ataxia, hind-limb paresis, loss of extensor reflex, and tail stiffening.
Analysis of PrP by immunoblotting
Total protein content from brain extracts was determined by bicinchoninic acid (BCA) assay (Pierce Biotechnology Inc., Rockford, IL). In experiments involving PK digestion of brain homogenates, sarkosyl was added to a final concentration of 2% and samples containing 50–100 μg total protein were incubated for 1 h at 37°C at a PK concentration of 20 μg/ml unless otherwise stated. In experiments involving PK digestion on ice followed by NaPTA precipitation, previously published methods were followed (Tremblay et al, 2004) except that 300 μg of total protein was used instead of 1 ml of clarified 10% brain homogenate. Protease digestions were terminated by the addition of phenyl methyl sulfonyl fluoride (PMSF) to a final concentration of 2 mM. Samples were boiled in an equal volume of 2 × nonreducing sodium dodecyl sulfate (SDS) loading buffer for 5 min and resolved by SDS–polyacrylamide gel electrophoresis (PAGE) using a 12% discontinuous gel. Proteins were electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes and incubated for 10 min in 5% (w/v) nonfat milk in Tris-buffered saline containing Tween 20 (TBST), incubated with appropriate primary and secondary antibodies, developed using enhanced chemiluminescent (ECL) detection (Amersham), and exposed to X-ray film.
Immunoprecipitation
A 300 μl portion of Protein G Dynabeads (Dynal) or mouse anti-IgM Dynabeads was coated with 10 μg Mab 15B3, Mab 6H4, or the respective isotype control antibody. Fifty μl of coated Dynabeads and 5 μl of a 10% (w/v) brain homogenate were added to 1 ml of Tris-buffered saline (TBS) containing 0.3% Sarcosyl and incubated on a rotating wheel for 1 h. For PK treatment, immune complexes on beads were suspended in 50 μl PBS containing 2% sarkosyl and incubated for 1 h at 37°C with 20 μg/ml PK. Digestion was terminated by the addition of PMSF to a final concentration of 2 mM. Beads were washed three times with TBS containing 0.25% Sarcosyl. For deglycosylation of immunoprecipitated PrP, beads were treated with Peptide:N glycosidase F (PNGase F) for 3 h at 37°C, as specified by the supplier (New England Biolabs, Beverly, MA). Beads were suspended in 20 μl of 2 × nonreducing SDS loading buffer, heated for 5 min at 96°C, and immunoprecipitated proteins were resolved by SDS–PAGE using a 12% discontinuous gel. Immunoprecipitated proteins were electrophoretically transferred to PVDF membranes and incubated for 10 min in 2% (w/v) blocking solution provided in the Advanced ECL detection kit (Amersham Biosciences, Piscataway, NJ) in TBST. Membranes were probed with Mab 6H4 conjugated to horseradish peroxidase (HRP) at a 1:5000 dilution in TBST, developed using Advanced ECL detection kit (Amersham Biosciences, Piscataway, NJ), and exposed to X-ray film.
Sucrose gradient centrifugation
A 1.5 ml portion of 5, 10, 15, 20, 30, 40, and 50% sucrose solutions prepared in PBS were loaded into centrifugation tubes adapted for the Surespin™630 rotor of the Discovery 90SE ultracentrifuge (Sorvall) to form a zonal gradient. Mouse brain homogenates (10% w/v) prepared in PBS were centrifuged at 50 000 g for 30 min at 4°C and 200 μl of supernatant was loaded on top of the gradient and centrifuged at 100 000 g for 4 h at 4°C. A total of 12 1 ml fractions were collected from the top of the gradient and proteins were precipitated by the addition of 10 volumes of ice-cold methanol. After centrifugation at 4000 g for 15 min, the pellet was dissolved in 100 μl of homogenization buffer. Aliquots (20 μl) from each fraction were immunoprecipitated using Mab 15B3 or 6H4 and analyzed by Western blotting as described above.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported in part by grants from the US Public Health Service RO1 NS/AI40334 from the National Institute of Neurological Disorders and Stroke and N01-AI-25491 from the National Institute of Allergy and Infectious Diseases.
References
- Bessen RA, Marsh RF (1992) Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 66: 2096–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen RA, Marsh RF (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68: 7859–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC (1994) Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35: 513–529 [DOI] [PubMed] [Google Scholar]
- Chandler RL (1961) Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet 1: 1378–1379 [DOI] [PubMed] [Google Scholar]
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L (1995) Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 270: 19173–19180 [DOI] [PubMed] [Google Scholar]
- Chesebro B (1998) BSE and prions: uncertainties about the agent. Science 279: 42–43 [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA (1998) Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21: 1339–1351 [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Ghetti B, Harris DA (2003) Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol 77: 7611–7622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen FE, Pan K-M, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB (1994) Structural clues to prion replication. Science 264: 530–531 [DOI] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15: 1255–1264 [PMC free article] [PubMed] [Google Scholar]
- Fischer MB, Roeckl C, Parizek P, Schwarz HP, Aguzzi A (2000) Binding of disease-associated prion protein to plasminogen. Nature 408: 479–483 [DOI] [PubMed] [Google Scholar]
- Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott J, Prusiner SB (1989) Linkage of a prion protein missense variant to Gerstmann–Sträussler syndrome. Nature 338: 342–345 [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Groth D, Scott M, Yang S-L, Serban H, Rapp D, Foster D, Torchia M, DeArmond SJ, Prusiner SB (1994) Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc Natl Acad Sci USA 91: 9126–9130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB (1990) Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 250: 1587–1590 [DOI] [PubMed] [Google Scholar]
- Kaneko K, Ball HL, Wille H, Zhang H, Groth D, Torchia M, Tremblay P, Safar J, Prusiner SB, DeArmond SJ, Baldwin MA, Cohen FE. (2000) A synthetic peptide initiates Gerstmann–Straussler–Scheinker (GSS) disease in transgenic mice. J Mol Biol 295: 997–1007 [DOI] [PubMed] [Google Scholar]
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT Jr, Caughey B (1994) Cell-free formation of protease-resistant prion protein. Nature 370: 471–474 [DOI] [PubMed] [Google Scholar]
- Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, Parchi P, Gambetti P, Will R, Ironside J, Heinrich C, Tremblay P, DeArmond SJ, Prusiner SB (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse–human prion protein transgene. Proc Natl Acad Sci USA 100: 4784–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, Schulz-Schaeffer W, Kretzschmar H, Raeber A, Braun U, Ehrensperger F, Hornemann S, Glockshuber R, Riek R, Billeter M, Wuthrick K, Oesch B (1997) Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 389: 74–77 [DOI] [PubMed] [Google Scholar]
- Lasmezas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, Fournier JG, Hauw JJ, Rossier J, Dormont D (1997) Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 275: 402–405 [DOI] [PubMed] [Google Scholar]
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB (2004) Synthetic mammalian prions. Science 305: 673–676 [DOI] [PubMed] [Google Scholar]
- Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, Somerville R, Ironside J, Will R, Sy MS, Melton DW, Hope J, Bostock C (1999) A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J 18: 6855–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ, Prusiner SB (1999) Prion protein conformation in a patient with sporadic fatal insomnia. N Engl J Med 340: 1630–1638 [DOI] [PubMed] [Google Scholar]
- Morel N, Simon S, Frobert Y, Volland H, Mourton-Gilles C, Negro A, Sorgato MC, Creminon C, Grassi J (2004) Selective and efficient immunoprecipitation of the disease-associated form of the prion protein can be mediated by nonspecific interactions between monoclonal antibodies and scrapie-associated fibrils. J Biol Chem 279: 30143–30149 [DOI] [PubMed] [Google Scholar]
- Moroncini G, Kanu N, Solforosi L, Abalos G, Telling GC, Head M, Ironside J, Brockes JP, Burton DR, Williamson RA (2004) Motif-grafted antibodies containing the replicative interface of cellular PrP are specific for PrPSc. Proc Natl Acad Sci USA 101: 10404–10409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SBH, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE, Prusiner SB, Weissmann C (1985) A cellular gene encodes scrapie PrP 27–30 protein. Cell 40: 735–746 [DOI] [PubMed] [Google Scholar]
- Paramithiotis E, Pinard M, Lawton T, LaBoissiere S, Leathers VL, Zou WQ, Estey LA, Lamontagne J, Lehto MT, Kondejewski LH, Francoeur GP, Papadopoulos M, Haghighat A, Spatz SJ, Head M, Will R, Ironside J, O'Rourke K, Tonelli Q, Ledebur HC, Chakrabartty A, Cashman NR (2003) A prion protein epitope selective for the pathologically misfolded conformation. Nat Med 9: 893–899 [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB (2001) Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412: 739–743 [DOI] [PubMed] [Google Scholar]
- Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216: 136–144 [DOI] [PubMed] [Google Scholar]
- Rhie A, Kirby L, Sayer N, Wellesley R, Disterer P, Sylvester I, Gill A, Hope J, James W, Tahiri-Alaoui A (2003) Characterization of 2′-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J Biol Chem 278: 39697–39705 [DOI] [PubMed] [Google Scholar]
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB (1998) Eight prion strains have PrPSc molecules with different conformations. Nat Med 4: 1157–1165 [DOI] [PubMed] [Google Scholar]
- Sayer NM, Cubin M, Rhie A, Bullock M, Tahiri-Alaoui A, James W (2004) Structural determinants of conformationally selective, prion-binding aptamers. J Biol Chem 279: 13102–13109 [DOI] [PubMed] [Google Scholar]
- Shaked Y, Engelstein R, Gabizon R (2002) The binding of prion proteins to serum components is affected by detergent extraction conditions. J Neurochem 82: 1–5 [DOI] [PubMed] [Google Scholar]
- Soto C, Castilla J (2004) The controversial protein-only hypothesis of prion propagation. Nat Med 10 (Suppl): S63–S67 [DOI] [PubMed] [Google Scholar]
- Tateishi J, Kitamoto T (1995) Inherited prion diseases and transmission to rodents. Brain Pathol 5: 53–59 [DOI] [PubMed] [Google Scholar]
- Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB (1996a) Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 10: 1736–1750 [DOI] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB (1996b) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274: 2079–2082 [DOI] [PubMed] [Google Scholar]
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83: 79–90 [DOI] [PubMed] [Google Scholar]
- Tremblay P, Ball HL, Kaneko K, Groth D, Hegde RS, Cohen FE, DeArmond SJ, Prusiner SB, Safar JG (2004) Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J Virol 78: 2088–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G, Gabizon R, Taraboulos A (2002) Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41: 12868–12875 [DOI] [PubMed] [Google Scholar]
- Uptain SM, Lindquist S (2002) Prions as protein-based genetic elements. Annu Rev Microbiol 56: 703–741 [DOI] [PubMed] [Google Scholar]
- Yadavalli R, Guttmann RP, Seward T, Centers AP, Williamson RA, Telling GC (2004) Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J Biol Chem 279: 21948–21956 [DOI] [PubMed] [Google Scholar]
- Zou WQ, Zheng J, Gray DM, Gambetti P, Chen SG (2004) Antibody to DNA detects scrapie but not normal prion protein. Proc Natl Acad Sci USA 101: 1380–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information