

Abstract
Salivary gland tumors, a group of histologically diverse benign and malignant neoplasms, represent a challenging problem for diagnosis and treatment. A specific recurring t(11;19)(q21;p13) translocation is associated with two types of salivary gland tumors, mucoepidermoid carcinomas and Warthin's tumors. This translocation generates a fusion protein comprised of the N-terminal CREB (cAMP response element-binding protein)-binding domain of the CREB regulator MECT1 (Mucoepidermoid carcinoma translocated-1) and the C-terminal transcriptional activation domain of the Notch coactivator Mastermind-like 2 (MAML2). Here, we demonstrate that the MECT1-MAML2 fusion protein induces expression of multiple genes known to be CREB transcriptional targets. MECT1-MAML2 was found to bind to CREB, recruit p300/CBP into the CREB complex through a binding domain on MAML2, and constitutively activate CREB-dependent transcription. The transforming activity of MECT1-MAML2 was markedly reduced by blocking CREB DNA binding. Thus, this fusion oncogene mimics constitutive activation of cAMP signaling, by activating CREB directly. This study has identified a novel, critical mechanism of transformation for an oncogene associated very specifically with salivary gland tumors, and identified potential targets for the development of novel therapies.
Keywords: CREB, MECT1-MAML2 fusion, mucoepidermoid carcinoma, transformation, Warthin's tumor
Introduction
Salivary gland tumors are a group of highly heterogeneous benign and malignant neoplasms arising from the major or minor salivary glands, and account for approximately 5% of all head-and-neck cancers. The clinical management of these tumors is complicated by their varied biological behaviors and relative infrequency (McGurk and Renehan, 2001). Understanding the molecular changes in different types of salivary gland tumors is important for both the treatment and diagnosis of these diseases. Mucoepidermoid carcinomas (MEC) are the most common human malignant salivary gland and the second most common bronchial gland tumors, while Warthin's tumors are the second most common benign parotid gland tumors. Although these two types of tumors are histologically distinct, both are associated with a specific recurring chromosomal t(11;19)(q14–21;p12–13) translocation, suggesting that the chromosomal rearrangement is directly involved in their pathogenesis (Martins et al, 1996, 1997, 2004).
The genes involved in the t(11;19) translocation in these tumors have been identified, and consist of a novel gene at 19p13 termed Mucoepidermoid carcinoma translocated-1 (MECT1, also called TORC1) and a member of the Mastermind-like gene family (MAML2) at 11q21 (Conkright et al, 2003; Tonon et al, 2003; Enlund et al, 2004). Intriguingly, the MECT1 gene product was recently characterized as a binding protein for CREB (cAMP response element-binding protein) transcription factor (Conkright et al, 2003; Iourgenko et al, 2003). CREB belongs to the class of basic domain-leucine zipper transcription factors and binds as a dimer to cAMP-responsive elements (CRE), 8-bp palindromic sequences TGACGTCA (Johannessen et al, 2004). In response to cAMP signaling and activation of protein kinase A (PKA), CREB is phosphorylated at Ser133 resulting in the recruitment of the coactivators CREB-binding protein (CBP) and p300, thereby activating the transcription of cAMP-responsive genes. In normal cells, MECT1 acts as a CREB regulator, functioning by binding to CREB and enhancing transcription of a CRE-containing reporter independently of CREB phosphorylation (Conkright et al, 2003; Iourgenko et al, 2003). The MAML2 gene, on the other hand, encodes a transcriptional coactivator for Notch receptors (Wu et al, 2002). Notch signaling is an evolutionarily conserved mechanism in which cell–cell interactions may determine cell fates, and is important in the development of many tissues (Artavanis-Tsakonas et al, 1999). MAML2 is part of the Notch transcriptional complex, which involves the transcription factor CSL and the nuclear activated form of Notch receptor (ICN) after ligand stimulation, and is required for transcriptional activation of the Notch target genes (Wu et al, 2002). The consequence of this chromosome translocation is the creation of a novel fusion protein, MECT1-MAML2, which is composed of the 42 aa CREB-binding domain from MECT1 with the 981 aa transcriptional activation domain (TAD) from MAML2 (Tonon et al, 2003; Enlund et al, 2004).
The MECT1-MAML2 fusion has transforming activity, as indicated by the finding that it independently induces colony formation in the E1A-immortalized rat kidney epithelial cell line, RK3E (Tonon et al, 2003). However, the mechanism of transformation is unknown. Interestingly, several known Notch target genes are induced independent of Notch ligand stimulation, including Hairy-Enhancer of Split-1 (HES-1) and HES-related protein (HERP). Since previous studies indicate that Notch signaling is important in salivary gland development in Drosophila (Haberman et al, 2003), it was postulated that altered Notch signaling might affect gland cell differentiation and therefore contribute to the transforming activity of MECT1-MAML2. However, the mechanism of activation of Notch target gene expression by MECT1-MAML2 is unknown, and we have previously shown that this activity does not require the normal Notch transcription factor (CSL)-binding sites in the Notch target HES-1 promoter (Tonon et al, 2003).
While activation of Notch target genes is still an attractive mechanism responsible for its transforming activity, Conkright et al (2003) suggested that interference of the CREB pathway by the MECT1-MAML2 fusion may also be important. First, the fusion retains the CREB-binding domain from the MECT1 translocation partner and is able to bind CREB. Second, the fusion induces activation of a reporter containing the CRE site, and a dominant-negative CREB expression plasmid (A-CREB), known to interfere with the CREB DNA binding, blocked such activation. Since the CREB complex mediates basic cellular processes including cell growth and survival in response to a variety signals, and is also implicated in a number of cancers including endocrine tumors, melanoma, and leukemias (Yu and Melmed, 2001; Poser and Bosserhoff, 2004; Shankar and Sakamoto, 2004), targeting of the CREB pathway might also be critical in mediating MECT1-MAML2 fusion's transforming activity.
In preliminary studies with expression arrays, we found that MECT1-MAML2 induced expression of multiple genes previously shown to be responsive to cAMP signaling in epithelial cells. In an effort to understand the role of the CREB pathway in transformation by the fusion protein, we have examined the role of the CREB pathway in cellular transformation and analyzed the mechanism of activation of this pathway by MECT1-MAML2.
Results
Transforming activity of the MECT1-MAML2 fusion protein requires both the CREB-binding domain from MECT1 and the TAD domain from MAML2
MECT1-MAML2 is a fusion of the N-terminal CREB-binding domain of MECT1 (42 aa) with the C-terminal TAD of the Notch coactivator MAML2 (981 aa). Previously, we showed that the MECT1-MAML2 fusion efficiently induces foci formation in E1A-immortalized RK3E cells, while the fusion partner MAML2 has no activity (Tonon et al, 2003). To determine if the other fusion partner, MECT1, might have transforming activity, MECT1 was introduced into RK3E cells and focus formation was quantified. In contrast to MECT1-MAML2, the MECT1 gene product itself was unable to induce focus formation in RK3E cells (Figure 1A). Therefore, only the MECT1-MAML2 fusion, but not the two genes involved in the translocation, is able to transform.
Figure 1.
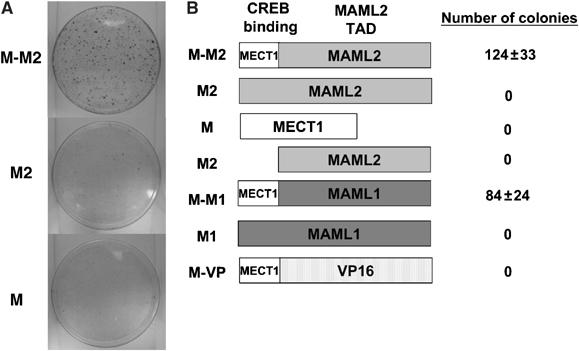
Both the CREB-binding domain of MECT1 and the TAD domain of MAML2 are required for the MECT1-MAML2 fusion to induce foci formation in RK3E cells. (A) RK3E cells were transfected with the indicated expression plasmids, and stained with crystal violet for foci formation at 3 weeks post-transfection. (B) Diagram of the constructs used in focus assays, and the number of foci generated from the transfection of each of these constructs. The number of foci shown was obtained from the 10 cm plates based on the triplicate experiments. M-M2, M2, M, M1, M-M1, and M-VP stand for MECT1-MAML2 fusion, MAML2, MECT1, MAML1, MECT1-MAML1, and MECT1-VP16, respectively. All these genes were cloned into the pFLAG-CMV2 expression vector, and expressed as FLAG-tagged proteins.
The motifs within MECT1-MAML2 fusion required for transformation were evaluated by using chimeric proteins including MECT1-MAML1 and MECT1-VP16, generated by replacing the TAD domain of MAML2 in MECT1-MAML2 fusion with the corresponding TAD domain of MAML1, another member of the MAML family (Wu et al, 2002), and the unrelated activation domain of VP16, respectively (Tonon et al, 2003). Colony formation was observed for MECT1-MAML1, but not for MECT1-VP16 nor the full-length MAML1 (Figure 1B). These results indicate that the MAML1 TAD domain can functionally replace MAML2 TAD, suggesting that this TAD domain of the MAML family has unique properties contributing to MECT1-MAML2-mediated transformation. Moreover, the TAD domain of wild-type MAML2 did not produce colonies, indicating that the domain is not sufficient for transformation; therefore, the N-terminal 42 residues of the MECT1 gene are also required for the transforming activity of the fusion protein. In conclusion, the transforming activity of MECT1-MAML2 fusion results from the unique activity generated by both the MECT1 CREB-binding domain and the MAML2 TAD, which are brought together by the t(11;19) translocation.
Microarray analysis identified downstream targets differentially regulated by the MECT1-MAML2 fusion oncoprotein including those normally regulated by cAMP signaling
For a systematic analysis of potential MECT1-MAML2 target genes contributing to transformation, we used Affymetrix human U133A array to compare the transcription profiles of HeLa cells that were transiently transfected with the MECT1-MAML2 fusion, MAML2, MECT1, or vector. Expression of MECT1-MAML2 fusion and its fusion partners was confirmed (Supplementary Figure S1). With at least two-fold changes at the transcript levels considered as significant, we found that there is a unique set of genes differentially regulated by the MECT1-MAML2 fusion compared to expression of either fusion partner alone (see Table I). These genes are clearly involved in many critical cellular processes including cell growth, differentiation, and apoptosis, suggesting that the fusion protein may interfere with multiple cellular processes that lead to cell transformation. The identified set of target genes (Table I) included seven genes previously reported to be regulated by cAMP, including the transcription factors, ATF3 (Hai and Hartman, 2001), CREM (Daniel et al, 2000), FOS (Ahn et al, 1998), and two members of the nuclear receptor subfamily 4, NR4A1 (Nur77) and NR4A2 (Nurr1) (Kovalovsky et al, 2002); inhibitor of DNA-binding protein, Id2 (Scobey et al, 2004); and stanniocalcin 1 (Wong et al, 2002).
Table 1.
Target genes regulated by the MECT1-MAML2 fusion oncoprotein in HeLa cells
| Gene symbol | Description | M-M2 fold change | MAML2 fold change | MECT1 fold change | Family |
|---|---|---|---|---|---|
| ADAMTS5 | A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motif, 5 (aggrecanase-2) | 2.5 | −0.5 | 1.6 | Protease |
| ATF3 | Activating transcription factor 3 | 6.6 | 1.5 | 1.3 | Transcription regulator |
| BNIP3L | BCL2/adenovirus E1B 19 kDa interacting protein 3-like | 3.7 | 0.2 | −0.5 | — |
| CARD14 | Caspase recruitment domain family, member 14 | 2.8 | 0.1 | 1.2 | — |
| CLK1 | CDC-like kinase 1 | 3.0 | 0.4 | 1.7 | Kinase |
| CREM | cAMP responsive element modulator | 2.0 | −0.8 | 1.8 | Transcription regulator |
| CYP26B1 | Cytochrome P450, family 26, subfamily B, polypeptide 1 | 2.5 | 0.0 | 1.9 | Cytochrome P450 |
| CYR61 | Cysteine-rich, angiogenic inducer, 61 | 2.5 | 2.1 | 0.0 | — |
| DKFZp451J1719 | Hypothetical DKFZp451J1719 | 5.6 | 0.6 | −0.1 | — |
| DNAJB9 | DnaJ (Hsp40) homolog, subfamily B, member 9 | 7.6 | −1.2 | −1.6 | Transcription regulator |
| DUSP1 | Dual specificity phosphatase 1 | 2.2 | 1.0 | −1.0 | Phosphatase |
| FKBP8 | FK506 binding protein 8, 38 kDa | 2.7 | 1.4 | 1.6 | — |
| FOS | v-fos FBJ murine osteosarcoma viral oncogene homolog | 3.6 | −2.3 | 1.3 | Transcription regulator |
| FOSB | FBJ murine osteosarcoma viral oncogene homolog B | 6.2 | −0.7 | 0.1 | Transcription regulator |
| FOSL2 | FOS-like antigen 2 | 2.4 | −0.7 | 1.1 | Transcription regulator |
| GABARAPL1 | GABA(A) receptor-associated protein like 1 | 3.0 | 0.8 | −1.1 | — |
| GEM | GTP binding protein overexpressed in skeletal muscle | 3.9 | 0.5 | 1.6 | Enzyme |
| HOXA5 | Homeo box A5 | 6.4 | −0.5 | 1.2 | Transcription regulator |
| ID2 | Inhibitor of DNA binding 2, dominant negative helix–loop–helix protein | 4.6 | −0.3 | 1.4 | — |
| INSL4 | Insulin-like 4 (placenta) | 2.4 | 0.6 | 1.5 | — |
| KLF4 | Kruppel-like factor 4 (gut) | 2.8 | 0.7 | 1.3 | Transcription regulator |
| KRT17 | Keratin 17 | 2.5 | −0.4 | 1.4 | — |
| MT1X | Metallothionein 1X | 2.3 | 1.3 | 1.3 | — |
| NDRG1 | N-myc downstream regulated gene 1 | 2.8 | −0.1 | 0.1 | — |
| NR4A1 | Nuclear receptor subfamily 4, group A, member 1 (Nur77) | 3.9 | 0.8 | −1.6 | Ligand-dependent nuclear receptor |
| NR4A2 | Nuclear receptor subfamily 4, group A, member 2 (Nurr1) | 6.7 | −0.1 | 1.7 | Ligand-dependent nuclear receptor |
| PIGA | Phosphatidylinositol glycan, class A (paroxysmal nocturnal hemoglobinuria) | 4.1 | 0.7 | 0.2 | Enzyme |
| PMP22 | Peripheral myelin protein 22 | 2.1 | −0.7 | 1.3 | — |
| SAP18 | sin3-associated polypeptide, 18 kDa | 2.6 | 0.6 | 0.1 | Transcription regulator |
| SLC16A6 | Solute carrier family 16 (monocarboxylic acid transporters), member 6 | 2.4 | 1.1 | 1.4 | — |
| SLC20A1 | Solute carrier family 20 (phosphate transporter), member 1 | 2.0 | 1.1 | 0.2 | Transporter |
| SLC2A3 | Solute carrier family 2 (facilitated glucose transporter), member 3 | 2.4 | 1.3 | 1.8 | Transporter |
| SNRK | SNF-1 related kinase | 2.6 | 1.4 | 1.1 | Kinase |
| STC1 | Stanniocalcin 1 | 5.2 | 1.3 | 1.9 | — |
| TFF1 | Trefoil factor 1 (breast cancer, estrogen-inducible sequence expressed) | 10.1 | 1.6 | 0.2 | Growth factor |
| TM4SF1 | Transmembrane 4 superfamily member 1 | 2.1 | 0.6 | 1.2 | — |
| YPEL5 | Yippee-like 5 (Drosophila) | 3.0 | −0.3 | 1.3 | — |
| ZBED1 | Zinc finger, BED domain containing 1 | 2.3 | −0.7 | 1.5 | — |
| The bold lines indicate genes previously reported to be regulated by camp. |
To validate these expression array data, we performed real-time PCR assays for five target genes, including two known cAMP-responsive genes, Nurr1 and ATF3, and three genes previously unknown to be regulated by cAMP, DUSP1, TFF1, and NDRG1, and showed that all these genes are indeed upregulated when the fusion is expressed (Figure 2A).
Figure 2.
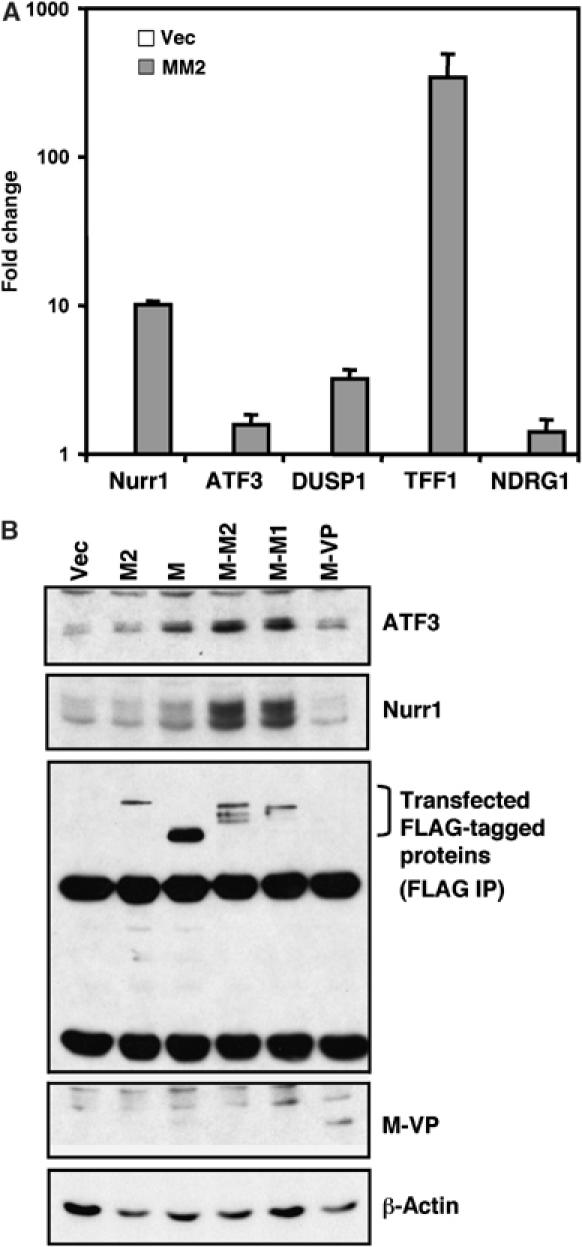
MECT1-MAML2 fusion induces expression of CREB transcriptional target genes. (A) Expression levels of five target genes (both known and previously unknown regulated by cAMP signaling) in MECT1-MAML2-transfected cells, relative to the empty vector-transfected cells, were determined by SYBR green real-time PCR. (B) Induction of two known CREB targets, ATF3 and Nurr1 genes, wasanalyzed by Western blot analyses in human immortalized parotid HSY cells transfected with the indicated expression constructs. Transfected FLAG-tagged proteins except MECT1-VP16 (its band overlaps with IgG light chain, and the expression is shown on the whole-cell lysate blot) were detected after immunoprecipitation (IP) with anti-FLAG antibodies. M2, M, M-M2, M-M1, and M-VP stand for MAML2, MECT1, MECT1-MAML2, MECT1-MAML1, and MECT1-VP16, respectively.
MECT1-MAML2 induces expression of multiple targets of the cAMP signaling pathway; thus, we investigated mechanisms of activation of the cAMP signaling pathway via the fusion oncogene. Consistent with the microarray and real-time PCR data, Western blot analysis revealed that the MECT1-MAML2 fusion induced the expression of these two known target genes in the CREB pathway, Nurr1 and ATF3, at much higher levels than did either MAML2 or MECT1 alone in the human immortalized parotid cell line HSY (Figure 2B). Overexpression of the MECT1 gene caused only a modest induction of Nurr1 and ATF3 genes, despite the fact that its expression level was much higher than that of MECT1-MAML2. This might be explained in part by the difference in their subcellullar distribution: MECT1 is primarily localized in the cytoplasm (Supplementary Figure S2), while MECT1-MAML2 is localized in the nucleus when expressed as GFP-tagged proteins (Tonon et al, 2003). It was recently shown that increasing intracellular cAMP or calcium levels induces nuclear transport of MECT1 (TORC1), which is subsequently sufficient to activate CRE-dependent transcription (Bittinger et al, 2004). Thus, these data suggest that MECT1-MAML2 is a constitutive activator for CREB-mediated transcription, while MECT1 requires additional signaling for such transcriptional activation. Interestingly, while the chimeric protein MECT1-MAML1 activates expression of Nurr1 and ATF3 at high levels, the MECT1-VP16 chimeric protein does not (Figure 2B). These data were consistent with the differential activities of these proteins to activate the CRE promoter reporter (Supplementary Figure S3).
Combined with our results from the colony formation studies (Figure 1), the ability of the MECT1-MAML2 fusion to activate the CREB pathway appears to correlate with its ability to induce colonies in RK3E cells.
Disruption of CREB activity reduces the transforming activity of MECT1-MAML2
The importance of activation of the CREB pathway in mediating MECT1-MAML2 transforming activity was then evaluated using a well-characterized dominant-negative mutant, A-CREB, which specially blocks CREB binding to DNA by heterodimerizing with CREB (Ahn et al, 1998). RK3E cells transduced with A-CREB viruses (expressing A-CREB plus GFP), or control viruses (expressing GFP only) were transfected with MECT1-MAML2 to examine the expression of CREB target genes at 48 h post-transfection and to score for colonies after 3 weeks. We first confirmed by Western blot analysis that A-CREB is expressed in RK3E cells infected with A-CREB viruses (Figure 3A). The induction of Nurr1 and ATF3 proteins by MECT1-MAML2 was significantly reduced in RK3E cells expressing A-CREB, as compared to the control cells (Figure 3B). Importantly, expression of A-CREB also led to a significantly decreased number of colonies (Figure 3C). However, we found that A-CREB expression did not affect the number of colonies induced by an activating mutation of B-RAF proto-oncogene (V599E). Thus, these data indicate that interference of CREB activity reduced the activation of the CREB pathway, and also decreased the transforming activity of the fusion protein. Moreover, by knocking down the endogenous CREB level with RNAi in 293T cells, we found by Western blot analysis and/or real-time PCR that there are reduced expression levels of known CREB target genes, Nurr1 and ATF3, as well as three other target genes, DUSP1, TFF1, and NDRG1, previously unknown to be regulated by CREB, suggesting that induction of these genes might be dependent on CREB (Figures 3D and E). Taken together, CREB activity is important for activation of the CREB pathway and cellular transformation via the MECT1-MAML2 fusion.
Figure 3.
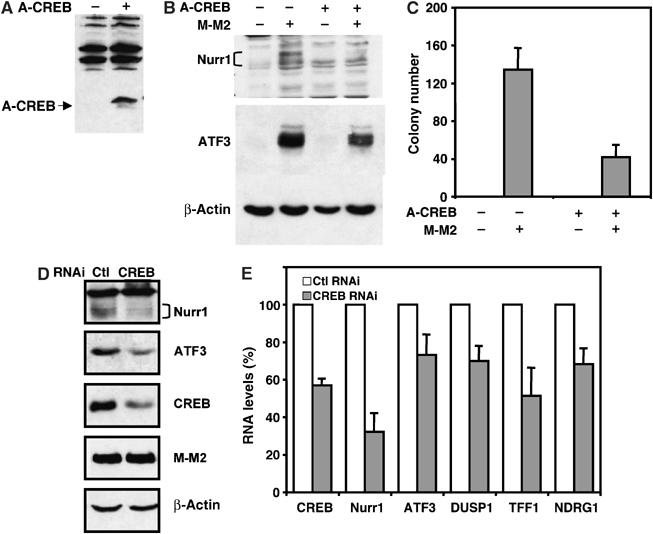
Disruption of CREB activity reduced the abilities of the MECT1-MAML2 fusion to activate the CREB pathway and to induce colony formation in RK3E. (A) Expression of A-CREB in RK3E cells transduced with A-CREB viruses was determined by Western blot analysis. (B) RK3E cells expressing A-CREB and GFP, and the control cells expressing GFP alone, were transfected with either vector or the MECT1-MAML2 expression construct. The expression levels of two CREB target genes, ATF3 and Nurr1, were analyzed by Western blot analysis. (C) A-CREB-expressing RK3E cells and the control cells were transfected with either vector only or the expression construct encoding MECT1-MAML2 fusion, and the colonies were scored 3 weeks after transfection. (D) 293T cells were cotransfected with the MECT1-MAML2 expression vector and CREB RNAi (or control RNAi), and the levels of ATF3, Nurr1, CREB, and transfected MECT1-MAML2 fusion were analyzed 48 h after transfection by Western blotting. (E) Expression levels of CREB, and five fusion target genes in 293T cells cotransfectd with MECT1-MAML2 and CREB RNAi, relative to cells cotransfected with MECT1-MAML2 and control RNAi were determined by SYBR green real-time PCR.
Next, we asked if CREB activity is essential for the growth of MECT1-MAML2 fusion-expressing MEC cell lines. Two MECT1-MAML2-expressing MEC cell lines, H292 and H3118, along with the immortalized MECT1-MAML2-negative cell line HSY were transduced either with A-CREB or control GFP viruses. The multiplicity of infection (MOI) was optimized to obtain an infection rate of 30–50%, and then the percentage of GFP-positive populations was determined at 3 or 4 days intervals, over a total time period of 15 days. As shown in Figure 4A, the percentage of MECT1-MAML2-positive H3118 and H292 cells that expressed A-CREB was progressively reduced, while their control, GFP-only-expressing cells did not have a growth disadvantage. In contrast, the percentage of HSY cells (lacking expression of MECT1-MAML2) that expressed A-CREB reduced only modestly. These results suggest that expression of A-CREB in MECT1-MAML2-positive MEC cells, but not in MECT1-MAML2-negative cells, resulted in a negative effect on cellular growth. In order to directly compare growth rates, GFP-positive cells were purified by fluorescence activated cell sorting. We found that A-CREB expression inhibited the growth of the MECT1-MAML2-positive MEC cells, but not the control HSY cells (Figure 4B). Finally, we investigated the effect of A-CREB expression on the cell cycle and apoptosis. We found that A-CREB expression caused an increased percentage of cells in the G1 phase in both H3118 and H292, as compared with their GFP control cells, where no significant difference was seen with HSY cells (Figure 4C). In addition, A-CREB expression did not lead to apoptosis in both M-M2-negative and -positive cells (not shown). Therefore, the inhibition of CREB activity via the dominant-negative A-CREB resulted in a significant inhibition of growth in the MECT1-MAML2-positive cells, but only marginally affected MECT1-MAML2-negative cells. These results demonstrate that CREB activity is critical for the growth of MECT1-MAML2-positive MEC cells.
Figure 4.
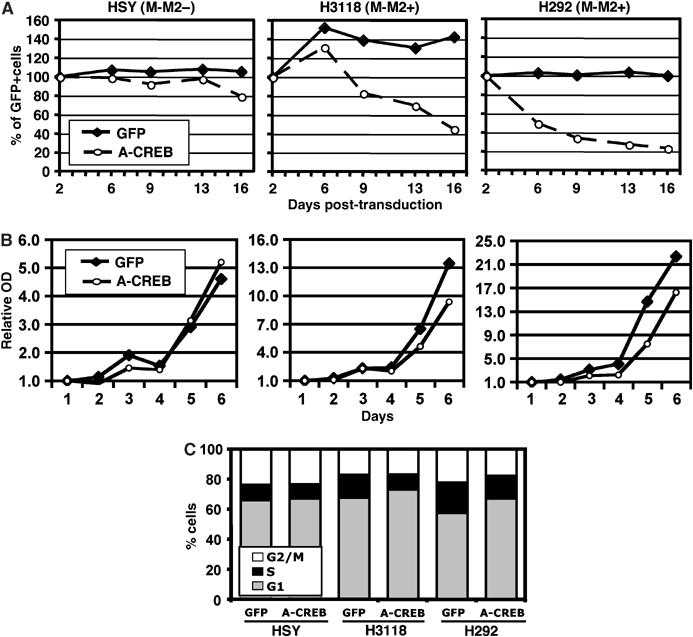
Disruption of CREB activity significantly suppressed the growth of two human MECT1-MAML2-positive MEC cell lines. (A) A representative diagram showing the changes in the percentage of GFP-positive cells between cell populations expressing A-CREB and GFP and their control counterparts expressing GFP only. Two MECT1-MAML2-expressing MEC cell lines, H292 and H3118, along with the immortalized normal parotid cell line HSY (MECT1-MAML2 negative), were transduced with A-CREB or control GFP viruses. The percentages of GFP-positive cells were determined by FACS analysis at 3–4 days intervals, for a total of 15 days. The percentage of GFP-positive cells at day 2 postinfection was considered as 100%, and the remaining data were normalized. (B) A representative growth curve of cells expressing A-CREB plus GFP versus controls expressing GFP only. The cell number was determined using crystal blue staining daily for 6 days. (C) Cell cycle distribution of HSY, H3118, and H292 cells transduced with A-CREB viruses or vector control viruses as determined by propidium iodide straining followed by FACS analysis.
The ability of the MECT1-MAML2 fusion to recruit p300/CBP to the CRE transcriptional complex is partially responsible for activation of the CREB pathway and its transforming activity
Signal-mediated activation of the cAMP/PKA pathway leads to phosphorylation of CREB at Ser133, which subsequently recruits p300/CBP coactivators (Johannessen et al, 2004). The recruitment of p300/CBP is a critical event for the CREB complex to activate transcription. We therefore considered the possibility that either MECT1-MAML2 leads to the phosphorylation of CREB, allowing the recruitment of p300/CBP, or that MECT1-MAML2 itself recruits transcriptional coactivators such as p300/CBP to CREB. Either of these mechanisms would allow the fusion to bypass cAMP signaling-dependent phosphorylation events. We found that the overexpression of the fusion in 293T cells did not induce phosphorylation of CREB at Ser133 (Supplementary Figure S4). Therefore, phosphorylation of CREB is unlikely to be the mechanism by which MECT1-MAML2 activates the CREB pathway.
To test the possibility that MECT1-MAML2 recruits p300/CBP, we first determined whether p300/CBP colocalizes with the MECT1-MAML2 fusion. We found that p300 is localized in the nucleus with a diffuse staining pattern when expressed alone in U20S cells. However, p300 showed a nuclear dot pattern, and colocalized with the fusion protein when coexpressed with MECT1-MAML2 (Figure 5A).
Figure 5.
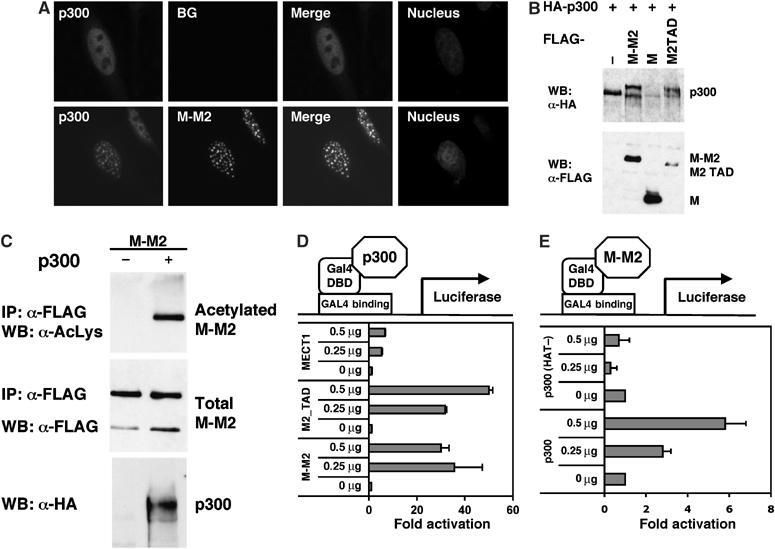
MECT1-MAML2 interacts with the CREB pathway regulatory molecule p300. (A) MECT1-MAML2 and p300 colocalize in the nuclei. U20S cells were transfected with the GFP-tagged MECT1-MAML2 and HA-tagged p300, and stained with an anti-HA antibody for p300 expression. The DAPI staining labels the nuclei of the same cells. (B) MECT1-MAML2 post-translationally modifies p300. 293T cells were transfected with HA-tagged p300 and either empty vector, FLAG-tagged MECT1-MAML2, MECT1, or MAML2 TAD (173-1153). p300 expression was detected by IP with anti-HA antibodies followed by Western blot analysis with anti-HA. The expression levels of the FLAG-tagged proteins were determined by probing with anti-FLAG antibodies. (C) p300 modifies MECT1-MAML2 via acetylation. 293T cells were transfected with FLAG-tagged MECT1-MAML2 and either empty vector or HA-tagged p300. Expression levels of the acetylate, total MECT1-MAML2 fusion proteins, and p300 were analyzed by Western blot analyses followed by immunoprecipitation. (D) Expression of MECT1-MAML2 enhances p300 transcriptional activity. U20S cells were transfected with 0.5 μg of a pG5luc firefly luciferase construct containing GAL4-binding sites, 0.5 μg of the plasmid encoding DB fused to p300, increasing amounts of expression constructs expressing FLAG-tagged MECT1-MAML2 (or MAML2-TAD, or MECT1), and 10 ng of a pRL-TK control plasmid expressing Renilla luciferase. pG5luc reporter firefly luciferase activity, corrected for Renilla luciferase activity, is expressed as fold activation relative to cells not expressing MECT1-MAML2. (E) Expression of p300, not the HAT defective mutant, enhances MECT1-MAML2 transcriptional activity. U20S cells were transfected with 0.5 μg of pG5luc reporter and 0.5 μg of plasmid encoding DB fused to MECT1-MAML2 and increasing amounts of expression constructs expressing p300 or p300 with deficient HAT activity. The firefly luciferase activity, normalized to Renilla luciferase expressed from the pBIND plasmid, was expressed as fold activation relative to cells not expressing p300.
Interestingly, coexpression of MECT1-MAML2 and p300 resulted in a mobility shift of the p300 species (Figure 5B). This mobility shift in p300 was also observed when the MAML2 TAD domain is expressed, but not full-length MECT1, indicating that the MAML2 TAD domain in the fusion protein is responsible for p300 modification. Furthermore, the p300 mobility shift was reduced by incubation of the p300 immune complex with calf intestinal alkaline phosphatase (not shown), suggesting that the modification of p300 may be due to phosphorylation. Conversely, p300 was found to acetylate MECT1-MAML2 (Figure 5C). Therefore, our results indicate that MECT1-MAML2 and p300 interact, resulting in mutual post-translational modifications.
Previously, it was shown that p300 phosphorylation can be induced by various transcription factors including c/EBPβ, and c/EBPβ-mediated phosphorylation of p300 is required for full activity of p300 as a coactivator for c/EBPβ (Schwartz et al, 2003). We therefore asked if expression of the MECT1-MAML2 fusion affects p300 transcriptional activity. p300 was expressed as a fusion protein with a GAL4 DNA-binding domain, and its ability to activate a luciferase reporter that contains four GAL4-binding sites in the promoter was examined. We found that overexpression of either MECT1-MAML2 or the MAML2 TAD, but not MECT1, strongly stimulates p300-mediated transcription. These results indicate that MECT1-MAML2 strongly enhances p300 transcriptional activity, and this enhancement is mainly contributed by the TAD domain of MAML2 (Figure 5D). Combined with the data showing that the MAML2 TAD is sufficient to cause p300 modification, it is possible that MECT-MAML2 leads to p300 modification, and thereby enhances its transcriptional activity, or that MECT-MAML2 recruits additional cofactors to the p300 complex that further enhance p300-dependent transcription.
The acetylation of MECT1-MAML2 when associated with p300 is of great interest, since acetylation of transcription factors can have either positive or negative effects on function (Freiman and Tjian, 2003). Since p300 has histone acetyltransferase (HAT) activity, it is most likely that p300 itself is responsible for this modification. Using similar assay described above, we found that full-length p300, but not a p300 mutant with a defective HAT domain, enhances MECT1-MAML2 transcriptional activity, suggesting that the HAT activity of p300 is required for this transcriptional enhancement (Figure 5E).
The MECT1-MAML2 fusion and both exogenous and endogenous CBP were also observed to interact by colocalization and co-immunoprecipitation studies (Supplementary Figure S5). Therefore, the results here suggest an important interaction of p300/CBP and MECT1-MAML2, an idea supported by a change in the nuclear localization of p300/CBP upon coexpression with MECT1-MAML2, co-immunoprecipitation of MECT1-MAML2 and CBP/p300, and post-translational modification of both p300 and MECT1-MAML2 upon their coexpression. This interaction appears to enhance transcriptional activity of both p300 and MECT1-MAML2.
We then asked if the interaction of p300/CBP and MECT1-MAML2 fusion is important for CREB activation and cellular transforming activity. We first mapped the p300-binding domain on MECT1-MAML2, and found that a deletion mutant of MECT1-MAML2 lacking aa 48–222 (corresponding to aa 177–351 of the MAML2 protein) was unable to bind to p300, and was not acetylated by p300, even when an additional nuclear localization signal from SV40 was added to ensure nuclear localization (Figure 6A and B). The deletion mutant (aa 48–176), although having a very weak binding activity to p300, seems to be sufficient to become acetylated by p300. Thus, the p300-binding site on MECT1-MAML2 is located with aa 48–222. This p300-binding-deficient mutant showed a reduced ability to activate the CREB pathway (by Western blotting analysis of the expression of Nurr1 and ATF3), and to induce foci formation in RK3E, as compared to the full-length MECT1-MAML2 (Figure 6C and D). Interestingly, while this mutant was completely defective in p300 binding, it was only partially defective in the induction of Nurr1 and ATF3 expression, and colony formation, suggesting that p300 binding contributes substantially to MECT1-MAML2 transforming activity, and that other events are also relevant.
Figure 6.

p300-binding-deficient mutant exhibits a reduction in the ability to activate the CREB pathway and to induce colony formation in RK3E. (A) The MECT1-MAML2 mutant, Δ48–222, is unable to immunoprecipitate with p300. 293T cells were transfected with constructs expressing HA-tagged p300 and FLAG-tagged full-length or truncation mutants of MECT1-MAML2. The anti-HA immunoprecipitates were immunoblotted with anti-FLAG antibodies. (B) The MECT1-MAML2 (Δ48–222) mutant is not acetylated by p300. 293T cells were transfected with constructs expressing HA-tagged p300 and FLAG-tagged full-length or truncation mutants of MECT1-MAML2. The anti-FLAG immunoprecipitates were immunoblotted with anti-acetylated lysine and anti-FLAG antibodies. (C) The p300-binding-deficient mutant, MECT1-MAML2 (Δ48–222), induced less Nurr1 and ATF3 expression, as compared to the full-length MECT1-MAML2. (D) The p300-binding-deficient MECT1-MAML2 mutant generated a reduced number of colonies in RK3E colony formation assays. The data were based on three independent experiments.
Lastly, in order to assess whether p300/CBP is recruited to the transcriptional complex bound to the CRE site on the genes regulated by the MECT1-MAML2 fusion, we performed chromatin immunoprecipitation (ChIP) analysis of a cAMP-responsive promoter in 293T cells transiently transfected with the MECT1-MAML2 fusion, compared to the mock-transfected control cells. The ChIPs were carried out for endogenous p300, CBP, CREB, and transfected fusion protein, and then the associated DNA was used as a template for the PCR amplification of the Nurr1 promoter sequence containing the CRE site. The intensity of the PCR amplification bands would indicate the levels of binding of these proteins to this Nurr1 promoter region. As shown in Figure 7, the level of endogenous CREB binding to the promoter is similar in the absence and presence of the expression of MECT1-MAML2, suggesting that CREB constitutively binds to the Nurr1 promoter. When the MECT1-MAML2 fusion is expressed, it is recruited to the transcriptional complex on the Nurr1 promoter, and also greatly enhanced p300 and CBP recruitment to the promoter. Furthermore, we found no detectable PCR amplification product for the promoter of the GAPDH gene, which is not a CREB-mediated gene. These results therefore demonstrate that p300/CBP are recruited to the transcriptional complex bound to the CRE of the Nurr1 promoter upon the expression of the MECT1-MAML2 fusion.
Figure 7.
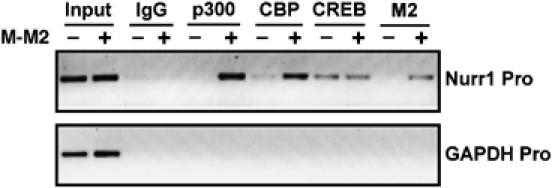
MECT1-MAML2 enhances the recruitment of p300/CBP to the transcriptional complex bound to the CRE site of the Nurr1 promoter. 293T cells were transfected with empty vector or FLAG-tagged MECT1-MAML2 for 24 h. Cells were then fixed with formaldehyde and chromatin lysates were prepared. Chromatin lysates were subjected to immunoprecipitation with antibodies against p300, CBP, CREB, and anti-FLAG (M2) antibodies. Rabbit IgG was used as a negative control. The DNA associated with immunoprecipitates was isolated and used as templates to amplify the Nurr1 promoter region containing CRE. PCR amplification of the GAPDH promoter was used as a negative control. The PCR products were resolved by agarose gel electrophoresis and stained with ethidium bromide.
The results from the studies described above along with the results of Conkright et al (2003) allow us to propose a novel mechanism for constitutive activation of the CREB pathway by the MECT1-MAML2 fusion protein (Figure 8). MECT1-MAML2 activates transcription of target genes of the CREB pathway that subsequently mediate at least part of its transforming activity. Two domains of the fusion oncoprotein are important for its activity: the CREB-binding domain, which allows for localization to the CREB complex on the promoters of the target genes; and the MAML2 TAD domain, which allows for strong CREB-mediated transcription by interacting with p300/CBP and likely, other undefined factors.
Figure 8.
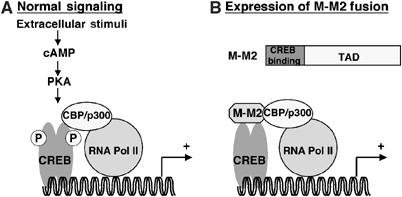
A proposed model to explain the mechanism by which the MECT1-MAML2 fusion protein constitutively activates the CREB pathway. (A) In a signal-dependent fashion, signal-mediated activation of the cAMP/PKA pathway leads to phosphorylation of CREB at Ser133, which allows the recruitment of p300/CBP and subsequently enhances transcriptional activation. (B) The MECT1-MAML2 fusion contains two functional domains, the CREB-binding domain from MECT1 and the TAD domain from MAML2. The CREB-binding domain enables the MECT1-MAML2 fusion to localize to the CREB complex on the promoters of target genes, and the TAD domain is responsible for subsequent strong transcription by interacting with p300/CBP coactivators and other factors. Thus, the fusion protein is able to bypass signaling requirements and activate transcription of CREB target genes that mediate its transforming activities.
Discussion
The novel MECT1-MAML2 fusion encoded by the t(11;19) translocation is associated with both malignant mucoepidemoid carcinomas and benign Warthin's tumors, and is potentially important in cell transformation of these salivary tumors (Martins et al, 1996, 1997, 2004). Here, we have investigated the molecular basis for the oncogenic activities of the MECT1-MAML2 fusion. Prior work has demonstrated that the MECT1-MAML2 fusion oncoprotein activates Notch signaling independent of ligand stimulation, suggesting that deregulation of Notch signaling might have a role in epithelial cell transformation (Tonon et al, 2003; Enlund et al, 2004). When MECT1 was identified as a ‘transducer of regulated CREB activity' (TORC1), it was suggested that the MECT1-MAML2 fusion might also deregulate the CREB pathway, and MECT1-MAML2 was shown to activate the transcription of a CRE-containing promoter reporter (Conkright et al, 2003). In this study, we support this hypothesis by demonstrating that the MECT1-MAML2 fusion constitutively and potently activates the CREB pathway, and that this is a critical mechanism by which the MECT1-MAML2 fusion mediates its cell transforming activity in vitro.
CREB mediates cellular responses to a variety of stimuli including hormones, growth factors, cytokines, and stress, by regulating the expression of numerous genes that subsequently modulate cell proliferation, differentiation, and apoptosis (Mayr and Montminy, 2001). Abnormal CREB activity has been implicated in growth and progression of a number of cancers, implying that deregulated CREB activation might be a frequent mechanism by which tumors evade normal control for growth and survival. Here, we demonstrate a critical role for CREB activation in MECT1-MAML2-mediated cell transformation. First, the ability of the MECT1-MAML2 fusion to activate the CREB pathway is correlated with its ability to induce foci in RK3E epithelial cells. Second, interference of CREB activity significantly reduced the ability of the fusion to induce the expression of CREB target genes and its transforming activity in RK3E cells. Third, interference of CREB activity leads to growth suppression of fusion-positive MEC cells, but not negative cells. These data indicate that the MECT1-MAML2 fusion is able to activate the CREB pathway, which plays an essential role in cell transformation and possibly maintenance of tumor phenotypes.
Currently, it is still not clear how constitutively elevated CREB activity leads to tumorigenesis. To address this question, it is necessary to identify key CREB-regulated genes that mediate MEC and Warthin's tumor initiation and progression. Since expression of CREB-mediated genes may be cell type specific, it is thus essential to study the target cells naturally transformed by the fusion oncoprotein in vivo in order to identify such mediators. Our studies showed that the MECT1-MAML2 fusion induced multiple CREB-mediated genes in cultured epithelial cells. For example, among the known CREB-mediated genes we identified, Id2 (Ruzinova and Benezra, 2003), FOS (Tulchinsky, 2000), and ATF3 (Perez et al, 2001) have known transforming activity, and the two members of nuclear receptor subfamily 4, NR4A1–2, has been implicated in cellular responses to hormones and growth factors (Maruyama et al, 1998). Intriguingly, one well-known Notch target gene, HES-1, previously reported to be upregulated by the fusion and expressed at elevated levels in primary MEC tumors, did not appear as a target gene in our microarray study. It is possible that the discrepancy is due to the time point we examined. Nonetheless, HES-1 was shown to be a CREB-regulated gene (Herzig et al, 2003). It is therefore possible that activation of Notch target genes associated with MECT1-MAML2 occurs through activation of CREB, at least in part. This is consistent with our previous observation that MECT1-MAML2-induced expression of HES-1 is independent of CSL-binding sites in the promoter (Tonon et al, 2003). Therefore, target genes for the MECT1-MAML2 fusion, mediated by CREB, affect multiple cellular processes that might contribute to carcinogenesis.
In light of the role of CREB activation in MECT1-MAML2 cell transformation, it is conceivable that inhibition of CREB activity might interfere with the formation, progression, and maintenance of MECT1-MAML2-associated tumors, which would open a new avenue for therapeutic intervention. In our study, the expression of dominant-negative A-CREB significantly reduced MEC cell growth in vitro and warrants investigation in vivo. Our results also have implications for the diagnosis and prognosis for MEC and Warthin's tumors. The presence of the MECT1-MAML2 fusion seems to be closely linked to the MEC phenotype and may be used as a diagnostic marker for MEC tumors (Martins et al, 2004). The status of CREB activation might serve as another important diagnostic marker and may have prognostic value as well.
One important finding in this study is that MECT1-MAML2 elicits potent CREB activation at least in part through the recruitment of p300/CBP coactivators. p300/CBP is generally believed to function as a physical bridge between a number of transcription factors and the basal transcriptional machinery. In this study, we showed that there is an interaction of p300/CBP and MECT1-MAML2, and this interaction leads to post-translational modifications as well as increased transcriptional activities of both p300 and MECT1-MAML2. Various transcription factors including C/EBP family members were previously shown to trigger phosphorylation of p300 through unknown mechanisms, which seems to modulate p300 as a transcriptional coactivator (Schwartz et al, 2003). It is still unknown if phosphorylation of p300 regulates its HAT activity, which could be one mechanism for transcriptional regulation. Modification, possibly phosphorylation, of p300 by MECT1-MAML2 might have a significant impact on intrinsic p300 HAT activity, and thus on the biological activities it regulated.
The acetylation of the MECT1-MAML2 fusion is of equal interest. To date, acetylation of non-histone regulatory proteins has been reported to regulate multiple cellular events, including gene transcription, chromatin remodeling, protein trafficking, and apoptosis (Freiman and Tjian, 2003; Cohen and Yao, 2004). p300/CBP has been shown to acetylate several transcription factors, thereby either stimulating or inhibiting their functions by modulating protein–protein interactions or DNA binding. Therefore, an important question that remains to be addressed is whether acetylation plays any role in transcriptional regulation and cellular transformation via this MECT1-MAML2 fusion. The p300-binding site is located in the MAML2 TAD domain of the fusion, while a similar binding site (aa 75–301) was also identified in another MAML family member, MAML1, and found to be necessary for in vitro chromatin-dependent transcription of a promoter reporter with multiple CSL-binding sites (Fryer et al, 2002). Therefore, the binding of p300/CBP seems to be a common function for the MAML family, and studying the interactions of p300/CBP and the MAML family in detail will help elucidate the general role of this family as transcriptional coactivators.
We also identified other target genes of the MECT1-MAML2 fusion in addition to the known CREB-regulated genes (see Table I). Although more conclusive data are required to show whether the expression of these genes also is mediated by CREB, it is possible that the MECT1-MAML2 fusion activates some target genes independently of CREB via unknown mechanisms, and some of their activities also contribute to cell transformation.
In summary, we have demonstrated that MECT1-MAML2-induced activation of CREB is a critical mechanism for cell transformation. Therefore, the MECT1-MAML2 fusion is able to undermine at least two signaling pathways, CREB and Notch, which could be potentially important in cancer development. Although it has been suggested that one of the Notch targets, HES-1, is regulated by CREB, it is still not known if MECT1-MAML2-induced Notch activation is completely mediated by CREB. Therefore, it will be important to evaluate the contribution of both signaling pathways in cell transformation via the MECT1-MAML2 fusion. In vivo evaluation of the MECT1-MAML2 fusion using mouse models will be important in understanding the direct roles of this fusion and its induced CREB-mediated genes in tumorigenesis in vivo.
Materials and methods
Plasmids
The A-CREB sequence was cloned from the CMV500_A-CREB construct (Ahn et al, 1998) into the retroviral vector MSCV-IRES-GFP. Full-length MECT1 cDNA was cloned into pFLAG-CMV2 (Sigma) and pEGFP-C3 (Clontech) expression vectors from KIAA0616 cDNA clone (obtained from KAZUSA DNA Research Institute in Japan). MECT1-MAML2 mutants, Δ48–222 and Δ48–176 were cloned into the pFLAG-CMV2 vector. pRevTRE vectors expressing bicistronic message RNAs for both HA-tagged MAML2 (or MECT1, or MECT1-MAML2) and GFP were also made. The details of cloning are available upon request. Expression constructs for MECT1-MAML2, MECT1-MAML1 (1–42 aa of MECT1 and 97–1016 aa of MAML1), MECT1-VP16 (1–42 aa of MECT1 and VP16 TAD domain), MAML2, and MAML1 in pFLAG-CMV2, MECT1-MAML2 in pBIND, HA-tagged CREB in pcDNA 3.1, Gal4-p300, HA-tagged p300 in pCMV, p300 WT and p300 HAT mutant in pcDNA3.1, and Myc-tagged human CBP in CMV vector have all been described previously (Eckner et al, 1994; Borrow et al, 1996; Chen et al, 2002; Wu et al, 2002; Conkright et al, 2003; Tonon et al, 2003).
Antibodies
The following antibodies were used: αFlag (M2), αMyc (9E10), αβ-actin (Sigma); αp300 (c-20), αCBP (C-1), αATF3 (C-19), αNurr1 (M196) (Santa Cruz); αHA (clone HA.11) (Babco); αacetylated-lysine (Cell Signaling); Alexa Fluor 594-conjugated anti-mouse (or rabbit) IgG (Molecular Probes).
Cell lines, transfections, and retroviral transduction
RK3E, HeLa tetON (Clontech), 293T, U20S, HSY, H3118, and H292 were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% inactivated fetal calf serum and penicillin/streptomycin. Cells were grown at 37°C under 5% CO2. Transfections with plasmids or CREB siRNA (SMART pool, Upstate) were carried out using Lipofectamine 2000 Reagent (Gibco) according to the manufacturer's instructions. Viral production, concentration, and titration were carried out as described previously (Carlesso et al, 1999). Cells were infected with the viruses in fresh complete medium plus Polybrene (8 μg/ml; Sigma) for 6–8 h, washed, and replaced with fresh medium. Flow cytometric detection of GFP-expressing cells was performed using a FACS machine (Cytomics FC 500; Becton Dickinson).
Gene chip analyses
HeLa tetOn cells were transfected with the pRevTRE vectors expressing bicistronic message RNAs for both HA-tagged MAML2 (or MECT1, or MECT1-MAML2) and GFP, and induced for expression in the presence of 200 ng/ml doxycyclin for 24 h. GFP-positive cells were sorted, and the total RNA extracted using Trizol reagents (Life Technologies) and purified via the columns from the SV 96 Total RNA Isolation kit (Promega). Hybridizations with Affymetrix U133A microarray chips were performed in our central core facility, and data were analyzed using d-Chip analysis software (Li and Wong, 2001). Two-fold changes at the transcript levels were considered significant from two independent experiments. The assignment of the gene family was obtained by SwissPort classification analysis. The microarray data were deposited in the ArrayExpress database (accession number #E-MEXP-340).
Real-time polymerase chain reaction
RNA was isolated by the Trizol method, and cDNA was generated (GeneAmp RNA PCR kit, Applied Biosystems) from 5 μg of total RNA. PCR was performed on the cDNA samples using an ABI PRISM 7700 sequence detector (PE Applied Biosystems) and the formation of PCR products monitored using the SYBR green method (PE Biosystems). All samples were amplified in triplicate. The relative changes in the amount of transcripts in each sample were determined by normalizing for the GAPDH mRNA expression levels, as described previously (ABI PRISM sequence detection system user bulletin no. 2, PE Applied Biosystems). The sequences for the primers used are listed in Supplementary Table 1.
Western blot analyses and immunoprecipitation
Western blotting and IP were performed as described previously (Wu et al, 2002).
Cell cycle analysis
Cells were fixed with ethanol, and stained with propidium iodide. DNA content was measured by a flow cytometer (Cytomics FC 500; Becton Dickinson), and cell cycle distribution determined by Multicycle software (Phoenix Flow Systems). A minimum of 10 000 events were collected to create each DNA content histogram.
RK3E foci formation and growth curve assay
RK3E foci formation was performed as described previously (Tonon et al, 2003). For growth curve assays, cells were plated on six-well plates at 1 × 104 cells/well in duplicate, and staining was performed daily for the next 6 days. Specifically, cells were fixed with 4% formalin for 10 min, stained with 0.1% crystal violet for 20 min, washed with H2O 10 times, air-dried, and wrapped with an aluminum foil. When all the staining was completed, 1 ml of 1% acetic acid solution was added to each well to dissolve the dye, and absorbance at a wavelength of 590 was measured. The amount of dye taken up by the cells was used as an indication of cell number.
Immunofluorescence staining
Staining was performed as described previously (Wu et al, 2002).
Reporter assays
Reporter assays were performed as described previously (Wu et al, 2002).
ChIP assay
ChIP assay was performed using a ChIP assay kit (Upstate) with minor modifications. Briefly, 293T cells were transfected with either empty vector or the MECT1-MAML2 expression vector for 24 h, and fixed with formaldehyde to crosslink the DNA to chromatin-associated protein complexes. Nuclei were isolated for further preparation of chromatin lysates. IPs were then performed: about 5 μg of antibodies was used for each IP; the aliquot of chromatin equivalent to 500 μg of DNA was used for IP with rabbit IgG, or p300, or CBP antibodies, and 100 μg of DNA for IP with CREB or anti-FLAG (for transfected MECT1-MAML2) antibodies. Immunoprecipitate complex was washed extensively and DNA was purified. PCR was performed using the primers flanking the CREB-binding site of the Nurr1 promoter. The primer sequences are listed in Supplementary Table 1.
Supplementary Material
Supplementary Materials
Acknowledgments
We are grateful to Drs M Montminy, C Vinson, WC Greene, and DM Livingston for reagents. This work was supported in part by NIH RO1 CA036167 (JDG) and NIH RO1 CA097148 (LW).
References
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C (1998) A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol 18: 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284: 770–776 [DOI] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M (2004) Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol 14: 2156–2161 [DOI] [PubMed] [Google Scholar]
- Borrow J, Stanton VP Jr, Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dube I, Frischauf AM, Horsman D, Mitelman F, Volinia S, Watmore AE, Housman DE (1996) The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet 14: 33–41 [DOI] [PubMed] [Google Scholar]
- Carlesso N, Aster JC, Sklar J, Scadden DT (1999) Notch1-induced delay of human hematopoietic progenitor cell differentiation is associated with altered cell cycle kinetics. Blood 93: 838–848 [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC (2002) Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J 21: 6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen T, Yao TP (2004) AcK-knowledge reversible acetylation. Sci STKE 2004: pe42. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M (2003) TORCs: transducers of regulated CREB activity. Mol Cell 12: 413–423 [DOI] [PubMed] [Google Scholar]
- Daniel PB, Rohrbach L, Habener JF (2000) Novel cyclic adenosine 3′,5′-monophosphate (cAMP) response element modulator theta isoforms expressed by two newly identified cAMP-responsive promoters active in the testis. Endocrinology 141: 3923–3930 [DOI] [PubMed] [Google Scholar]
- Eckner R, Arany Z, Ewen M, Sellers W, Livingston DM (1994) The adenovirus E1A-associated 300-kD protein exhibits properties of a transcriptional coactivator and belongs to an evolutionarily conserved family. Cold Spring Harb Symp Quant Biol 59: 85–95 [DOI] [PubMed] [Google Scholar]
- Enlund F, Behboudi A, Andren Y, Oberg C, Lendahl U, Mark J, Stenman G (2004) Altered Notch signaling resulting from expression of a WAMTP1-MAML2 gene fusion in mucoepidermoid carcinomas and benign Warthin's tumors. Exp Cell Res 292: 21–28 [DOI] [PubMed] [Google Scholar]
- Freiman RN, Tjian R (2003) Regulating the regulators: lysine modifications make their mark. Cell 112: 11–17 [DOI] [PubMed] [Google Scholar]
- Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA (2002) Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev 16: 1397–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman AS, Isaac DD, Andrew DJ (2003) Specification of cell fates within the salivary gland primordium. Dev Biol 258: 443–453 [DOI] [PubMed] [Google Scholar]
- Hai T, Hartman MG (2001) The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273: 1–11 [DOI] [PubMed] [Google Scholar]
- Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F, Montminy M (2003) CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature 426: 190–193 [DOI] [PubMed] [Google Scholar]
- Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, Orth AP, Miraglia L, Meltzer J, Garza D, Chirn GW, McWhinnie E, Cohen D, Skelton J, Terry R, Yu Y, Bodian D, Buxton FP, Zhu J, Song C, Labow MA (2003) Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA 100: 12147–12152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U (2004) What turns CREB on? Cell Signal 16: 1211–1227 [DOI] [PubMed] [Google Scholar]
- Kovalovsky D, Refojo D, Liberman AC, Hochbaum D, Pereda MP, Coso OA, Stalla GK, Holsboer F, Arzt E (2002) Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol Endocrinol 16: 1638–1651 [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH (2001) Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA 98: 31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins C, Cavaco B, Tonon G, Kaye FJ, Soares J, Fonseca I (2004) A study of MECT1-MAML2 in mucoepidermoid carcinoma and Warthin's tumor of salivary glands. J Mol Diagn 6: 205–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins C, Fonseca I, Roque L, Pinto AE, Soares J (1996) Malignant salivary gland neoplasms: a cytogenetic study of 19 cases. Eur J Cancer B 32B: 128–132 [DOI] [PubMed] [Google Scholar]
- Martins C, Fonseca I, Roque L, Soares J (1997) Cytogenetic characterisation of Warthin's tumour. Oral Oncol 33: 344–347 [DOI] [PubMed] [Google Scholar]
- Maruyama K, Tsukada T, Ohkura N, Bandoh S, Hosono T, Yamaguchi K (1998) The NGFI-B subfamily of the nuclear receptor superfamily (review). Int J Oncol 12: 1237–1243 [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609 [DOI] [PubMed] [Google Scholar]
- McGurk M, Renehan AG (2001) Controversies in the Management of Salivary Gland Disease. New York: Oxford University Press [Google Scholar]
- Perez S, Vial E, van Dam H, Castellazzi M (2001) Transcription factor ATF3 partially transforms chick embryo fibroblasts by promoting growth factor-independent proliferation. Oncogene 20: 1135–1141 [DOI] [PubMed] [Google Scholar]
- Poser I, Bosserhoff AK (2004) Transcription factors involved in development and progression of malignant melanoma. Histol Histopathol 19: 173–188 [DOI] [PubMed] [Google Scholar]
- Ruzinova MB, Benezra R (2003) Id proteins in development, cell cycle and cancer. Trends Cell Biol 13: 410–418 [DOI] [PubMed] [Google Scholar]
- Schwartz C, Beck K, Mink S, Schmolke M, Budde B, Wenning D, Klempnauer KH (2003) Recruitment of p300 by C/EBPbeta triggers phosphorylation of p300 and modulates coactivator activity. EMBO J 22: 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scobey MJ, Fix CA, Walker WH (2004) The Id2 transcriptional repressor is induced by follicle-stimulating hormone and cAMP. J Biol Chem 279: 16064–16070 [DOI] [PubMed] [Google Scholar]
- Shankar DB, Sakamoto KM (2004) The role of cyclic-AMP binding protein (CREB) in leukemia cell proliferation and acute leukemias. Leuk Lymphoma 45: 265–270 [DOI] [PubMed] [Google Scholar]
- Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, O'Neil K, Stover K, El-Naggar A, Griffin JD, Kirsch IR, Kaye FJ (2003) t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet 33: 208–213 [DOI] [PubMed] [Google Scholar]
- Tulchinsky E (2000) Fos family members: regulation, structure and role in oncogenic transformation. Histol Histopathol 15: 921–928 [DOI] [PubMed] [Google Scholar]
- Wong CK, Yeung HY, Mak NK, DiMattia GE, Chan DK, Wagner GF (2002) Effects of dibutyryl cAMP on stanniocalcin and stanniocalcin-related protein mRNA expression in neuroblastoma cells. J Endocrinol 173: 199–209 [DOI] [PubMed] [Google Scholar]
- Wu L, Sun T, Kobayashi K, Gao P, Griffin JD (2002) Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol 22: 7688–7700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Melmed S (2001) Oncogene activation in pituitary tumors. Brain Pathol 11: 328–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials