

Summary
The intricate development and functionality of the mammalian heart are influenced by the heterogeneous nature of cardiomyocytes (CMs). In this study, single-cell and spatial transcriptomics were utilized to analyze cells from neonatal mouse hearts, resulting in a comprehensive atlas delineating the transcriptional profiles of distinct CM subsets. A continuum of maturation states was elucidated, emphasizing a progressive developmental trajectory rather than discrete stages. This approach enabled the mapping of these states across various cardiac regions, illuminating the spatial organization of CM development and the influence of the cellular microenvironment. Notably, a subset of transitional CMs was identified, characterized by a transcriptional signature marking a pivotal maturation phase, presenting a promising target for therapeutic strategies aimed at enhancing cardiac regeneration. This atlas not only elucidates fundamental aspects of cardiac development but also serves as a valuable resource for advancing research into cardiac physiology and pathology, with significant implications for regenerative medicine.
Subject areas: Cell biology, Omics, Transcriptomics
Graphical abstract
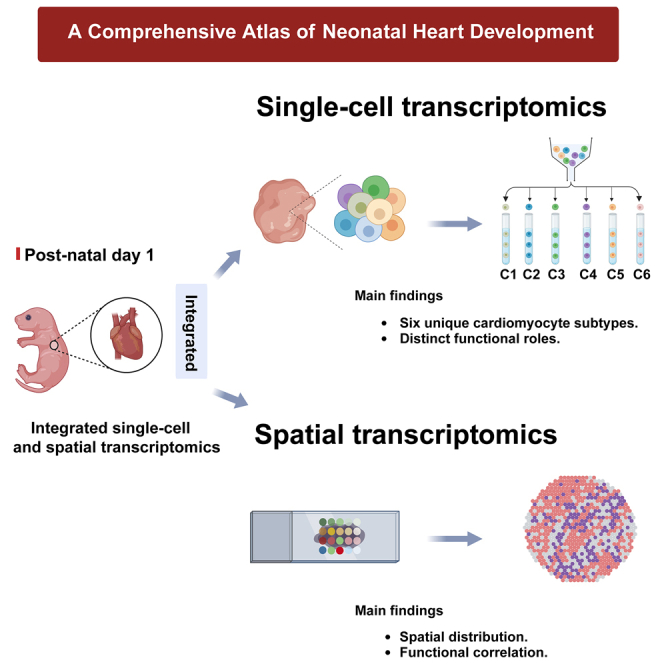
Highlights
-
•
Comprehensive atlas constructed using single-cell and spatial transcriptomics
-
•
Continuum of cardiomyocyte maturation states identified, not discrete stages
-
•
Unique transitional cardiomyocytes reveal targets for enhancing cardiac regeneration
Cell biology; Omics; Transcriptomics
Introduction
Cardiac development and maturation are essential processes during the neonatal period, characterized by the precise orchestration of cellular differentiation and maturation. Central to this developmental process, cardiomyocytes (CMs) undergo differentiation through a series of transcriptional and epigenetic modifications, resulting in distinct cellular phenotypes within the cardiac environment.1,2 The nascent post-natal phase is crucial as it establishes the foundational heart function and structure that persist into adulthood.3 Disruptions in these developmental sequences contribute to congenital heart diseases, a primary cause of neonatal morbidity and mortality, emphasizing the need to understand and address these defects.4
Recent advancements in high-throughput sequencing technologies have revolutionized our ability to deconstruct the heart’s cellular composition with remarkable detail and precision. Notably, single-cell RNA sequencing (scRNA-seq) has been transformative, uncovering cardiac cell types and elucidating the intricate transcriptional networks underlying their functional characteristics.5,6 For instance, previous studies have profiled CMs at various developmental stages in both mouse and human models, significantly enhancing our understanding of cardiac biology.7,8,9 These investigations have provided extensive data on the transcriptional identities and functionalities of CMs, particularly during the pivotal postnatal day 1 (P1) stage in mice.
Complementing scRNA-seq, spatial transcriptomics adds a layer of anatomical specificity, linking gene expression profiles with specific regions of the heart.10,11 Together, these methodologies offer a comprehensive view of the cardiac cellular ecosystem from the earliest stages of life. Our previous 2023 JAHA publication focused more on the developmental changes in heart cell proportions across days 1, 2, 4, and 6. In contrast, the present study aims to reveal the physiological status of heart cells specifically on the first day of postnatal development. By combining scRNA-seq with spatial transcriptomics, we can not only identify distinct cell types and their transcriptional profiles but also map these profiles to their precise anatomical locations within the heart. This integrated approach allows us to better understand the spatial organization and functional heterogeneity of CMs, offering insights that would be unattainable with either method alone.12
Despite the substantial body of published work, our current study aims to expand upon these foundations by integrating scRNA-seq with spatial transcriptomic analyses to map the multifaceted nature and maturation trajectory of CMs within neonatal mouse hearts. Our focus on P1 C57BL/6 mice aims to elucidate whether there are significant findings from this critical developmental period and how our dataset compares to previously published datasets.
Previous investigations have highlighted the diversity of CMs during heart development, revealing subpopulations with transcriptional identities that fulfill specialized myocardial functions, including contraction, conduction, and metabolic homeostasis.13,14,15 Recognizing this CM diversity underscores the complexity of cardiac ontogeny and suggests potential for targeted therapeutics for pediatric cardiac disorders.
Our study categorizes the distinct clusters of CMs that emerge during this critical developmental period, revealing their transcriptional signatures and spatial organization. This enhanced understanding of the roles these subpopulations play in shaping cardiac structure and function can pave the way for innovative treatments targeting congenital heart disease and strategies for cardiac regeneration.
An integral component of cardiac maturation is the complex network of intercellular communication that orchestrates the harmonious integration of varied cell types.16,17,18 This investigation delves into the intricacies of this communicative network, particularly within the ventricular and atrial chambers, highlighting the signaling pathways pivotal for cardiac development and CM maturation.
In summary, our research enriches our comprehension of the cellular landscape of the neonatal heart. By applying cutting-edge transcriptomic technologies and comprehensive data analysis, we delineate the principal elements and processes involved in the maturation and spatial coordination of CMs. This approach not only builds upon existing knowledge but also propels advancements in the field of cardiac developmental biology, potentially leading to therapeutic strategies.
Results
Single-cell transcriptomic analysis unveils cardiac cell diversity
To elucidate the cellular heterogeneity and molecular underpinnings governing early cardiac ontogeny, myocardial specimens from C57BL/6 mice at P1 were incorporated into our scRNA-seq framework. This assay facilitated the dissection of transcriptional landscapes within nascent cardiac cell subtypes, thereby exposing the intricate gene regulatory networks presiding over this essential phase of cardiac maturation (Figure 1A). For quality control (QC) in our study, cells with fewer than 200 or more than 6,000 detected genes were excluded. Additionally, cells with mitochondrial gene expression exceeding 10% were removed. After removal of low-quality cells, 8,652 cells were retained for biological analysis, which detected a median of 2,699 genes and 8,337 transcripts per cell (Figure S1). Subsequent normalization of gene expression levels and principal-component analysis (PCA) preceded the adoption of a graph-based clustering algorithm (Figure 1B), which segregated the cellular array into 10 discernible clusters. Marker gene expression facilitated the assignment of these clusters to ten well-established cardiac cell lineages (Figures 1C–1E): CMs (3,409 cells, 39.40%, identified by Ttn, Ryr2, and Mybpc3), fibroblasts (2,366 cells, 27.30%, marked by Dcn, Pdgfra, and Col1a1), endothelial cells (1,787 cells, 20.70%, distinguished by Vwf, Cdh5, and Pecam1), smooth muscle cells (221 cells, 2.60%, labeled with Acan, Myh11, and Lmod1), mesothelial cells (209 cells, 2.40%, characterized by Msln, Bnc1, and Lrrn4), pericytes (196 cells, 2.30%, indicated by Rgs5, Kcnj8, and Pdgfrb), myeloid cells (264 cells, 3.10%, recognized by Cd14, Cd68, and C1qa), neutrophils (94 cells, 1.10%, detected by Cd177, Cxcr2, and Ly6g), B cells (59 cells, 0.70%, marked with Cd79b, Ms4a1, and Cd79a), and neuronal cells (47 cells, 0.50%, identified by Gfra3, Plp1, and Kcna1).
Figure 1.

Cardiac cellular atlas of postnatal day 1
(A) Schematic diagram for the generation of single-cell RNA sequencing (scRNA-seq) data. Nine C57/BL6 mouse hearts were collected.
(B) t-stochastic neighbor embedding (t-SNE) plots showing cell types for the cells.
(C) The proportion of each cell type in 9 samples.
(D) Dot plot illustrating the expression levels and patterns of selected cell-type-specific genes of major clusters. Three signature genes were shown for each cluster.
(E) t-SNE plots showing the expression levels of canonical marker genes for ten cell types. See also Figure S1.
Transcriptional heterogeneity in cardiomyocyte clusters
Building upon this comprehensive cellular characterization, further in-depth analysis was conducted to probe the transcriptional and functional nuances of the CM cluster, which comprised 39.40% of the total cell population. t-stochastic neighbor embedding (t-SNE) dimensionality reduction was applied to delineate distinct CM subpopulations. Six clusters emerged, each marked by distinctive gene expression signatures indicative of their specialized roles within the cardiac landscape (Figure 2A). Detailed analysis revealed a set of core genes for each subtype, which were subsequently correlated to their specialized functions (Figures 2B and 2C). Representative genes for these clusters included Lars2 for cluster C1, S100a10 for C2, Abcc9 for C3, Bmp10 for C4, Sfrp1 for C5, and Ube2c for C6.
Figure 2.

Single-cell transcriptomic analysis identified multiple cell types in postnatal day 1
(A) t-SNE visualization of cardiomyocytes, colored by cell types. (cluster 1, C1; cluster 2, C2; cluster 3, C3; cluster 4, C4; cluster 5, C5; cluster 6, C6.)
(B) Dot plot showing the smoothed expression distribution of marker genes in six cell types.
(C) t-SNE plots showing the expression levels of canonical marker genes for nine cell types.
(D)The lollipop chart illustrating the biological processes and related functions associated with the core genes of each subtype. See also Tables S1, S2, S3, S4, S5, and S6.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses were utilized to clarify the function profiles of each cell cluster (Tables S1, S2, S3, S4, S5, and S6), with the involvement of core genes in biological processes detailed in Figure 2D. Lars2 primarily participated in functions related to the mitochondrial proton-transporting ATP synthase complex and mitochondrial matrix. S100a10 was linked to the collagen-containing extracellular matrix, ubiquitin ligase inhibitor activity, Salmonella infection in Mus musculus, and the regulation of supramolecular fiber organization. Abcc9 contributed to the generation of precursor metabolites and energy, contractile fiber, sarcolemma, regulation of heart contraction, intracellular chemical homeostasis, and cardiac conduction. Bmp10 was implicated in contractile fiber, heart development, regulation of system process, cellular anatomical entity morphogenesis, regulation of heart growth, regulation of developmental growth, and tissue morphogenesis. Sfrp1 participated in myofibril formation, regulation of monoatomic ion transmembrane transporter activity, and positive regulation of the apoptotic process. Ube2c was involved in the mitotic cell cycle, regulation of cell cycle processes, regulation of chromosome organization, positive regulation of chromosome separation, and cell cycle phase transition. These analyses provided a detailed understanding of the complex transcriptional landscape and functional heterogeneity present within the CM population, emphasizing the specialized roles played by individual subclusters in cardiac function and health. The identification of these specialized functions suggests the potential for targeted therapeutic strategies tailored to address the specific demands of CM biology. It is important to note that while Lars2 appears to be broadly expressed across the CM subclusters, additional markers and functional validation are necessary to fully elucidate the roles of each sub-cluster.
Insights into neonatal cardiomyocyte differentiation and maturity
Expanding upon the foundational transcriptional characterizations established through t-SNE analysis for CM subpopulations, a subsequent pseudotime analysis was conducted, revealing a subtle yet significant finding: differentiation pathways among neonatal CMs, with the notable exception of the C2 subgroup, appeared to converge, suggesting a continuum of maturation rather than discrete differentiated states at the time of birth (Figure 3A). To interrogate this continuum further, a heatmap was constructed to elucidate the expression dynamics of characteristic genes within subgroups C1 to C6 (Figure 3B). This analysis uncovered key genes instrumental to the maturation of CM functionality. Notably, the C6 subgroup expressed genes such as Mki67 and Top2a, hallmarks of an immature CM phenotype, potentially signifying a propensity for continued cellular proliferation. In contrast, the C3 subgroup demonstrated enhanced expression of genes relevant to sarcomere architecture, metabolic proficiency, and mechanisms of excitation-contraction coupling, indicative of a more mature phenotype with the establishment of quintessential CM functions. These comprehensive analyses collectively illuminate the complex developmental landscape of neonatal mouse CMs, thereby advancing our understanding of their maturation continuum and the nuances of functional specialization. Additionally, the conclusions drawn from trajectory analysis should be validated by analysis of additional time points before and after P1 in future research.
Figure 3.

Transcriptomic profiling of neonatal cardiomyocyte subpopulations
(A) Pseudotime analysis visualization of neonatal cardiomyocyte differentiation pathways, highlighting the distinct maturation trajectory of the subgroup.
(B) Heatmap representation of gene expression dynamics across cardiomyocyte subgroups C1–C6, pinpointing essential genes that mark different phases of cardiomyocyte maturation and specialization.
Cardiomyocyte cluster C3: A central role in cardiac function and metabolism
Given the subtle differences presented by the pseudotime analysis, we redirected our focus toward a comparative intersectional gene analysis among the cell groups. This strategic shift aimed to elucidate both shared and distinct elements of gene expression that may define common developmental pathways or subgroup-specific traits. Specifically, C4, C5, C3, C1, and C6 demonstrated concordance in over 20 differentially expressed genes (Figure 4A; Table S7). Cluster C3 has been identified as a potential transcriptional nexus, with preliminary findings indicating overlaps in gene expression profiles. Nonetheless, further empirical validation is requisite to substantiate its functional role. (Figure 4B). The shared biological processes involving C3 were manifold: “heart process” and “heart development” pathways were commonly expressed with C4 and C5, suggesting C3’s integral role during the early developmental orchestration of cardiac functionality and structure. The process of “striated muscle cell differentiation,” shared with C5, further highlighted C3’s contribution to the differentiation and maturation of cardiac muscle fibers. Moreover, metabolic pathways such as “oxidative phosphorylation” and “generation of precursor metabolites and energy,” shared with clusters C1 and C6 respectively, implicated C3 in the metabolic circuitry essential for cardiac energetics and function (Figure 4C).
Figure 4.

Intersectional gene expression analysis reveals cardiomyocyte subpopulation C3 as a transcriptional nexus in cardiac development and function
(A) Venn diagram showcasing shared and distinct gene counts across cardiomyocyte subgroups C1, C2, C3, C4, C5, and C6.
(B) Bar chart ranking of shared gene counts among cardiomyocyte subgroups C1, C2, C3, C4, C5, and C6.
(C) Detailed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of shared biological processes between C3 and other clusters.
(D) Key genes and their functional roles in C3. See also Table S7.
Pertinently, our analysis identified a cohort of genes expressed in C3, revealing a transcriptional signature that potentially confers distinct functions to this cluster within the cardiac cellular network (Figure 4D). This CM subpopulation exhibits a distinct gene expression profile indicating a potent capacity for energy metabolism, primarily through optimized beta-oxidation of fatty acids and enhanced ATP production. Specific genes such as Mybpc3 and Des are directly linked to myocardial contraction and structural integrity, ensuring the heart muscle operates efficiently. Ion channels and transport proteins, exemplified by Gja1, facilitate effective electrical signal transmission between heart muscle cells. Additionally, genes like Nppb and Prkaa2 play roles in blood pressure regulation and energy sensing, while stress response proteins like Hspa9 underscore this subgroup’s ability to manage cellular stress. Ultimately, this highlights the critical role of this CM subpopulation in heart physiology, especially in sustaining heartbeats and maintaining cardiac structural stability. While our current study provides compelling bioinformatic evidence, additional validation and functional experiments are required to support the argument that subcluster 3 is a transcriptional nexus in cardiac development and function. Future investigations are planned to incorporate targeted gene knockdown or overexpression experiments, contingent upon the identification of specific markers for the C3 cluster, to rigorously validate our findings.
Spatial transcriptomic analysis elucidates cardiomyocyte distribution and function
To dissect the intersection of transcriptional identity and spatial localization within the cardiac landscape, we expanded our research framework to integrate spatial transcriptomic sequencing. This additional analytical dimension enabled a nuanced correlation between gene expression signatures and the anatomical positioning of CM subtypes. The resulting spatial transcriptomic data disclosed a distinct cellular topography within the heart, with certain regions showcasing specific subtype enrichments (Figure 5A).
Figure 5.

Spatial transcriptomic profiling of cardiomyocyte subtypes across the cardiac landscape
(A) Schematic cross-section of the heart illustrating anatomical regions for spatial transcriptomic analysis. The diagram visually delineates the heart’s anatomical regions—apex, ventricle, atrium, and upper atrium.
(B) High-resolution spatial transcriptomic images depicting cellular composition in heart regions. The images are accompanied by a color-coded key that identifies the distribution and localization of different types of cardiomyocytes and other cardiac cells across the segmented areas.
(C) Bar graph depicting cell population changes in the final sections of each cardiac region.
The ventricular myocardium, particularly at the apex, was characterized by a high density of subtype C3 cells (Figure 5B). This local predominance aligns with the subtype’s transcriptional propensity for contractile functionality, implicating C3 as a key player in myocardial contraction. The interspersion of subtype C2 cells and fibroblasts throughout these ventricular zones suggests an auxiliary role in sustaining structural support and ensuring ventricular resilience.
A transition to the atrial sections revealed a more diverse cellular composition. Notably, the presence of subtype C5 cells, along with a higher abundance of subtype C2 cells, fibroblasts, and endothelial cells, suggested a multifaceted involvement in atrial-specific functions, potentially encompassing contributions to atrial contraction, structural integrity, and vascular maintenance.
In the atrial upper chambers, the cellular constituency was marked by a decline in C3 and C2 cells, while subtype C5 and C4 cells, along with fibroblasts, displayed an increased prevalence. This distribution pattern may denote specialized regulatory functions tailored to the electrophysiological and hemodynamic requirements of the upper atria (Figure 5C).
Collectively, these spatially resolved patterns of cellular distribution, detailed in Figure 4, illuminate the intricate dependency of cell type and function on anatomical location within the heart. The consonance between cellular specialization and topological placement supports the notion that cardiac anatomy is a key determinant of functional zonation—an essential aspect of the heart’s complex physiology. This integrative analysis underscores the significance of anatomical context in shaping the cardiac cellular milieu and contributes to our holistic understanding of cardiac structure and function.
Intercellular communication networks and cardiac functional maturation
The spatial transcriptomic analyses of cardiac slices have illuminated the prominent role of the C3 CM cluster, which is abundantly present across the heart and likely essential for postnatal cardiac functional maturation. In an attempt to delineate the intercellular communication networks within the heart, we focused on the terminal sections of each cardiac region, hypothesizing that interactions involving C3 cells are pivotal to this process (Figure 6A).
Figure 6.

Spatial transcriptomics uncovers intercellular communication networks in the apex, ventricle, and atrium of the heart
(A) Cardiomyocyte diversity, intercellular communication networks, and ligand-receptor relationships at the cardiac apex.
(B) Cardiomyocyte diversity, intercellular communication networks, and ligand-receptor relationships at the ventricle.
(C) Cardiomyocyte diversity, intercellular communication networks, and ligand-receptor relationships at the atrium.
Within the ventricular apex, the communication network is dominated by interactions between C3 and C2 cells. Specifically, signaling in the apex is orchestrated through the Ptn-Ncl receptor-ligand pair, emanating from C2 cells and implicated in the regulation of local growth and structural organization. In the ventricular myocardium, C3 cells utilize the Apln-Aplnr pathway to communicate with C2 cells, a pathway known to modulate cardiovascular development and cardiac contractile dynamics (Figure 6B).
As the analysis extends to the atrial compartments, marked by increased cellular heterogeneity, the complexity of intercellular communication proportionally increases. C3 cells engage in signaling with the C5 subtype and endothelial cells through the Igf2-Igf2r axis, a pathway crucial for cellular proliferation, differentiation, and angiogenesis (Figure 6C). In the atria’s upper chambers, the Igf2-Igf2r pathway continues to facilitate communication between C3 and C5 cells (Figures 7A–7C), while endothelial cells and fibroblasts join this communicative circuit, potentially coordinating the nuanced electrophysiological and contractile functions of the atria.
Figure 7.

Spatial transcriptomics reveals intercellular communication networks in the upper atrium of the heart
(A) Cardiomyocyte diversity at the upper atrium of the heart.
(B) Intercellular communication networks at the upper atrium of the heart.
(C) Ligand-receptor relationships at the upper atrium of the heart.
This dissection of the cardiac intercellular communication networks, supported by precise anatomical localization, underscores the importance of cellular dialog in the refinement of cardiac functions. These findings highlight the complex interplay between localized cell populations and their signaling pathways, which collectively contribute to the regional specialization and functional maturation of the heart. The integration of spatial and molecular dimensions in our analysis offers a sophisticated understanding of how cell-to-cell interactions are choreographed within the cardiac microenvironment, exemplifying the intricate relationship between anatomical structure and cellular function. Functional validation of the results concerning the signaling pathways communicating between different subclusters is essential for a complete understanding. Future studies should include evidence that the relevant encoded proteins are expressed as predicted by the transcriptomic data, using methods such as western blotting or flow cytometry.
Discussion
This investigation has substantially enhanced our comprehension of CM heterogeneity and spatial organization in the developing cardiac milieu of the neonatal murine heart. By utilizing scRNA-seq in conjunction with spatial transcriptomics, we have elucidated the diverse transcriptional states of CMs and their correlation with specific anatomical regions, which are pivotal for postnatal heart development and function. The identification of ten discrete cardiac cell populations, including six CM subclusters, corroborates and extends the findings of seminal high-resolution studies, such as those by Feng W et al., and our previous work, thereby enriching our understanding of the cellular mechanisms that underlie functional adaptability and robustness within the neonatal heart.12,19
The concept that the heart comprises specialized cells, each fulfilling an important role within the cardiac apparatus, is further refined by our observations, aligning with the emerging consensus from single-cell cardiac studies.9,20 The dynamic continuum of developmental states among neonatal CMs underscores the fluid nature of cardiac maturation. Particularly noteworthy is the transcriptional maturity and specialized function of the C3 cluster, highlighting its significant role in cardiac performance due to its enhanced contractile and metabolic capabilities. This finding is consistent with the insights of Guo Y et al., who also emphasized the cardinal role of mature CMs in heart development.21
Our spatial transcriptomic analysis offers critical insights into the interplay of transcriptional diversity and anatomical localization. Specifically, the spatial distribution of the C3 cluster, with its prominent presence in the ventricular apex, underscores its role in meeting the mechanical demands of the myocardium, thus supporting the concept of functional myocardial zonation.22,23 This spatial organization suggests that specific CM subpopulations are strategically positioned to optimize cardiac efficiency and resilience. Furthermore, this study illuminates the intricate intercellular communication networks that drive cardiac development and maturation. Pathways such as Ptn-Ncl and Apln-Aplnr were particularly notable, exemplifying the complex signaling interactions necessary for coordinated heart function.24,25 The intercellular communication in the atrial compartments, facilitated by pathways such as the Igf2-Igf2r axis, highlights the complexity of cardiac signaling networks (Figures 7A–7C). The importance of the Igf2-Igf2r axis in vascular development is further supported by studies highlighting its critical function in placental microvasculature expansion and the recruitment of endothelial progenitor cells.26,27 These mechanisms likely parallel those in cardiac tissue, where IGF2 may regulate essential vascular and cellular growth for heart development. These pathways not only elucidate the regulatory mechanisms of heart development but also suggest potential therapeutic targets. For instance, targeting specific metabolic pathways within the C3 cluster could enhance cardiac regeneration and address heart failure, providing an avenue for therapeutic intervention.28,29,30
Regarding sequencing methodologies, scRNA-seq provides a comprehensive view of the entire transcriptome within individual cells, capturing detailed gene expression profiles. However, due to the large size of CMs, scRNA-seq may introduce biases in the selection of cells, as larger cells can be more challenging to capture and analyze. This can lead to an underrepresentation of certain CM subpopulations. On the other hand, snRNA-seq, which isolates and sequences RNA from the nuclei of cells, can mitigate some of these biases. This approach is particularly advantageous for CMs, as it allows for the analysis of transcriptionally active regions while avoiding the technical difficulties associated with the large size and structural complexity of whole cells. By focusing on nuclear RNA, snRNA-seq can provide complementary insights into the transcriptional landscape of the heart, enhancing our understanding of gene expression regulation, especially in large and complex cells like CMs. Future studies should consider integrating both scRNA-seq and snRNA-seq to obtain a more comprehensive and unbiased view of the cardiac transcriptome. One of the major strengths of our study is the integration of scRNA-seq with spatial transcriptomics, which allowed us to resolve the identity of various cardiac cell types and map them spatially within the neonatal murine heart. This dual approach enabled us to elucidate how diverse cardiac cell types spatially coordinate to create the complex morphological structures crucial for heart function. Similar methodologies have been employed in earlier studies to map the human heart,31 revealing cell subpopulations exclusive to specific anatomical regions and their roles in forming cellular communities. Our findings align with these studies, particularly in identifying specialized subpopulations of ventricular CMs that display complex organizational patterns akin to those observed in human heart development. These findings underscore the significance of spatial organization in the functional specialization of cardiac tissues.
In conclusion, our study offered a comprehensive view of CM diversity and maturation within the neonatal mouse heart, setting a foundation for further investigations into cardiac regeneration and disease. Through integrated cellular and molecular analyses, we had revealed key aspects of CM heterogeneity, developmental trajectories, and spatial distribution. These findings collectively contribute to our understanding of cardiac function maturation and suggest avenues for therapeutic interventions.
Limitations of the study
However, our study has certain limitations. While the integration of scRNA-seq and spatial transcriptomics offers high-resolution insights, the reliance on neonatal murine models may not fully encapsulate the complexity and diversity of human cardiac development. Furthermore, our study predominantly emphasizes transcriptional profiles and spatial mapping, with less focus on functional validation through in vivo models or in vitro systems, such as human pluripotent stem cell-derived CMs.
At present, the experimental validation of the C3 cluster’s role presents challenges due to the incomplete characterization of specific markers. The dependence on a singular marker complicates the precision of cell sorting. In light of these obstacles, we are undertaking additional bioinformatics analyses utilizing existing datasets to reinforce our conclusions. These efforts will inform and facilitate the design of future experimental validations. Future research should prioritize validating these findings in human tissues and delving into the molecular mechanisms driving the differentiation and functional specialization of C3 CMs. Incorporating genetic mouse models and human cell systems could yield deeper insights into the multicellular signaling pathways and their roles in cardiac morphogenesis. Additionally, investigating how these cells adapt to various physiological challenges, such as hypoxia or increased energetic demands, is imperative. Such studies will pave the way for developing targeted therapies that harness the properties of specific CM clusters to enhance heart function under pathological conditions.
Resource availability
Lead contact
Requests for further information should be directed to and will be fulfilled by the lead contact, Yanfei Li (liyf@sumhs.edu.cn).
Materials availability
All materials used in this study will be made freely available upon request and the completion of applicable material transfer agreements.
Data and code availability
-
•
All data needed to evaluate the conclusions in the paper are present in the paper and/or the The raw data have been deposited in the Gene Expression Omnibus (GEO: GSE232466) and Mendeley Data: https://data.mendeley.com/datasets/4zfdygb9bz/1, and they are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The R code to analyze the data used in this study is available online: https://github.com/Lizhiyongjuan/scRNA_ST.git.
-
•
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.
Acknowledgments
We are grateful for the guidance and assistance of the Shanghai Biotechnology Corporation’s staff in sample processing, and for the support of single cell and spatial transcriptomics program. This study was supported by the Natural Science Foundation of Shanghai (21ZR1428400) and the Shanghai Medical Science and Technology Support Project (21S11901700).
Author contributions
All authors contributed to the study conception and design. Y.L. and L.M. designed the study. X.W. and L.C. wrote the initial version of the manuscript and revised the final version. X.W., L.C., R.C., and J.S. performed the experiment and analyzed the data together and analyzed the data. Y.L. and L.M. supervised the work. All authors contributed to the manuscript and approved the submitted version.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase/Dispase | Roche | 11097113 001 |
| Red Blood Cell Lysis Solution | Thermo Fisher | 00-4333-57 |
| Trypan Blue | Thermo Fisher | 14190144 |
| TissueTek O.C.T. Compound | SAKURA | 4583 |
| Methanol,for HPLC, ≥99.9% | Millipore Sigma | 34860 |
| Tris Base | Thermo Fisher Scientific | BP152-500 |
| Acetic acid, ≥99.9% | Millipore Sigma | A6283 |
| Hematoxylin, Mayer’s | Agilent | S330930-2 |
| Bluing Buffer, Dako | Agilent | CS70230-2 |
| Sodium dodecyl sulfate (SDS) solution, 10% in water | Millipore Sigma | 71736 |
| Hydrochloric Acid Solution, 0.1N | Fisher Chemical | SA54-1 |
| Critical commercial assays | ||
| Chromium Next GEM Chip G Single Cell Kit | 10xGenomics | 1000120 |
| Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit v3.1 | 10xGenomics | 1000121 |
| Chromium i7 Multiplex Kit | 10xGenomics | 120262 |
| SPRIselect Reagent Kit | Beckman Coulter | B23318 |
| ExKubit dsDNA HS | ExCell Bio | NGS00-3012 |
| High Sensitivity DNA Kit | Agilent | 5067–4626 |
| Visium Spatial Gene Expression Slide & Reagents Kit | 10xGenomics | 1000184 |
| Visium Spatial Tissue Optimization Slide & Reagents Kit | 10xGenomics | 1000193 |
| Deposited data | ||
| Single-cell RNA-sequencing datasets | This paper | GEO: GSE232466 |
| Spatial transcriptomics data | This paper | https://data.mendeley.com/datasets/4zfdygb9bz/1 |
| The R code | This paper | https://github.com/Lizhiyongjuan/scRNA_ST.git |
| Software and algorithms | ||
| Cell Ranger (version 2.1.0) | 10x Genomics | https://www.10xgenomics.com/support/software/cell-ranger |
| Seurat (version 3.1.2) | Stuart et al.32 | https://github.com/satijalab/seurat/releases/tag |
| scDblFinder (version 1.8.0) | Germain et al.33 | https://github.com/orgs/catg-umag/packages/container/package/scdblfinder |
| Harmony (version 0.1.0) | Ilya Korsunsky et al.34 | https://github.com/immunogenomics/harmony |
| spacexr (version 2.2.1) | Cable et al.10 | https://github.com/dmcable/spacexr |
| Monocle (version 2.22.0) | Rao et al.35 | https://cole-trapnell-lab.github.io/monocle-release/papers/ |
| CellPhoneDB (version 1.6.1) | Liu et al.36 | https://github.com/ventolab/CellphoneDB |
Experimental model and study participant details
Mice
A total of 10 C57BL/6 mice (Postnatal Day 1) were purchased from Shanghai Slake Experimental Animal Co, Ltd. All animal experiments in this study have been approved by the Shanghai University of Medicine and Health Sciences under the approval number 2021-SZR-05-410482. The mice were maintained under strict laboratory conditions, adhering to the highest standards of animal welfare. All experimental procedures involving mice were conducted in accordance with ethical guidelines and approved by the relevant institutional animal care and use committee.
Method details
Cell preparation for single-cell sequencing
In preparing cells for scRNA-seq from the hearts of C57BL/6 neonatal mice, tissues were finely minced and subjected to enzymatic digestion using 0.75% Collagenase/Dispase (Roche, 11097113 001). This method facilitates the release of individual cells while preserving cell surface markers essential for downstream applications. Post-dissociation, we applied 1× Red Blood Cell Lysis Solution (Thermo Fisher, 00-4333-57) to remove erythrocytes, which could otherwise interfere with the accurate identification and sorting of target cell populations. Following RBC lysis, cells were washed with phosphate-buffered saline (PBS) containing 2% FBS, pelleted via centrifugation, and resuspended in a sorting buffer to maintain cell viability and minimize nonspecific binding. Cell viability was assessed using Trypan Blue (Thermo Fisher, 14190144) staining. Cells were prepared with a viability of 90% as measured using the Countess II Automated Cell Counter. Regarding the cell count, the single-cell suspension contained approximately 844,000 viable cells. Achieving a viability of over 90% made the cells suitable for proceeding with the single-cell sequencing steps.
Single-cell RNA sequencing
Sorted cells were prepared for scRNA-seq by encapsulating them into droplets using the Chromium Next GEM Chip G Single Cell Kit (10xGenomics, 1000120) and Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit v3.1 (10xGenomics, 1000121). Each droplet contained a barcoded bead for capturing mRNA from individual cells. Following cell lysis within the droplets, mRNA was reverse-transcribed into cDNA, which was subsequently amplified and prepared into sequencing libraries. The Chromium i7 Multiplex Kit (10xGenomics, 120262) was used during library preparation. The heart cells from the 9 mice used in our single-cell sequencing were combined and captured in a single run to ensure consistency and reduce batch effects.
The quality of the sequencing libraries was assessed using bioanalytical instruments, such as the Agilent Bioanalyzer 2100 with the High Sensitivity DNA Kit (Agilent, 5067-4626). Libraries that passed QC were sequenced on an Illumina NovaSeq 6000 platform using 2 × 150 chemistry. The total number of targeted reads was approximately 15,000. Raw reads were processed using the Cell Ranger (2.1.0), which included demultiplexing, alignment to the GRCm38 mouse genome using the STAR algorithm, and UMI-based quantification of transcripts. QC measures were taken to exclude low-quality cells and doublets, ensuring a dataset composed of high-quality single-cell transcriptomes. Cells with fewer than 200 or more than 6,000 detected genes were excluded, and cells with mitochondrial gene expression exceeding 10% were removed, based on ref.37,38 and the characteristics of our own data. Initially, 14,684 cells were captured, and after QC, 8,652 cells were retained for analysis. Additionally, the scDblFinder package (version 1.8.0) was used to identify doublets. Raw sequencing data were preprocessed using a standardized pipeline that included trimming, alignment, and quantification of gene expression levels. Batch effects were minimized, and data normalization was carried out to allow comparison across cells.
Heart tissue section preparation for spatial transcriptomic analysis
Heart was excised from euthanized neonatal C57BL/6 mice following approved institutional protocols for animal care and use. The excised heart was immediately rinsed in cold phosphate-buffered saline (PBS) to remove excess blood and maintained on ice to preserve tissue integrity. Immediately after extraction, the heart tissue was fixed to preserve cellular structure and prevent degradation. The tissue was submerged in 4% paraformaldehyde (PFA) for 48 h at room temperature. Subsequently, the heart tissue was dehydrated and embedded in paraffin, after which the paraffin block containing the tissue was sectioned using a microtome.
The heart was anatomically divided into four regions: apex, ventricle, atrium, and upper atrium. Each region was sectioned into six pieces, resulting in a 24-piece heart model for comprehensive spatial analysis. Each tissue section was first subjected to histological staining using TissueTek O.C.T. Compound (SAKURA, 4583) to preserve and highlight cellular and structural features, thus facilitating the identification of distinct anatomical areas for subsequent transcriptomic profiling. Following staining, each piece underwent careful processing for spatial transcriptomic sequencing.
Spatial transcriptomic sequencing
Tissue sections were mounted on spatially barcoded microarray slides, specifically the Visium Spatial Gene Expression Slide & Reagents Kit (10xGenomics, 1000184), designed for capturing mRNA transcripts while retaining their spatial information. The slides featured an array of oligo-dT primers, each carrying a unique spatial barcode and a poly (T) sequence to capture polyadenylated mRNA molecules directly from the tissue. Before sequencing, an in situ reverse transcription reaction was performed on the slides, converting the captured mRNA into cDNA while preserving the spatial barcodes. This step was followed by the amplification of the cDNA directly on the slide, ensuring that sufficient material was generated for high-quality sequencing without the loss of spatial resolution.
The amplified cDNA was then subjected to library preparation using the Visium Spatial Tissue Optimization Slide & Reagents Kit (10xGenomics, 1000193), where sequencing adapters and additional indexes were added. The prepared libraries were quantified, and their quality was validated using a high-sensitivity DNA chip on a Bioanalyzer or a similar instrument. Once verified, the libraries were pooled and sequenced on a next-generation sequencing platform capable of reading the spatial barcodes along with the transcriptomic information.
Post-sequencing, the raw data were processed using specialized bioinformatics pipelines. This involved demultiplexing the reads according to their spatial barcodes, aligning the sequences to the reference genome, and annotating the transcripts to their respective genes. The resulting data provided a gene expression profile for each spatially barcoded spot on the microarray slide.
Quantification and statistical analysis
Single-cell RNA data analysis
The single-cell data processing was accomplished using the Seurat package (version 3.1.2). Initially, for the entire heart dataset, the data were normalized, and principal components (PCs) were selected using an Elbow Plot, focusing on PCs 1 to 26. These PCs were then used to construct a nearest neighbor graph. During clustering analysis, resolution values ranging from 0.1 to 1.2 were explored, with a final resolution of 1.2 being chosen. Based on this, t-SNE and UMAP were performed for nonlinear dimensional reduction, and the results were visualized by both cluster and sample grouping (Figures 1 and S1).
For the CM subpopulations, data normalization and feature selection were similarly conducted, with 2000 highly variable genes identified using the FindVariableFeatures function. Subsequently, PCA was performed, and batch effect correction was applied using the Harmony package (version 0.1.0). An Elbow Plot was used again to select PCs 1 to 20, which were utilized to build a nearest neighbor graph. During clustering, resolution values from 0.1 to 1.2 were tested, with a final choice of resolution at 0.1. Using the harmony corrected results, t-SNE and UMAP analyses were conducted, and visualizations were created based on clustering and sample groupings (Figure 2).
Spatial gene expression mapping and integration with single-cell RNA sequencing data
Initially, the expression matrix and spatial image data were loaded using the Read10X and Read10X_Image functions to create a Seurat object. The SpatialFeaturePlot function was then employed to visualize the spatial expression of specific genes. Subsequently, the gene expression matrix from the "Spatial" assay was extracted, and the number of locations with non-zero expression for each gene was calculated. As part of QC, genes expressed in more than 10 locations were retained. The percentage of mitochondrial gene expression was also calculated, and both mitochondrial and ribosomal genes were removed. Thereafter, data normalization was performed using the SCTransform function, followed by PC analysis with RunPCA. Clustering analysis was conducted using FindNeighbors and FindClusters, while dimensionality reduction and visualization were achieved with RunUMAP, dividing the data into several cell subpopulations. Deconvolution with single-cell RNA-seq data was then used to annotate the cell types within the spatial transcriptomics data.
The spacexr package (version 2.2.1) was employed for deconvolution analysis, aiming to determine the cellular composition in spatial transcriptomics data. Initially, scRNA and stRNA data were loaded. A reference object was constructed by extracting RNA counts and cell type information from the scRNA data. Spatial coordinates and count matrices were obtained from the stRNA data, and a spatial RNA object was created using the SpatialRNA function. Finally, deconvolution analysis was performed with the create. RCTD and run. RCTD functions to infer the cell type composition at each spatial location, and the SpatialDimPlot function was used to visualize the spatial distribution of different cell types.39
This integration of spatial and scRNA-seq data provided a multi-layered understanding of the cellular composition, gene expression patterns, and functional states within the neonatal mouse heart. This combined approach enabled the exploration of intricate relationships between gene expression, cell type distribution, and anatomical organization, offering insights into the developmental and functional architecture of the heart (Figure 5).
Pseudotime trajectory analysis
To infer the developmental trajectory of CM subtypes in the neonatal mouse heart, we employed the Monocle package (version 2.22.0) to construct pseudotime trajectories. This computational approach allows for the ordering of individual cells based on their transcriptional profiles, thus providing insights into the temporal progression of cellular differentiation. By aligning single-cell transcriptomic data along a constructed trajectory, we can observe the emergence of distinct cellular states and the functional specialization of CMs (Figure 3).
Analysis of intercellular communication
The elucidation of ligand-receptor interactions among cell types and subtypes was performed using the CellPhoneDB package (version 1.6.1). This computational method predicts potential intercellular communications by identifying significant ligand-receptor pairs. The analysis was conducted by collating single-cell transcriptomic data, which provided a detailed expression profile for each cell type present in the developing murine heart. Subsequently, the inferred interactions were overlaid onto spatial transcriptomic maps, allowing for the localization of these interactions within the tissue architecture. This integrative approach enabled the identification of spatially resolved communication networks, which are crucial for understanding the functional specialization and heterogeneity of cells in the neonatal mouse heart (Figures 6 and 7).
Functional enrichment and pathway analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were executed to interpret the biological functions and processes associated with the identified gene expression profiles (Figures 2 and 4).
Published: December 13, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111596.
Contributor Information
Linlin Ma, Email: linlinma1986@gmail.com.
Yanfei Li, Email: liyf@sumhs.edu.cn.
Supplemental information
References
- 1.Zhou P., VanDusen N.J., Zhang Y., Cao Y., Sethi I., Hu R., Zhang S., Wang G., Ye L., Mazumdar N., et al. Dynamic changes in P300 enhancers and enhancer-promoter contacts control mouse cardiomyocyte maturation. Dev. Cell. 2023;58:898–914. doi: 10.1016/j.devcel.2023.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paige S.L., Plonowska K., Xu A., Wu S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015;116:341–353. doi: 10.1161/circresaha.116.302752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang H., Pei L., Ouyang Z., Wang H., Chen X., Jiang K., Huang S., Jiang R., Xiang Y., Wei K. AP-1 activation mediates post-natal cardiomyocyte maturation. Cardiovasc. Res. 2023;119:536–550. doi: 10.1093/cvr/cvac088. [DOI] [PubMed] [Google Scholar]
- 4.Katz J.A., Levy P.T., Butler S.C., Sadhwani A., Lakshminrusimha S., Morton S.U., Newburger J.W. Preterm congenital heart disease and neurodevelopment: the importance of looking beyond the initial hospitalization. J. Perinatol. 2023;43:958–962. doi: 10.1038/s41372-023-01687-4. [DOI] [PubMed] [Google Scholar]
- 5.Paik D.T., Cho S., Tian L., Chang H.Y., Wu J.C. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 2020;17:457–473. doi: 10.1038/s41569-020-0359-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kürten C.H.L., Kulkarni A., Cillo A.R., Santos P.M., Roble A.K., Onkar S., Reeder C., Lang S., Chen X., Duvvuri U., et al. Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing. Nat. Commun. 2021;12:7338. doi: 10.1038/s41467-021-27619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z., Cui M., Shah A.M., Tan W., Liu N., Bassel-Duby R., Olson E.N. Cell-Type-Specific Gene Regulatory Networks Underlying Murine Neonatal Heart Regeneration at Single-Cell Resolution. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.109211. [DOI] [Google Scholar]
- 8.Martini E., Kunderfranco P., Peano C., Carullo P., Cremonesi M., Schorn T., Carriero R., Termanini A., Colombo F.S., Jachetti E., et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation. 2019;140:2089–2107. doi: 10.1161/circulationaha.119.041694. [DOI] [PubMed] [Google Scholar]
- 9.DeLaughter D.M., Bick A.G., Wakimoto H., McKean D., Gorham J.M., Kathiriya I.S., Hinson J.T., Homsy J., Gray J., Pu W., et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell. 2016;39:480–490. doi: 10.1016/j.devcel.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cable D.M., Murray E., Shanmugam V., Zhang S., Zou L.S., Diao M., Chen H., Macosko E.Z., Irizarry R.A., Chen F. Cell type-specific inference of differential expression in spatial transcriptomics. Nat. Methods. 2022;19:1076–1087. doi: 10.1038/s41592-022-01575-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asp M., Giacomello S., Larsson L., Wu C., Fürth D., Qian X., Wärdell E., Custodio J., Reimegård J., Salmén F., et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell. 2019;179:1647–1660. doi: 10.1016/j.cell.2019.11.025. [DOI] [PubMed] [Google Scholar]
- 12.Shen J., Ma L., Hu J., Li Y. Single-Cell Atlas of Neonatal Mouse Hearts Reveals an Unexpected Cardiomyocyte. J. Am. Heart Assoc. 2023;12 doi: 10.1161/jaha.122.028287. [DOI] [Google Scholar]
- 13.Tucker N.R., Chaffin M., Fleming S.J., Hall A.W., Parsons V.A., Bedi K.C., Jr., Akkad A.D., Herndon C.N., Arduini A., Papangeli I., et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020;142:466–482. doi: 10.1161/circulationaha.119.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Litviňuková M., Talavera-López C., Maatz H., Reichart D., Worth C.L., Lindberg E.L., Kanda M., Polanski K., Heinig M., Lee M., et al. Cells of the adult human heart. Nature. 2020;588:466–472. doi: 10.1038/s41586-020-2797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakano H., Nakano A. The role of metabolism in cardiac development. Curr. Top. Dev. Biol. 2024;156:201–243. doi: 10.1016/bs.ctdb.2024.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed R.E., Tokuyama T., Anzai T., Chanthra N., Uosaki H. Sarcomere maturation: function acquisition, molecular mechanism, and interplay with other organelles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2022;377 doi: 10.1098/rstb.2021.0325. [DOI] [Google Scholar]
- 17.Coulombe K.L.K., Bajpai V.K., Andreadis S.T., Murry C.E. Heart regeneration with engineered myocardial tissue. Annu. Rev. Biomed. Eng. 2014;16:1–28. doi: 10.1146/annurev-bioeng-071812-152344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voges H.K., Foster S.R., Reynolds L., Parker B.L., Devilée L., Quaife-Ryan G.A., Fortuna P.R.J., Mathieson E., Fitzsimmons R., Lor M., et al. Vascular cells improve functionality of human cardiac organoids. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.112322. [DOI] [Google Scholar]
- 19.Feng W., Bais A., He H., Rios C., Jiang S., Xu J., Chang C., Kostka D., Li G. Single-cell transcriptomic analysis identifies murine heart molecular features at embryonic and neonatal stages. Nat. Commun. 2022;13:7960. doi: 10.1038/s41467-022-35691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanemaru K., Cranley J., Muraro D., Miranda A.M.A., Ho S.Y., Wilbrey-Clark A., Patrick Pett J., Polanski K., Richardson L., Litvinukova M., et al. Spatially resolved multiomics of human cardiac niches. Nature. 2023;619:801–810. doi: 10.1038/s41586-023-06311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Y., Pu W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020;126:1086–1106. doi: 10.1161/circresaha.119.315862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian J., Liao J., Liu Z., Chi Y., Fang Y., Zheng Y., Shao X., Liu B., Cui Y., Guo W., et al. Reconstruction of the cell pseudo-space from single-cell RNA sequencing data with scSpace. Nat. Commun. 2023;14:2484. doi: 10.1038/s41467-023-38121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L., Hua K., Zhang N., Wang J., Meng J., Hu Z., Gao H., Li F., Chen Y., Ren J., et al. Multifaceted Spatial and Functional Zonation of Cardiac Cells in Adult Human Heart. Circulation. 2022;145:315–318. doi: 10.1161/circulationaha.121.055690. [DOI] [PubMed] [Google Scholar]
- 24.Lin C., Chen Y., Zhang F., Zhu P., Yu L., Chen W. Single-cell RNA sequencing reveals the mediatory role of cancer-associated fibroblast PTN in hepatitis B virus cirrhosis-HCC progression. Gut Pathog. 2023;15:26. doi: 10.1186/s13099-023-00554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Liu W., Xiao Y., Yuan H., Wang F., Jiang P., Luo Z. Association of Apelin and Apelin Receptor Polymorphisms With the Risk of Comorbid Depression and Anxiety in Coronary Heart Disease Patients. Front. Genet. 2020;11:893. doi: 10.3389/fgene.2020.00893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandovici I., Georgopoulou A., Pérez-García V., Hufnagel A., López-Tello J., Lam B.Y.H., Schiefer S.N., Gaudreau C., Santos F., Hoelle K., et al. The imprinted Igf2-Igf2r axis is critical for matching placental microvasculature expansion to fetal growth. Dev. Cell. 2022;57:63–79. doi: 10.1016/j.devcel.2021.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maeng Y.S., Choi H.J., Kwon J.Y., Park Y.W., Choi K.S., Min J.K., Kim Y.H., Suh P.G., Kang K.S., Won M.H., et al. Endothelial progenitor cell homing: prominent role of the IGF2-IGF2R-PLCbeta2 axis. Blood. 2009;113:233–243. doi: 10.1182/blood-2008-06-162891. [DOI] [PubMed] [Google Scholar]
- 28.Lopaschuk G.D., Karwi Q.G., Tian R., Wende A.R., Abel E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021;128:1487–1513. doi: 10.1161/circresaha.121.318241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto T., Maurya S.K., Pruzinsky E., Batmanov K., Xiao Y., Sulon S.M., Sakamoto T., Wang Y., Lai L., McDaid K.S., et al. RIP140 deficiency enhances cardiac fuel metabolism and protects mice from heart failure. J. Clin. Invest. 2023;133 doi: 10.1172/jci162309. [DOI] [Google Scholar]
- 30.Ritterhoff J., Tian R. Metabolic mechanisms in physiological and pathological cardiac hypertrophy: new paradigms and challenges. Nat. Rev. Cardiol. 2023;20:812–829. doi: 10.1038/s41569-023-00887-x. [DOI] [PubMed] [Google Scholar]
- 31.Farah E.N., Hu R.K., Kern C., Zhang Q., Lu T.Y., Ma Q., Tran S., Zhang B., Carlin D., Monell A., et al. Spatially organized cellular communities form the developing human heart. Nature. 2024;627:854–864. doi: 10.1038/s41586-024-07171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Germain P.L., Sonrel A., Robinson M.D. pipeComp, a general framework for the evaluation of computational pipelines, reveals performant single cell RNA-seq preprocessing tools. Genome Biol. 2020;21:227. doi: 10.1186/s13059-020-02136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rao Y., Zhong D., Qiu K., Cheng D., Li L., Zhang Y., Mao M., Pang W., Li D., Song Y., et al. Single-Cell Transcriptome Profiling Identifies Phagocytosis-Related Dual-Feature Cells in A Model of Acute Otitis Media in Rats. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.760954. [DOI] [Google Scholar]
- 36.Liu Z., Sun D., Wang C. Evaluation of cell-cell interaction methods by integrating single-cell RNA sequencing data with spatial information. Genome Biol. 2022;23:218. doi: 10.1186/s13059-022-02783-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao M., Wang X., Guo G., Wang L., Chen S., Yin P., Chen K., Chen L., Zhang Z., Chen X., et al. Resolving the intertwining of inflammation and fibrosis in human heart failure at single-cell level. Basic Res. Cardiol. 2021;116:55. doi: 10.1007/s00395-021-00897-1. [DOI] [PubMed] [Google Scholar]
- 38.Du L., Sun X., Gong H., Wang T., Jiang L., Huang C., Xu X., Li Z., Xu H., Ma L., et al. Single cell and lineage tracing studies reveal the impact of CD34(+) cells on myocardial fibrosis during heart failure. Stem Cell Res. Ther. 2023;14:33. doi: 10.1186/s13287-023-03256-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cable D.M., Murray E., Zou L.S., Goeva A., Macosko E.Z., Chen F., Irizarry R.A. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 2022;40:517–526. doi: 10.1038/s41587-021-00830-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data needed to evaluate the conclusions in the paper are present in the paper and/or the The raw data have been deposited in the Gene Expression Omnibus (GEO: GSE232466) and Mendeley Data: https://data.mendeley.com/datasets/4zfdygb9bz/1, and they are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The R code to analyze the data used in this study is available online: https://github.com/Lizhiyongjuan/scRNA_ST.git.
-
•
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.