

Abstract
The genetic code is established by the aminoacylation reactions of tRNA synthetases. Its accuracy depends on editing reactions that prevent amino acids from being assigned to incorrect codons. A group of class I synthetases share a common insertion that encodes a distinct site for editing that is about 30 Å from the active site. Both misactivated aminoacyl adenylates and mischarged amino acids attached to tRNA are translocated to this site, which, in turn, is divided into subsites—one for the adenylate and one for the aminoacyl moiety attached to tRNA. Here we report that a specific mutation in isoleucyl-tRNA synthetase prevents editing by blocking translocation. The mutation alters a widely conserved residue that is believed to tether the amino group of mischarged tRNA to its subsite for editing. These and other data support a model where editing is initiated by translocation of the misacylated amino acid attached to tRNA to create an “editing complex” that facilitates subsequent rounds of editing by translocation of the misactivated adenylate.
The coupling of an amino acid to its corresponding tRNA establishes the connection between the genome and the building blocks of proteins. The enzymes that catalyze this ligation, aminoacyl-tRNA synthetases, specifically recognize their respective amino acid cognates as substrates (1–3). If a tRNA is charged with a noncognate amino acid, the error will be propagated into a nascent polypeptide chain, possibly leading to an inactive or misfolded protein. To avoid such mistakes, nature has evolved a number of editing mechanisms that protect against the formation of mischarged tRNAs (4).
Isoleucine and valine differ by a single methylene group. Therefore, isoleucyl-tRNA synthetase (IleRS) must not only activate isoleucine but also work against misactivation of the off-target valine. To achieve high accuracy, IleRS uses two enzymatic functions that act as a “double sieve” (5). The enzyme aminoacylates tRNAIle with isoleucine and also hydrolyzes (edits) misactivated valine in a tRNAIle-dependent process (6). There is evidence that IleRS can edit both Val-AMP (pretransfer) and Val-tRNAIle (posttransfer) (Fig. 1A). Posttransfer editing was demonstrated directly through the rapid IleRS-catalyzed deacylation of exogenous Val-tRNAIle (7). Fersht interpreted the data from rapid quench experiments as suggesting that pretransfer editing could be a significant alternative editing pathway for IleRS (5). Further support for a pretransfer editing pathway comes from the isolation of a DNA aptamer that triggers the editing reaction but cannot be aminoacylated (8, 9).
Figure 1.

(A) The IleRS-catalyzed editing reaction in the presence of tRNAIle and valine. (B) Closeup view of the posttransfer editing site. D279 of T. thermophilus ValRS (which aligns with D342 of E. coli IleRS) is positioned to interact with a misactivated amino acid that would be attached to either the 2′-OH or the 3′-OH of A76 in a posttransfer complex. The D279 side chain is too far away to directly interact with the ribose hydroxyls or to interact with the misactivated aminoacyl-AMP in a possible pretransfer complex. Coordinates of the ValRS/tRNAVal complex were taken from Fukai et al. (19). ValRS and tRNAVal are colored by atom type (green, carbon; red, oxygen; blue, nitrogen). Hydrogen atoms and protein backbone oxygen atoms are not shown.
IleRS has two distinct active sites—one for aminoacylation and one for editing. The editing activity derives from a large insertion termed connective polypeptide 1 (CP1, 276 amino acids) (10–15). After amino acid activation, either Val-AMP or Val-tRNAIle must be translocated to the CP1 editing site, which sterically excludes the correctly activated isoleucine (13, 16). Mutagenesis of some conserved residues in CP1 yielded variants that possessed full aminoacylation activity but were altered in their ability to edit valine (13). Specific site-directed mutations in CP1 of IleRS (and ValRS) give proteins that synthesize mischarged tRNAs because of their impaired editing (17, 18).
The pre- and posttransfer editing pathways can be separated by mutation, suggesting the existence of distinct pre- and posttransfer subsites (T. L. Hendrickson, T.K.N., and P.S., unpublished results). This result is consistent with the crystal structure of the homologous Thermus thermophilus ValRS complexed with tRNAVal (19). The posttransfer subsite model described by Fukai et al. (Fig. 1B) contains an aspartate (D328 in T. thermophilus IleRS, D279 in T. thermophilus ValRS, D342 in Escherichia coli IleRS) that does not appear in the pretransfer subsite model (19). This aspartate is conserved in all published sequences of IleRS and is also conserved in the most closely related synthetases, i.e., ValRS and LeuRS (20). The carboxylate side chain of D342 is predicted to form a salt bridge interaction with the α-NH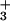 of Val-tRNAIle. By contrast, in the pretransfer model, D342 is not predicted to play a role in binding Val-AMP (19). Therefore, one prediction is that IleRS 342 mutants would be specifically deficient in posttransfer editing.
of Val-tRNAIle. By contrast, in the pretransfer model, D342 is not predicted to play a role in binding Val-AMP (19). Therefore, one prediction is that IleRS 342 mutants would be specifically deficient in posttransfer editing.
Earlier results showed that pretransfer editing can be selectively disrupted through mutation (T. L. Hendrickson, T.K.N., and P.S., unpublished results). However, no IleRS mutants that are specifically posttransfer deficient have been described. Therefore, it was not clear whether pretransfer editing could take place in the absence of a functional posttransfer machinery. In the current study, we set out to further isolate the pre- and posttransfer editing pathways by mutating the putative posttransfer residue D342. In addition, we sought to better understand the translocation process by analyzing mutant proteins that harbored substitutions at a series of conserved residues along a possible “translocation pathway” between the aminoacylation and editing sites.
Materials and Methods
Plasmid Construction.
All IleRS point mutants were constructed in the pBAD plasmid by using the QuikChange (Stratagene) system according to the manufacturer's instructions. The mutagenic primers (Invitrogen) used are available on request. All mutations were confirmed by sequencing over the mutated region.
Complementation Assays.
The plasmids containing mutations in ileS were transformed into E. coli strain MG1655 (21). Overnight cultures (200 μl) of the resulting strains were pelleted, resuspended in 100 μl of 10 mM Mg SO4/5 mM CaCl2, and infected with 50 μl of P1 phage stock carrying the ileS∷kanr marker for 30 min at 37°C. Sodium citrate was added (200 μl, 1 M), and the cultures were incubated at 22°C. After 30 min, the entire culture was plated on LB/agar/50 μg/ml of Amp/25 μg/ml of Kan/0.02%arabinose and incubated for 3 days at 22°C. Kanamycin-resistant colonies were isolated twice and used to express IleRS variants.
Protein Expression and Purification.
Strains (ΔileS∷kanr) containing plasmids expressing the various IleRS mutants were grown overnight to saturation in LB/Amp/Kan/0.02% arabinose. The resulting cultures were diluted 1:100 and grown to OD600 ≈0.4–0.6. IleRS expression was induced by the addition of 0.2% arabinose for 4–5 h. IleRS and IleRS mutants were purified as previously described (22). IleRS mutants that were incapable of complementing the deletion of the wild-type gene (E199A, D532A, D564A, and R567L) were expressed from E. coli strain MI1 (17, 23, 24). This strain encodes a mutant IleRS that possesses an elevated KM for Ile (F570S, KM > 5 mM). Thus, the contaminating F570S IleRS (<10%) is silent under standard aminoacylation conditions ([Ile] = 8 μM).
Total Editing (ATPase) Assays.
Total editing assays were carried out essentially as described (25). For the measuring of the apparent KM for editing of tRNAIle, equivalent editing assays were carried out at 33.2 μM, 22.1 μM, 16.6 μM, 11.1 μM, 8.29 μM, and 5.53 μM tRNAIle.
Aminoacylation Assays.
Mischarging assays (with valine) were carried out at ambient temperature as previously described with 200 nM IleRS, 10 mg/ml of E. coli tRNA, and [3H]Val (9 μM, 1461 cpm/pmol) (17). Aminoacylation assays with isoleucine were performed in the same manner with the following changes: 20 nM IleRS, [3H]Ile (20 μM, 140 cpm/pmol), [tRNAIle] = 45.3 μM, 30.1 μM, 22.6 μM, 15.1 μM, 11.3 μM, 7.54 μM, 5.66 μM.
Resonance Energy Transfer Assay for Translocation.
Fluorescence measurements were made by using a Perkin–Elmer LS 50 Luminescence Spectrometer. Samples were stirred continuously at ambient temperature in 20 mM Hepes, 50 mM NaCl, and 1 mM MgCl2. IleRS (600 nM) was incubated with 30 μM N-methylanthraniloyl dATP (dATP†) and 480 nM chemically synthesized Val-AMP (16). The translocation of activated valine was initiated by the addition of 300 nM tRNAIle. Fractional occupation of the IleRS active site was monitored as previously described (17).
Deacylation of Val-tRNAIle.
E. coli tRNAIle (GAU) was produced from an overexpressing strain and purified as described (26). Val-tRNAIle was synthesized by incubating 10 μM tRNAIle, 6 μM T242P IleRS, 17.5 μM [3H]Val (0.57 μCi/pmol) at 37°C for 45 min under aminoacylation conditions (20 mM Hepes pH 7.5/0.1 mM EDTA/150 mM NH4Cl/10 μg/ml of BSA/2.5 mM ATP/5 mM MgCl2). The reaction mixture was phenol/chloroform (1:1) extracted and the misacylated tRNA was ethanol precipitated. Deacylation assays were performed as described with 10 nM IleRS or 20 μM of the isolated CP1 domain (25).
Results
Editing Activity of D342 Mutants.
Mutant proteins were cloned, purified, and then investigated for their editing activities. For this purpose, overall editing was measured by the hydrolysis of ATP when IleRS, tRNAIle, valine, and ATP are mixed. This assay monitors the sum of both pre- and posttransfer editing (Fig. 1A). Posttransfer editing activities were measured by monitoring the deacylation of exogenous Val-tRNAIle.
To probe the role of the aspartyl 342 side chain in editing, we generated alanine, glutamate, and asparagine mutants at position 342 (D342A, D342E, and D342N IleRS). These mutant enzymes demonstrated that the editing activity of IleRS is extremely sensitive to the identity of the chemical group at position 342 (Fig. 2A). D342A IleRS possesses a severely depressed editing rate (roughly 20- to 30-fold) that is difficult to estimate because it is close to the background rate observed with no tRNA in the reaction mixture. D342E IleRS, which differs from the wild type by one extra methylene group, has a 2- to 3-fold reduced total editing rate. D342N IleRS, which is isosteric with the wild-type enzyme but lacks the acidic functionality of Asp-342, is highly defective in total editing (10- to 15-fold). These results show that the carboxylate of Asp-342 is crucial for editing, and that slight displacement of the carboxylate can have a significant effect on the efficiency of editing.
Figure 2.

Total editing, mischarging, and deacylation activities of IleRS D342 mutants. (A) tRNA-dependent editing of misactivated valine at pH 7.5, 25°C, for wild-type (WT), D342A, D342E, and D342N IleRS. (B) Aminoacylation of E. coli tRNA with valine at pH 7.5, 25°C for D342A, D342E, and D342N IleRS. (C) Wild-type and D342 mutant IleRS enzymes (10 nM) were assayed for their ability to enzymatically deacylate Val-tRNAIle at pH 7.5, 25°C.
Mischarging Phenotypes of D342 Mutants.
Wild-type IleRS is incapable of generating Val-tRNAIle in appreciable amounts because of its editing activity (17). We tested whether amino acid substitutions at position 342 could render IleRS capable of mischarging tRNAIle with valine (Fig. 2B). D342N and D342A IleRS can produce substantial quantities of mischarged Val-tRNAIle, supporting the observation that a carboxyl group is required at position 342 for the proper editing functions of IleRS. In contrast, D342E IleRS is capable of producing only small amounts of Val-tRNAIle. This small amount of mischarging is consistent with its ability to edit misactivated valine at a rate that is only 2-fold lower than that of the wild-type enzyme.
Posttransfer Editing Activity of D342 Mutants.
D342 was predicted by Fukai et al. (19) to function specifically in the IleRS posttransfer editing subsite. To probe the role of D342 in posttransfer editing, we measured the abilities of the D342 mutants to enzymatically deacylate exogenous Val-tRNAIle. We found that D342A and D342N IleRS are highly deficient in their deacylation activities. At 10 nM IleRS, the wild-type enzyme rapidly deacylated Val-tRNAIle efficiently, whereas D342A and D342N IleRS only partially deacylated Val-tRNAIle after extended incubation times (Fig. 2C). D342E deacylated Val-tRNAIle at a rate of approximately one-half the rate of the wild-type enzyme. D342A and D342N IleRS were capable of efficiently deacylating Val-tRNAIle only at enzyme concentrations of 10- to 20-fold higher than used for wild-type and D342E IleRS (data not shown).
Fluorescence Resonance Energy Transfer Analysis of Misactivated Valine Translocation.
To further investigate the molecular basis for the editing defects of the 342 mutants, we used a fluorescence-based translocation assay that measures the reoccupation of the aminoacylation active site on translocation of misactivated valine to the editing site of IleRS (Fig. 3A) (16). [Earlier work established that translocation occurs in cis, that is, without dissociation of misactivated substrate from the enzyme (16)]. This assay utilizes the fluorescent nucleotide N-methylanthraniloyl dATP (dATP†) that binds to the aminoacylation site of IleRS (Kd ≈ 2 μM). When tRNAIle is added to a 1:1 IleRS/Val-AMP complex (Kd ≈ 10 nM), Val-AMP is translocated to the editing site, allowing dATP† to bind immediately at the aminoacylation site. The excitation maximum of dATP† (≈350 nm) overlaps the emission spectrum of the indole group of tryptophan side chains. The emission spectrum of dATP† is centered at 440 nm. Therefore, 295 nm excitation of active site tryptophans of the IleRS/dATP† complex gives rise to 440 nm emission by energy transfer. The level of 440 nm fluorescence indicates the fractional occupation of the active site with dATP†. The rate of this emission increase provides a direct measure of the kinetics of translocating misactivated valine from the aminoacylation site to the editing site.
Figure 3.

tRNA-dependent translocation of misactivated valine by wild-type IleRS and D342 mutants as monitored by resonance energy transfer (16). (A) Schematic diagram of the fluorescence assay used to measure IleRS translocation rates. The binding of tRNAIle to a complex of IleRS and misactivated valine (Val* as Val-AMP or Val-tRNAIle) triggers the translocation and hydrolysis of Val*. Translocation of Val* to the editing site allows for the rapid binding of fluorescent dATP† at the synthetic site of IleRS. This binding event is measured by the change in fluorescence (shown by white-to-black transition in synthetic site). (B) Chemically synthesized Val-AMP was bound to the aminoacylation site at pH 7.5, 25°C. Translocation from the synthetic site to the editing site was induced by the addition of tRNAIle (t = 0 sec), resulting in increased emission at 440 nm. Under single turnover conditions, the rate of increased emission is directly related to the rate of translocation (17).
D342A and D342N IleRS are severely deficient in their ability to translocate misactivated valine (Fig. 3B). Unlike wild-type IleRS, no burst of fluorescence increase is observed when tRNAIle is added to the D342A (or D342N) IleRS/Val-AMP complex. The initial rates of dATP† binding on addition of tRNAIle are reduced at least 10- to 20-fold for these “noncarboxylate” mutants. In good agreement with the data on total editing, D342E IleRS has a moderately reduced translocation rate. These data suggest that the carboxylate of Asp-342 plays a direct role in translocating misactivated valine to the editing active site.
Analysis of the Cloned Editing Domain.
D342A and D342N are the first translocation-deficient mutants of IleRS to be identified. This result is surprising in that D342 is located within the posttransfer editing site, not along a possible translocation pathway between the aminoacylation and editing sites. To test whether the D342 mutants had an inherent deficiency in their deacylation activities, we generated D342A CP1. The CP1 domain of IleRS can be expressed as a stable polypeptide that is capable of deacylating Val-tRNAIle (12). This truncated protein contains no aminoacylation active site. We found that D342A CP1 does not deacylate Val-tRNAIle, whereas wild-type CP1 deacylates mischarged tRNAIle (Fig. 4). These data suggest that D342 has a direct role in deacylation of Val-tRNAIle. Because it is unlikely that a single carboxyl would function in both a dynamic translocation event and the resulting hydrolytic cleavage, it seems most probable that the role of D342 is to bind misactivated valine at the conclusion of the translocation process.
Figure 4.

Deacylation defect of D342A CP1. Wild-type and D342A CP1 (IleRS residues 180–419) were assayed for their ability to enzymatically deacylate Val-tRNAIle at pH 7.5, 25°C.
Kinetic Analysis of D342E IleRS Editing.
From the above data, we hypothesized that a major function of D342 is to orient Val-tRNAIle at the end of the translocation process. Thus, the editing defects of D342E, D342A, and D342N could be because of a reduction in affinity of Val-tRNAIle for the editing active site. If this were true, then we would expect that the apparent Michaelis constant (KM app) for tRNAIle in the editing reaction would be higher in the D342 mutants than in the wild-type enzyme. To test this idea, we measured the editing rates of wild-type and D342E IleRS at various tRNAIle concentrations (for D342A and D342N, the editing rates are too low to perform the equivalent experiment). Lineweaver–Burke analysis of the resulting data revealed that the KM app for D342E IleRS is ≈4- to 5-fold higher than for the wild-type enzyme (Fig. 5; wild-type KM app ≈ 8.0 μM, D342E KM app ≈ 36 μM). This result is specific to the editing reaction, as the D342E mutation has no effect on the value of KM for tRNAIle in the aminoacylation reaction with Ile (Fig. 5 Inset). This experiment confirms that the carboxyl group of D342 plays an important role in binding the acceptor stem of tRNAIle in the editing site. In addition, the Vmax of D342E IleRS is ≈2- to 3-fold lower than that of wild-type IleRS. Thus, even a small displacement of the carboxyl group, through an aspartate to glutamate mutation, has a substantial effect on the enzyme's efficiency in the editing reaction.
Figure 5.

Kinetic analysis of D342E IleRS editing. ATP hydrolysis editing reactions (as in Fig. 2A) were carried out at various concentrations of tRNAIle. The reciprocals of the cpm of hydrolyzed ATP were plotted against the reciprocal of tRNAIle concentration. KM app for tRNAIle was defined as the negative reciprocal of the x-intercept of the resulting lines. Inset shows the equivalent experiment for the IleRS catalyzed aminoacylation reaction (formation of Ile-tRNAIle) by using various concentrations of tRNAIle.
The Search for Other “Translocation Pathway” Mutants.
Little is known concerning the protein determinants that are important for translocation of misactivated valine in tRNAIle-mediated editing. In an attempt to identify other important chemical groups along the translocation pathway, we generated a panel of IleRSs that contained point mutations in regions that lie between the aminoacylation active site and the editing active site (E199A, Y202F, V323A, G339A, D532A, D564A, R567L; E. coli numbering). The selected mutation sites are strongly conserved among IleRSs from different organisms (Fig. 6A). In addition, the amino acid side chains of these sites are situated such that they could conceivably help to form a translocation channel between tRNAIle and the tRNA-interacting face of IleRS (Fig. 6B).
Figure 6.

(A) IleRS sequence alignment of regions that were mutated in this study. Mutations that disrupted the ability of the gene to complement the deletion of wild-type ileS are shown in red. Gene mutations that complement the wild-type deletion are shown in green. (B) Crystal structure of T. thermophilus IleRS (all numbering corresponds to E. coli IleRS) (13). Mutation sites are color coded as in A. For orientation, some previously identified aminoacylation site residues (P57, W534) are shown in magenta, and editing site residues (T242, T243) are shown in blue.
All seven IleRS mutants were tested for their ability to complement a strain bearing the ΔileS deletion. This test was accomplished by infecting E. coli strains containing the plasmid-borne ileS mutants with P1 phage that carry a ΔileS∷kanr marker, followed by selection for kanr colonies. Four IleRS point mutants (E199A, D532A, D564A, and R567L) failed to rescue the deletion of the wild-type gene. Of the seven mutation sites, these four are the most proximal to the aminoacylation site of IleRS (Fig. 6B), suggesting that these noncomplementing mutants may have reduced aminoacylation activity. To investigate this possibility, we overexpressed and purified the four mutants. As suspected, E199A, D532A, D564A, and R567L IleRS all showed substantial aminoacylation defects (data not shown) and thus were not investigated further. Three mutants (Y202F, V323A, and G339A) complemented the deletion at levels comparable to complementation with the wild-type gene. In agreement with the complementation experiments, the purified forms of these mutants were able to aminoacylate tRNAIle with activities comparable to wild-type IleRS (data not shown).
Editing Activity of Potential Translocation Pathway Mutants.
Translocation of misactivated valine to the editing site is the rate-limiting step of the editing process (16). Therefore, mutants of IleRS that possess altered translocation rates should demonstrate differing editing rates. The IleRS mutants Y202F, V323A, and G339A had similar total editing activities, as measured by their ability to hydrolyze ATP in the presence of valine and tRNAIle (Fig. 7). G339A IleRS edits valine at a rate indistinguishable from the wild-type enzyme. Y202F and V323A IleRS have only slight defects in total editing. These results suggest that none of these putative “translocation residues” play a direct role in the translocation process.
Figure 7.

Total editing activity of IleRS “translocation pathway” mutants as measured by hydrolysis of ATP. tRNA-dependent editing of misactivated valine at pH 7.5, 25°C, for wild-type (WT), Y202F, V323A, G339A, and D342A IleRS.
Discussion
D342A and D342N IleRS are the first severely translocation-deficient IleRS mutants to be identified. These enzymes are essentially “dead” in an assay that uses a fluorescent ATP-analogue to measure the reoccupation of the aminoacylation site on translocation of misactivated valine. However, it is clear from further data that D342 does not play a role in translocation independent of a role in deacylation of exogenous Val-tRNAIle. This deacylation defect is also present in D342A CP1, thus showing that D342A IleRS's inability to deacylate is not because of its translocation defect.
Taken together, the data presented here suggest that D342 does not play a role in the “translocation pathway.” Rather, this conserved aspartate acts by helping to bind misactivated valine at the end of translocation. Kinetic analysis revealed that D342E IleRS has a substantially heightened apparent editing KM for tRNAIle. This result suggests that D342 functions as a binding element for misactivated valine in the editing active site. The question of whether the translocation pathway is a true pathway that is guided by specific protein contacts remains a subject of future work.
The D342 mutants also provide new insights into the substantial literature concerning the relative contributions of the pretransfer and posttransfer pathways to the overall editing reaction. Early kinetic studies suggested that pretransfer editing is the dominant pathway and that the posttransfer activity of IleRS functions as a “cleanup” mechanism (5). However, the results in this work show that posttransfer editing is important for the overall editing reaction—pre- and posttransfer. In the IleRS posttransfer active site model of Fukai et al. (19), the residue corresponding to D342 makes a salt bridge interaction with the α-NH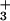 of Val-tRNAIle. D342 plays no role in the pretransfer active site model. Our results show that even subtle mutations at position 342 strongly affect total editing rates (overall ATPase activity; Fig. 1A). The isosteric D342N mutant enzyme edits valine at a rate roughly 5% of that of the wild-type enzyme. On the basis of the posttransfer subsite model, removal of the negatively charged aspartate should weaken a possible salt bridge between valine's α-NH
of Val-tRNAIle. D342 plays no role in the pretransfer active site model. Our results show that even subtle mutations at position 342 strongly affect total editing rates (overall ATPase activity; Fig. 1A). The isosteric D342N mutant enzyme edits valine at a rate roughly 5% of that of the wild-type enzyme. On the basis of the posttransfer subsite model, removal of the negatively charged aspartate should weaken a possible salt bridge between valine's α-NH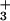 and the IleRS editing site. One would not predict that such a conservative mutation would disrupt pretransfer editing, as the putative pretransfer complex would not interact with D342. However, with all three D342 IleRS mutants, total editing rates drop by the same amount that the deacylation rate is reduced.
and the IleRS editing site. One would not predict that such a conservative mutation would disrupt pretransfer editing, as the putative pretransfer complex would not interact with D342. However, with all three D342 IleRS mutants, total editing rates drop by the same amount that the deacylation rate is reduced.
A new model for editing that accounts for the previous and current data proposes that each IleRS/tRNAIle complex undergoes one round of posttransfer editing before subsequent rounds of pretransfer editing can begin. Thus, the series of events in an in vitro editing reaction would be misacylation of tRNAIle with valine, translocation of the 3′ end of the Val-tRNAIle acceptor stem to the posttransfer editing site, followed by hydrolysis of Val-tRNAIle, and processive shuttling and hydrolysis of newly activated Val-AMP molecules to the pretransfer editing site (Fig. 8).
Figure 8.

A model for in vitro IleRS tRNA-mediated editing in which one round of posttransfer editing precedes multiple rounds of pretransfer editing. (i) tRNAIle binds to the IleRS/Val-AMP complex. (ii) Valine is covalently transferred to tRNAIle. (iii) The acceptor stem of Val-tRNAIle is translocated to the editing site. (iv) Val-tRNAIle is hydrolyzed and valine is released. A second molecule of Val-AMP is synthesized at the aminoacylation site. (v) Val-AMP is translocated to the editing site. (vi) Val-AMP is hydrolyzed at the editing site, and a third molecule of Val-AMP is synthesized at the aminoacylation site. (vii) Val-AMP is translocated to the editing site, and the cycle is repeated.
In this model, the translocation of the 3′ end of the Val-tRNAIle to the editing site serves to prime the enzyme for subsequent rounds of pretransfer editing. This “postpre-prepre” model is consistent with a number of observations that have until now been difficult to reconcile. These include the observations that the hydroxyl groups of the terminal A76 of tRNAIle are crucial for editing (6, 27), and that all deacylation-deficient IleRS mutants that have been discovered to date have strong defects in total editing. (If pretransfer editing comprised an independent and predominate pathway, it should be possible to isolate IleRS mutants that cannot deacylate Val-tRNAIle but retain high total editing activity.) The correlation between deacylation activity and total editing is most strikingly apparent in the various D342 mutants described here.
The postpre-prepre model of IleRS editing raises questions regarding the in vivo mechanisms of editing. If pretransfer editing must be preceded by a round of posttransfer editing, then it is likely that pretransfer editing has less biological significance in preventing the formation of mischarged Val-tRNAIle. IleRS binds isoleucine roughly 100 times more tightly than valine. Thus, the binding of two consecutive molecules of valine at the aminoacylation site would be rare except under conditions where intracellular pools of valine were high.
Acknowledgments
This work was supported by National Institutes of Health Grant GM15539 and by a fellowship from the National Foundation for Cancer Research. A.C.B. is supported by the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation Fellowship (DRG-1637).
Abbreviations
- IleRS
isoleucyl-tRNA synthetase
- CP1
connective polypeptide 1
References
- 1.Schimmel P. Annu Rev Biochem. 1987;56:125–158. doi: 10.1146/annurev.bi.56.070187.001013. [DOI] [PubMed] [Google Scholar]
- 2.Giegé R, Puglisi J D, Florentz C. Prog Nucleic Acid Res Mol Biol. 1993;45:129–206. doi: 10.1016/s0079-6603(08)60869-7. [DOI] [PubMed] [Google Scholar]
- 3.Carter C W., Jr Annu Rev Biochem. 1993;62:715–748. doi: 10.1146/annurev.bi.62.070193.003435. [DOI] [PubMed] [Google Scholar]
- 4.Jakubowski H, Goldman E. Microbiol Rev. 1992;56:412–429. doi: 10.1128/mr.56.3.412-429.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fersht A R. Biochemistry. 1977;16:1025–1030. doi: 10.1021/bi00624a034. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin A N, Berg P. J Biol Chem. 1966;241:839–845. [PubMed] [Google Scholar]
- 7.Eldred E W, Schimmel P R. J Biol Chem. 1972;247:2961–2964. [PubMed] [Google Scholar]
- 8.Hale S P, Schimmel P. Proc Natl Acad Sci USA. 1996;93:2755–2758. doi: 10.1073/pnas.93.7.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farrow M A, Schimmel P. Biochemistry. 2001;40:4478–4483. doi: 10.1021/bi0024052. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt E, Schimmel P. Science. 1994;264:265–267. doi: 10.1126/science.8146659. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt E, Schimmel P. Biochemistry. 1995;34:11204–11210. doi: 10.1021/bi00035a028. [DOI] [PubMed] [Google Scholar]
- 12.Lin L, Hale S P, Schimmel P. Nature (London) 1996;384:33–34. doi: 10.1038/384033b0. [DOI] [PubMed] [Google Scholar]
- 13.Nureki O, Vassylyev D G, Tateno M, Shimada A, Nakama T, Fukai S, Konno M, Hendrickson T L, Schimmel P, Yokoyama S. Science. 1998;280:578–582. doi: 10.1126/science.280.5363.578. [DOI] [PubMed] [Google Scholar]
- 14.Silvian L F, Wang J, Steitz T A. Science. 1999;285:1074–1077. [PubMed] [Google Scholar]
- 15.Mursinna R S, Lincecum T L, Martinis S A. Biochemistry. 2001;40:5376–5381. doi: 10.1021/bi002915w. [DOI] [PubMed] [Google Scholar]
- 16.Nomanbhoy T K, Hendrickson T L, Schimmel P. Mol Cell. 1999;4:519–528. doi: 10.1016/s1097-2765(00)80203-8. [DOI] [PubMed] [Google Scholar]
- 17.Hendrickson T L, Nomanbhoy T K, Schimmel P. Biochemistry. 2000;39:8180–8186. doi: 10.1021/bi0004798. [DOI] [PubMed] [Google Scholar]
- 18.Döring V, Mootz H D, Nangle L A, Hendrickson T L, de Crécy-Lagard V, Schimmel P, Marlière P. Science. 2001;292:501–504. doi: 10.1126/science.1057718. [DOI] [PubMed] [Google Scholar]
- 19.Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev D G, Yokoyama S. Cell. 2000;103:793–803. doi: 10.1016/s0092-8674(00)00182-3. [DOI] [PubMed] [Google Scholar]
- 20.Landes C, Perona J J, Brunie S, Rould M A, Zelwer C, Steitz T A, Risler J L. Biochimie. 1995;77:194–203. doi: 10.1016/0300-9084(96)88125-9. [DOI] [PubMed] [Google Scholar]
- 21.Blattner F R, Plunkett G, 3rd, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 22.Shepard A, Shiba K, Schimmel P. Proc Natl Acad Sci USA. 1992;89:9964–9968. doi: 10.1073/pnas.89.20.9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iaccarino M, Berg P. J Bacteriol. 1971;105:527–537. doi: 10.1128/jb.105.2.527-537.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Treiber G, Iaccarino M. J Bacteriol. 1971;107:828–832. doi: 10.1128/jb.107.3.828-832.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farrow M A, Nordin B E, Schimmel P. Biochemistry. 1999;38:16898–16903. doi: 10.1021/bi9920782. [DOI] [PubMed] [Google Scholar]
- 26.Glasfeld E, Landro J A, Schimmel P. Biochemistry. 1996;35:4139–4145. doi: 10.1021/bi9527810. [DOI] [PubMed] [Google Scholar]
- 27.von der Haar F, Cramer F. Biochemistry. 1976;15:4131–4138. doi: 10.1021/bi00663a034. [DOI] [PubMed] [Google Scholar]