

Abstract
para-Hydroxybenzoate hydroxylase catalyzes a two-step reaction that demands precise control of solvent access to the catalytic site. The first step of the reaction, reduction of flavin by NADPH, requires access to solvent. The second step, oxygenation of reduced flavin to a flavin C4a-hydroperoxide that transfers the hydroxyl group to the substrate, requires that solvent be excluded to prevent breakdown of the hydroperoxide to oxidized flavin and hydrogen peroxide. These conflicting requirements are met by the coordination of multiple movements involving the protein, the two cofactors, and the substrate. Here, using the R220Q mutant form of para-hydroxybenzoate hydroxylase, we show that in the absence of substrate, the large βαβ domain (residues 1–180) and the smaller sheet domain (residues 180–270) separate slightly, and the flavin swings out to a more exposed position to open an aqueous channel from the solvent to the protein interior. Substrate entry occurs by first binding at a surface site and then sliding into the protein interior. In our study of this mutant, the structure of the complex with pyridine nucleotide was obtained. This cofactor binds in an extended conformation at the enzyme surface in a groove that crosses the binding site of FAD. We postulate that for stereospecific reduction, the flavin swings to an out position and NADPH assumes a folded conformation that brings its nicotinamide moiety into close contact with the isoalloxazine moiety of the flavin. This work clearly shows how complex dynamics can play a central role in catalysis by enzymes.
Many enzyme reactions require that the reacting species be isolated from solvent. Proteins often accomplish this by rearranging the active site after substrates bind. With some enzymes, multistep reactions are catalyzed in which some steps require isolation from solvent, whereas others require access to it. Here, we describe the unique way that the flavoprotein para-hydroxybenzoate hydroxylase (PHBH; E.C. 1.14.13.2), which catalyzes the monooxygenation of p-hydroxybenzoate (p-OHB) to 3,4-dihydroxybenzoate (1), deals with these disparate requirements. A key intermediate in the catalytic cycle of PHBH is the flavin C4a-hydroperoxide, generated by reaction of molecular oxygen with the flavin after its reduction by NADPH (1). Because the nascent hydroperoxide readily reacts with water, forming oxidized flavin and hydrogen peroxide, it must be shielded from solvent (2). Nevertheless, access to NADPH is necessary to permit reduction of the flavin. These diverse requirements are met by moving the isoalloxazine moiety of FAD (3). Reduction of the flavin takes place when the isoalloxazine occupies a highly accessible position at the protein surface (4). The flexibility of the ribityl chain permits the reduced flavin to immediately swing back into the protein interior (3) where solvent is excluded. Thus, two conformations of the flavin exist, “out” (solvent exposed) and “in” (solvent excluded), and these have been observed both spectroscopically and crystallographically (3–7). Because no crystal structure of PHBH with NADP(H) bound was available, a clear understanding of how the flavin is reduced at the out position was previously lacking. Motion of the flavin to the out position is also believed to enhance accessibility of the binding pocket for p-OHB (3, 4), although a direct path for this substrate into the water-secluded protein interior could not be identified conclusively.
We now present a structural investigation of the R220Q mutant form of PHBH that discloses the binding mechanisms of NADPH and p-OHB and reveals that coordinated motions of the protein, the two cofactors, and the substrate are the basis for catalysis.
Methods
Data Collection, Structure Determination, and Refinement.
Crystals of substrate-free R220Q PHBH from Pseudomonas aeruginosa were obtained by vapor diffusion. Hanging drops containing 4 mg/ml protein/100 mM KPi, pH 7.0/0.05 mM glutathione/30 mM sodium sulfite/0.02 mM FAD/450 mM (NH4)2SO4 were equilibrated at 30°C for 7–10 days against a well solution of similar composition, but containing 900 mM (NH4)2SO4. Crystals of the NADPH-bound form of the enzyme were obtained by soaking in an aerobic environment substrate-free crystals in a holding solution of 100 mM KPi, pH 7.0/0.1 mM glutathione/30 mM Na2SO3/0.15 mM EDTA/0.008 mM FAD/16% (vol/vol) glycerol/2.8 M (NH4)2SO4/50 mM NADPH for a minimum of 2 h, with changes every 30 min. Although PHBH crystals do not undergo visible changes under these conditions, incubation with NADPH seems to increase both the static and dynamic disorder of the crystals as attested by the decreased diffraction limit and the significantly higher mean temperature factor 〈B〉 of the refined structure (see Table 1). Crystals of the p-OHB-bound form were obtained by soaking substrate-free crystals in a holding solution containing 100 mM p-OHB for 24 h with frequent changes. For each condition, diffraction data were collected from a single crystal at 100 K with an R-axis IV image plate detector at the CuKα wavelength. Oscillations were processed with hkl (8). Crystals belong to space group C2221 with unit cell dimensions a ≅ 72 Å, b ≅ 140 Å, c ≅ 92 Å, and diffract to ≈2.0 Å. The unit cell dimensions are essentially unchanged with respect to wild type, but the position and orientation of the enzyme in the unit cell are different. Consequently, the molecular replacement procedure was used to determine the structure of the mutant enzyme. All steps were carried out by using the CNS v. 1.0 suite of crystallographic programs (9). Model refinement was also carried out with CNS v. 1.0 using cross-validated maximum likelihood as the target function (10). Solvent molecules were added during the final stages of refinement after the protein model had stabilized. The crystallographic data and refinement statistics are shown in Table 1.
Table 1.
Data collection and refinement statistics
| — | NADPH | p-OHB | |
|---|---|---|---|
| Resolution, Å | 21.6 to 2.0 | 22.0 to 2.2 | 22.0 to 1.8 |
| Measurements | 261,632 | 262,880 | 557,623 |
| Unique reflections | 30,046 | 20,552 | 38,363 |
| 〈Redundancy〉 | 8.4 | 12.8 | 13.8 |
| Completeness, % | 96.5 | 86.6 | 90.6 |
| 〈I〉/σ〈I〉 | 19.0 | 15.4 | 28.3 |
| Rmerge,* % | 9.6 | 13.0 | 8.1 |
| Rcryst,† % | 20.9 | 22.1 | 20.7 |
| Rfree, % | 24.7 | 27.5 | 24.0 |
| Solvent molecules | 212 | 116 | 260 |
| 〈B〉, Å2 | 35.2 | 42.5 | 31.9 |
| rmsd bond lengths, Å | 0.007 | 0.008 | 0.007 |
| rmsd bond angles, deg | 1.3 | 1.4 | 1.3 |
| rmsd dihedral, deg | 22.9 | 22.5 | 22.3 |
| rmsd impropers, deg | 0.80 | 0.88 | 0.83 |
Rmerge = ∑h∑i|I(h)i − 〈I(h)〉|/∑h∑iI(h)i, where I(h)i is the ith measurement.
Rcryst = ∑|Fobs − Fcalc|/∑|Fobs|. Rfree was calculated on 10% of the data omitted from refinement. Mean B values were calculated from the refined models.
Modeling of the Reduction Step of the Catalytic Cycle.
The structure of mutant R220Q in complex with NADPH was used as the starting model. However, the FAD coordinates were replaced with those from the structure of the wild-type enzyme in complex with 2,4-dihydroxybenzoate (3), in which the isoalloxazine ring is not bent and occupies the out position. Repositioning of the nicotinamide ring of NADPH was obtained by manually modifying several torsion angles of the cofactor, after which, van der Waals overlaps were relieved by means of 5,000 cycles of conjugate gradient minimization with CNS v. 1.0 (9).
Results and Discussion
Protein Dynamics in PHBH.
The mutant enzyme R220Q has lower affinity for p-OHB than the wild-type enzyme (Table 2). Previous attempts to crystallize wild-type enzyme in the absence of substrate yielded poorly diffracting crystals (11). By contrast, R220Q PHBH crystallizes readily in the absence of substrate, and high-resolution structures could be determined (Table 1). PHBH is composed of a large globular domain (residues 1–180) with βαβ fold, where the flavin binds, a smaller domain consisting of a single helix and an antiparallel β-sheet (residues 180–270), where the substrate binds, and a third, exclusively helical domain (residues 270–394). The active site is at the interface of the three domains. Arg-220 occupies a central position in the active site, as its guanidinium group forms hydrogen bonds with Tyr-222, a residue directly involved in substrate binding, and via a water molecule, with the carbonyl of Arg-44, a residue immediately adjacent to the si side of the flavin ring (not shown). In R220Q, Gln-220 occupies approximately the position of the β through δ carbons of Arg-220, and a water molecule fills the space of the Arg-220 guanidinium group (Fig. 1). The hydrogen bond to the hydroxyl of Tyr-222 is lost (Fig. 1), but two new hydrogen bonds are gained to the backbone of Leu-188 and Tyr-221, respectively (not shown). Whereas the local changes associated with the R220Q substitution are relatively minor, more dramatic global effects are observed. In the absence of substrate, the sheet domain of R220Q PHBH rotates with respect to the other two domains, and the active site opens up (Fig. 2). As part of this process, the flavin ring takes an intermediate position between the in and out conformations, and an aqueous channel widens on its re side (Figs. 1 and 2). This channel connects the bulk solvent to the substrate-binding pocket, which appears filled by water molecules. In R220Q, the guanidinium group of Arg-214, which in the wild-type enzyme forms a salt-bridge with the carboxylate moiety of p-OHB, swings away from the substrate binding site in association with a backbone rearrangement of residues 212–218. Rearrangement of the protein backbone of residues 43–47 also occurs on the si side of the flavin ring (Fig. 2B). Together, these changes can be described in terms of an “open” active site in which the flavin occupies a more exposed position and the substrate pocket has lost a major element of specificity (Arg-214). It is difficult to rationalize why the substrate-free form of the enzyme can be observed in R220Q but not in the wild type. Perhaps Arg-220 is involved in modulating the dynamics of flavin movements, as suggested by the fact that it forms a hydrogen bond with O4 of the flavin ring, when FAD occupies the out position (3). However, in R220Q the flavin is partially out, and Gln-220 does not form a hydrogen bond with the flavin O4 (Fig. 1). One of the most interesting changes observed in substrate-free R220Q is the extended conformation of Arg-214 (Fig. 2B). Interestingly, under conditions in which the substrate pocket is filled (see below), Arg-214 resumes its normal conformation, as does the entire stretch of residues from 212 to 218. However, the substrate binding domain remains rotated with respect to the other two domains, the flavin is still in an intermediate position between in and out, and the backbone rearrangements around residues 43–47 are not reversed. Thus, the primary effect of the R220Q mutation is that of favoring the “open” conformation of the enzyme, regardless of the presence of substrate. By contrast, this conformation would occur only transiently in the wild-type enzyme, when substrate or product is exchanged.
Table 2.
Kinetic parameters for wild-type PHBH and mutant R220Q
| Enzyme | Relative activity |
K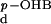 , mM , mM |
Without p-OHB
|
With p-OHB
|
||
|---|---|---|---|---|---|---|
K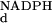 , mM , mM |
kred, s−1 |
K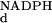 , mM , mM |
kred, s−1 | |||
| Wild type | 100 | 0.02 | 0.11 | 0.0003 | 0.2 | 50 |
| R220Q | 1 | 6 | 1.1 | 0.0003 | 44 | 0.37 |
Activities were measured under standard assay conditions at 25°C. Rate constants and Kd values were measured at pH 6.5 and 3°C as described (30). Values for the Kd of NADPH and for the rate constant of flavin reduction by NADPH were measured in the absence or presence of the substrate p-OHB at saturating concentration. Wild-type parameters are from ref. 31.
Figure 1.

The active site of R220Q PHBH. The Cα trace and side chains of the mutant enzyme are shown with salmon and ivory bonds, respectively; nitrogen is blue, and oxygen is red. The backbones of residues 44–47 and 297–300 are shown with sticks colored according to atom type. The Cα trace and the Arg-220 side chain of the wild-type enzyme are shown with transparent bonds. FAD is shown with green bonds, its re face pointing to the right. Hydrogen bonds are shown as dotted light blue lines. Coordinates for this figure are from PDB entry 1IUW (wild type) and 1K0L (R220Q PHBH, this study). Figures were generated with MOLSCRIPT (32), BOBSCRIPT (33), and RASTER3D (34).
Figure 2.

Protein dynamics in PHBH. (A) superposition of wild-type PHBH with bound p-OHB (light blue transparent trace) and of substrate-free R220Q mutant (salmon trace). Least-squares fitting was carried out to maximize the superposition of the FAD binding domain. FAD and p-OHB in the wild-type enzyme are shown as yellow bonds; FAD in the mutant enzyme is green. (B) Close-up and rotated view of the superposition shown in A. The Cα trace and side chains of the mutant enzyme are shown with salmon and ivory bonds, respectively; nitrogen is blue, and oxygen is red. The Cα trace and side chains of the wild type are shown with transparent bonds. Water molecules in the mutant enzyme appear as red spheres. The re face of FAD points to the right. Coordinates for this figure are as in Fig. 1.
Cofactor Dynamics in PHBH.
Addition of 50 mM NADPH under aerobic conditions causes crystals of R220Q PHBH to change from light green to light yellow. There is significant dichroism in the crystals, so it is not clear whether the observed color changes are due to a change in the flavin redox state or to the formation of a charge–transfer complex with NADP(H).¶ The crystal structure reveals that the isoalloxazine ring is bent and occupies the in position (Fig. 3A). The structure also contains electron density due to NADP(H). NADP(H) binds in an extended conformation at the enzyme surface in a groove that crosses the binding site of FAD (Fig. 3A). At one end of the groove, the adenosyl moiety of NADP(H) occupies a large pocket lined by the side chains of Arg-33, Gln-34, Tyr-38, Arg-42, Arg-44, and by the pyrophosphate and adenosyl moieties of FAD. A sulfate ion also fills the pocket. The adenine ring of NADP(H) intercalates between the si side of the flavin and the side chain of Arg-44 (Fig. 3A), with a hydrogen bond connecting the adenine N6 and the 2-OH of the FAD ribityl chain. The ribose phosphate of NADP(H) is held in place by a salt-bridge to the guanidinium moiety of Arg-44. Notably, in the absence of NADP(H) the position of the ribose 2′-phosphate is occupied by a lone phosphate or sulfate ion (not shown). At the opposite end of the groove, the pyridine nucleotide has more points of association with the protein. In particular, the pyrophosphate and nicotinamide moieties display multiple polar and hydrophobic interactions with the side chains of Phe-161, His-162, Gln-167, Pro-267, and Arg-269 (Fig. 3A). Additional interactions occur with Arg-128, Ile-164, Gln-167, and Ser-168 of a symmetry-related molecule (not shown). Despite these stabilizing contacts, the electron density of NADP(H) is progressively less defined toward the nicotinamide moiety, and only broken density marks the position of the nicotinamide (Fig. 3A). The binding mode of NADP(H) observed in R220Q is likely to represent closely the “true” binding mode of the nucleotide in the wild-type enzyme. In fact, essentially all of the residues that this study shows to be involved in interactions with the pyridine nucleotide were also identified as being important for binding NADPH on the basis of mutagenesis or chemical modification studies (12–18). In particular, there was strong evidence indicating that Arg-44 was involved in binding the adenosyl moiety and that His-162 and Arg-269 were involved in binding the pyrophosphate moiety (14). Tyr-38 and Arg-42 have also been implicated in binding the NADPH 2′-phosphate (12, 15). Interestingly, in the structure of R220Q PHBH, these two residues bind a sulfate or phosphate ion close to the 2′-phosphate of NADP(H) (Fig. 3A). Thus, either the effects of mutations in these residues on the binding of NADPH are indirect, or it might be that during the reduction reaction the 2′-phosphate of NADPH oscillates between the position observed in this study and the position marked by the isolated sulfate ion.
Figure 3.

Cofactor dynamics in PHBH. (A) Stereoview of the NADPH binding site. The Cα trace and side chains of the R220Q enzyme are shown with salmon and ivory bonds, respectively. FAD is shown with green bonds. NADPH is shown as ball-and-sticks with gold bonds, surrounded by a sigmaA-weighted omit electron density map contoured at 0.9 σ. Sulfate ions are shown as ball-and-sticks with yellow bonds. Nitrogen, blue; oxygen, red; sulfur, yellow; phosphorus, pale blue. Note how the FAD ring is slightly bent and occupies the “in” position. Coordinates are from PDB entry 1K0J (this study). (B) Modeling of the reduction step of the catalytic cycle. The FAD ring is “out,” and the nicotinamide moiety of NADPH swings “in” such that its R side faces the re side of the flavin. Only a few side chains are drawn for reference. Coordinates for this panel were derived by energy minimization after the flavin and nicotinamide rings had been manually repositioned.
Overall, the binding mode of NADP(H) revealed by this study clearly suggests how flavin reduction might take place. Biochemical and spectroscopic evidence indicates that reduction occurs with the isoalloxazine in the out position (4) and shows that hydride transfer takes place from the pro-R side of NADPH (at C4) to the re side of the flavin (at N5) (19–21). In the R220Q structure, the flavin occupies the in position, and C4 of the nicotinamide ring is 19 Å away from N5 of the flavin. Thus, for reduction to occur, there must be a simultaneous rotation of the flavin to the out position and a movement of the nicotinamide of NADPH to a position in which it is stacked with the isoalloxazine, so that C4NADPH is within 4 Å of N5FAD (Fig. 3B). In this process, NADPH would cradle the flavin in the out position during the time required for hydride transfer. After hydride transfer is completed, the nicotinamide moiety of NADP+ would swing out and the flavin ring would swing in, ready for reacting with oxygen and hydroxylating p-OHB.
How plausible is this model for the step of flavin reduction? First, significant protein conformational changes must occur to permit the nicotinamide of NADPH to reach the re side of the flavin. After manually modeling NADPH in its “folded” conformation, the structure was minimized to relieve van der Waals overlaps (see Methods). During the minimization, the active site expands to accommodate the nicotinamide group, with the largest positional displacements occurring along the direction perpendicular to the plane of the flavin ring (Fig. 3B). In particular, a full rotation of NADPH from the extended conformation to the folded conformation would normally be prevented by the stretch of residues 263–271, which are therefore expected to move significantly during the reduction step. A folded conformation of the pyridine nucleotide analogous to the one postulated in PHBH is also observed in flavin reductase P (22) and in catalases from Proteus mirabilis (23) and beef liver (24). In catalase, NADPH is stabilized by stacking the adenine moiety with the guanidinium group of an arginine that forms a salt bridge to the ribose phosphate. A similar interaction of NADPH with an arginine is also present in glutathione reductase (25) and in mercuric reductase (26); however, in these enzymes NADPH is in an extended rather than a folded conformation. These structures demonstrate a previously unrecognized structural motif for NADPH binding in which a key factor is the interaction of an arginine side chain with both the adenine and the 2′-phosphate of the pyridine nucleotide.
Previous attempts to identify crystallographically the binding site for NADPH in wild-type enzyme in the presence of p-OHB were unsuccessful. Why was it possible to observe NADP(H) bound to R220Q in the absence of substrate? First, the more open structure observed here for R220Q might better resemble the state of PHBH when NADPH is bound than does the closed conformation of the wild-type enzyme in complex with p-OHB. Second, flavin reduction in the wild-type enzyme in the presence of p-OHB is ≈200,000 times faster than in its absence (Table 2). Thus, it might be the longer residence time of NADPH in the active site of R220Q that allows its visualization. This hypothesis is supported by the observation that when NADPH and p-OHB are added together to crystals of the R220Q mutant, a condition in which flavin reduction is expected to undergo a substantial acceleration (Table 2), NADP(H) is no longer visible (not shown).
Substrate Dynamics in PHBH.
In substrate-free R220Q, an aqueous channel on the re side of the flavin connects the bulk solvent to the substrate pocket (Fig. 2B). This observation seems to discount an earlier suggestion that a channel on the si side of the flavin could allow substrate entry (3). It seems, however, that p-OHB does not enter the channel by chance; rather, it is guided into the channel by first binding at a low-affinity site located near the channel opening. This phenomenon was first recognized when crystals of R220Q were incubated in the presence of 100 mM p-OHB. This high concentration (≈17 times the Kd) was necessary to saturate the binding pocket because the mutant enzyme has low affinity for the substrate (Table 2). Under these conditions, two molecules of p-OHB bind to the enzyme, one at the standard site where hydroxylation takes place and the other at a low-affinity site. The latter is characterized by both polar and hydrophobic interactions with the side chains of Arg-269, Asp-286, and Ile-290 (Fig. 4). When crystals are incubated in the presence of only 20 mM p-OHB, the low-affinity site appears empty, suggesting that the Kd of p-OHB at this site is between 20 and 100 mM (not shown). In the high-affinity site, p-OHB displays all of the interactions that have been traditionally observed; in particular, the guanidinium group of Arg-214 forms a salt-bridge with the carboxylate moiety of the substrate (Fig. 4).
Figure 4.

Substrate dynamics in PHBH. The Cα trace and side chains of the R220Q enzyme are shown with salmon and ivory bonds, respectively. FAD is shown with green bonds; the re face is pointing up and to the right. Two molecules of p-OHB are shown as ball-and-stick models with yellow bonds, surrounded by a sigmaA-weighted omit electron density map contoured at 0.9 σ. Nitrogen, blue; oxygen, red. Water molecules appear as red spheres. Note the aqueous channel on the re side of the flavin connecting the low-affinity binding site of p-OHB (Lower) to the high-affinity site (Upper). Coordinates for this figure are from PDB entry 1K0I (this study).
p-OHB bound at the low-affinity site would clearly block the binding of NADPH when this cofactor binds in its extended conformation and, more importantly, would prevent the nicotinamide moiety from approaching the re side of the flavin ring for hydride transfer (see above). Thus, the p-OHB low-affinity site might exist only in the open conformation of the enzyme, when substrate is coming in or product is leaving, but not in the closed conformation. In fact, when crystals of the wild-type enzyme, which display the closed conformation, are incubated with 100 mM p-OHB, only one substrate molecule occupying the high-affinity site is observed (not shown). In particular, the conformation of the wild-type enzyme to which NADPH binds during the reduction of the flavin would differ from the conformation observed in R220Q because it would lack the p-OHB low-affinity site. This hypothesis is consistent with the observation that in the wild-type enzyme, steps in the hydroxylation reaction, but not the reduction reaction, are inhibited by high concentration of substrate (27, 28). By contrast, in the mutant R220Q, which favors the conformation containing both the binding site for NADPH and the low-affinity site for p-OHB, both the binding of NADPH and the reduction reaction are inhibited by high concentration of substrate (Table 2).
Conclusions
The structural snapshots presented here indicate a remarkable scenario for the dynamics of flavin reduction and substrate binding. First, the enzyme seems to be much more flexible than previously thought. In the absence of substrate, the large βαβ domain (residues 1–180) and the smaller sheet domain (residues 180–270) separate slightly, the flavin swings to a more exposed position, and an aqueous channel opens up from the bulk phase to the protein interior (Fig. 2). Recognition of this channel by the substrate is facilitated by the presence of a low-affinity site near the opening. The nature of the binding of p-OHB at this site suggests that the substrate enters the channel with the 4-hydroxyl first (Fig. 4). As p-OHB moves forward in the channel and reaches its high-affinity site, the βαβ and the sheet domains are expected to rotate and to close the active site onto the substrate. The subsequent stages of catalysis are dominated by the dramatic increase in the rate of flavin reduction by NADPH (Table 2), which is due to a shift of the equilibrium between the out and in conformations of the flavin in favor of the out conformation (4, 29). In this study, we show by model building how the flavin in the out conformation is reached by NADPH (Fig. 3B). The high mobility of the nicotinamide moiety of NADPH is essential in this process, as hydride transfer takes place from the flavin re side. Thus, whereas the flavin swings out, NADPH swings in, and flavin reduction occurs during a transient contact at the enzyme surface that finds its physical basis in the high flexibility of the two ligands. This view emphasizes how complex movements of the protein, the cofactors, and the substrate are all critically important to achieve catalysis.
Acknowledgments
We thank Drs. Sharon Ackerman and Martha Ludwig for critical evaluation of the manuscript. This research was supported by U.S. Public Health Service Grants AI42868 and AI43918 (to D.L.G), GM11106 (to V.M.), and GM20877 (to D.P.B.) and by Australian Research Council Grant A09906323 (to B.E.).
Abbreviations
- PHBH
para-hydroxybenzoate hydroxylase
- p-OHB
p-hydroxybenzoate
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1K0I, 1K0J, and 1K0L).
As the experiments were carried out aerobically, it cannot be certain whether the observed molecule is NADPH or NADP. Therefore, we designate it as NADP(H).
References
- 1.Entsch B, Ballou D P. Biochim Biophys Acta. 1989;999:313–322. doi: 10.1016/0167-4838(89)90014-9. [DOI] [PubMed] [Google Scholar]
- 2.Merenyi G, Lind J. J Am Chem Soc. 1991;113:3146. [Google Scholar]
- 3.Gatti D L, Palfey B A, Lah M S, Entsch B, Massey V, Ballou D P, Ludwig M L. Science. 1994;266:110–114. doi: 10.1126/science.7939628. [DOI] [PubMed] [Google Scholar]
- 4.Palfey B A, Ballou D P, Massey V. Biochemistry. 1997;36:15713–15723. doi: 10.1021/bi971427u. [DOI] [PubMed] [Google Scholar]
- 5.van Berkel W J, Eppink M H, Schreuder H A. Protein Sci. 1994;3:2245–2253. doi: 10.1002/pro.5560031210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreuder H A, Mattevi A, Obmolova G, Kalk K H, Hol W G, van der Bolt F J, van Berkel W J. Biochemistry. 1994;33:10161–10170. doi: 10.1021/bi00199a044. [DOI] [PubMed] [Google Scholar]
- 7.Zheng Y, Dong J, Palfey B A, Carey P R. Biochemistry. 1999;38:16727–16732. doi: 10.1021/bi9918893. [DOI] [PubMed] [Google Scholar]
- 8.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 9.Brunger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 10.Adams P D, Pannu N S, Read R J, Brunger A T. Proc Natl Acad Sci USA. 1997;94:5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Laan J M. Ph.D. thesis. The Netherlands: Univ. of Groningen; 1986. [Google Scholar]
- 12.Eppink M H, Overkamp K M, Schreuder H A, Van Berkel W J. J Mol Biol. 1999;292:87–96. doi: 10.1006/jmbi.1999.3015. [DOI] [PubMed] [Google Scholar]
- 13.Eppink M H, Bunthol C, Schreuder H A, van Berkel W J. FEBS Lett. 1999;443:251–255. doi: 10.1016/s0014-5793(98)01726-8. [DOI] [PubMed] [Google Scholar]
- 14.Eppink M H, Schreuder H A, van Berkel W J. J Biol Chem. 1998;273:21031–21039. doi: 10.1074/jbc.273.33.21031. [DOI] [PubMed] [Google Scholar]
- 15.Eppink M H, Schreuder H A, van Berkel W J. Eur J Biochem. 1998;253:194–201. doi: 10.1046/j.1432-1327.1998.2530194.x. [DOI] [PubMed] [Google Scholar]
- 16.Eppink M H, Schreuder H A, Van Berkel W J. Protein Sci. 1997;6:2454–2458. doi: 10.1002/pro.5560061119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eppink M H, Schreuder H A, Van Berkel W J. Eur J Biochem. 1995;231:157–165. doi: 10.1111/j.1432-1033.1995.0157f.x. [DOI] [PubMed] [Google Scholar]
- 18.van Berkel W J, Muller F, Jekel P A, Weijer W J, Schreuder H A, Wierenga R K. Eur J Biochem. 1988;176:449–459. doi: 10.1111/j.1432-1033.1988.tb14302.x. [DOI] [PubMed] [Google Scholar]
- 19.Ryerson C C, Ballou D P, Walsh C. Biochemistry. 1982;21:1144–1151. doi: 10.1021/bi00535a006. [DOI] [PubMed] [Google Scholar]
- 20.Manstein D J, Pai E F, Schopfer L M, Massey V. Biochemistry. 1986;25:6807–6816. doi: 10.1021/bi00370a012. [DOI] [PubMed] [Google Scholar]
- 21.Entsch B, Husain M, Ballou D P, Massey V, Walsh C. J Biol Chem. 1980;255:1420–1429. [PubMed] [Google Scholar]
- 22.Tanner J J, Tu S C, Barbour L J, Barnes C L, Krause K L. Protein Sci. 1999;8:1725–1732. doi: 10.1110/ps.8.9.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gouet P, Jouve H M, Dideberg O. J Mol Biol. 1995;249:933–954. doi: 10.1006/jmbi.1995.0350. [DOI] [PubMed] [Google Scholar]
- 24.Ko T P, Day J, Malkin A J, McPherson A. Acta Crystallogr D. 1999;55:1383–1394. doi: 10.1107/s0907444999007052. [DOI] [PubMed] [Google Scholar]
- 25.Karplus P A, Schulz G E. J Mol Biol. 1989;210:163–180. doi: 10.1016/0022-2836(89)90298-2. [DOI] [PubMed] [Google Scholar]
- 26.Schiering N, Kabsch W, Moore M J, Distefano M D, Walsh C T, Pai E F. Nature (London) 1991;352:168–172. doi: 10.1038/352168a0. [DOI] [PubMed] [Google Scholar]
- 27.Entsch B, Ballou D P, Husain M, Massey V. J Biol Chem. 1976;251:7367–7379. [PubMed] [Google Scholar]
- 28.Entsch B, Ballou D P, Massey V. J Biol Chem. 1976;251:2550–2563. [PubMed] [Google Scholar]
- 29.Palfey B A, Moran G R, Entsch B, Ballou D P, Massey V. Biochemistry. 1999;38:1153–1158. doi: 10.1021/bi9826613. [DOI] [PubMed] [Google Scholar]
- 30.Entsch B, Palfey B A, Ballou D P, Massey V. J Biol Chem. 1991;266:17341–17349. [PubMed] [Google Scholar]
- 31.Husain M, Massey V. J Biol Chem. 1979;254:6657–6666. [PubMed] [Google Scholar]
- 32.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 33.Esnouf R M. J Mol Graphics. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 34.Merrit E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]