

Abstract
Classical ligands bind to the extracellular surface of their cognate receptors and activate signaling pathways without crossing the plasma membrane barrier. We selectively targeted the intracellular receptor–G protein interface by using cell-penetrating membrane-tethered peptides. Attachment of a palmitate group to peptides derived from the third intracellular loop of protease-activated receptors-1 and -2 and melanocortin-4 receptors yields agonists and/or antagonists of receptor–G protein signaling. These lipidated peptides—which we have termed pepducins—require the presence of their cognate receptor for activity and are highly selective for receptor type. Mutational analysis of both intact receptor and pepducins demonstrates that the cell-penetrating agonists do not activate G proteins by the same mechanism as the intact receptor third intracellular loop but instead require the C-tail of the receptor. Construction of such peptide–lipid conjugates constitutes a new molecular strategy for the development of therapeutics targeted to the receptor–effector interface.
G protein-coupled receptors (GPCRs) play a vital role in the signaling processes that control cellular metabolism, cell growth, and motility, inflammation, neuronal signaling, and blood coagulation. Although remarkably diverse in sequence and function, all GPCRs share a highly conserved topological arrangement of a seven-transmembrane helical core domain joined by three intracellular loops, three extracellular loops, and N- and C-terminal domains (1). A key event for the switch from inactive to active receptor is ligand-induced conformational changes of transmembrane helices 3 (TM3) and 6 (TM6) (2). These helical movements in turn alter the conformation of the intracellular loops of the receptor to promote activation of associated heterotrimeric G proteins.
Mutagenesis studies (3–5) demonstrated that the third intracellular loop (i3) mediates a large part of the coupling between receptor and G protein. i3 loops expressed as minigenes have also been shown to directly compete with α1B-adrenergic receptors for Gq binding (6). Okamoto and colleagues (7) localized a G protein activator region in the C-terminal end of the third cytoplasmic loop of the human β2-adrenergic receptor. They showed that a soluble peptide corresponding to this region (R259-K273) activates Gs protein under cell-free conditions. Moreover, related peptides found in wasp venom, such as mastoparan, stimulate GDP–GTP exchange from purified G proteins (8). These amphiphilic cationic peptides act in the absence of receptors to directly stimulate Gi and Go and compete with intact receptor for the G protein α subunit (9). However, there are currently no effective strategies to directly study the mechanism of receptor–G protein coupling in a controlled fashion under in vivo conditions.
Here, we present an approach to study receptor-mediated G protein activation by using palmitoylated peptides as receptor-modulating agents based on the i3 loops of the protease-activated receptors (PAR), PAR1 and PAR2, and the melanocortin-4 receptor. This paper describes receptor-dependent cellular activation of phospholipase C-β (PLC-β), calcium-mobilizing pathways, and adenylate cyclase by tethered GPCR intracellular loop peptides for several distinct receptor structures. The peptide sequences display selectivity in their activation/antagonism of specific GPCR functions and can be used as in vivo tools to activate or block receptor signaling in intact cells or tissues.
Materials and Methods
Materials.
The agonist peptides SFLLRN (PAR1), SLIGKV (PAR2-specific), AYPGKF and GYPGKF (PAR4-specific) were synthesized as carboxyl amides and were purified by RP-HPLC to >95% purity. Human thrombin (3,000 NIH units/mg) was purchased from Hematologic Technologies (Essex Junction, VT). The BMS-200661 compound (10), trans-cinnamoyl-F(f)-F(Gn)L-R-Orn(propionyl)-NH2, was synthesized by Star Biochemicals (Torrance, CA).
Synthesis and Preparation of Palmitoylated Peptides.
Palmitoylated peptides were synthesized by standard Fmoc solid-phase synthetic methods with C-terminal amides. Palmitic acid was dissolved in 50% N-methyl pyrolidone/50% methylene chloride and coupled overnight to the deprotected N-terminal amine of the peptide. After cleavage from the resin, palmitoylated peptides were purified to >95% purity by C18 or C4 RP-HPLC. In some cases, fluorescein (Fluor) was conjugated to the i3 peptides by incubating equimolar concentrations of peptide and Fluor-5-EX-succinimidyl ester (Molecular Probes) for 2 h at 25°C in dimethylformamide/5% triethylamine. The conjugated peptide products were purified from reactants by using RP-HPLC. The composition of the conjugated peptides was confirmed by mass spectrometry.
Results and Discussion
Membrane-Tethered i3 Peptides Activate G Protein Signaling Pathways in Platelets.
Our strategy was to create i3 loop peptides (Fig. 1A) with N-terminal hydrophobic transmembrane residues that would partition the peptides into and across the lipid bilayer of whole cells. The hydrophobic residues would also serve to anchor the peptide in the lipid bilayer and increase the effective molarity for potential targets such as the receptor–G protein interface. If properly bound, the exogenous i3 peptide would then disrupt receptor–G protein interactions and cause inhibition of signaling. Initially, we synthesized an i3 peptide, designated P1-i3–40, containing the adjacent transmembrane α-helical amino acids from the TM5 of PAR1. As a primary screen for biological activity, we tested the ability of P1-i3–40 to inhibit platelet activation by monitoring intracellular Ca2+. Quite unexpectedly, instead of inhibiting platelet activation, the P1-i3–40 peptide caused a rapid intracellular Ca2+ transient (Ca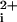 ) that mimics the Ca
) that mimics the Ca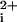 response generated by thrombin (Fig. 1B). The Ca
response generated by thrombin (Fig. 1B). The Ca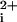 transient has no measurable lag phase (<5 s), and the maximum Ca
transient has no measurable lag phase (<5 s), and the maximum Ca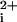 is saturable.
is saturable.
Figure 1.

Membrane-tethered PAR1 i3 loop peptides activate regulated Ca2+ signaling and aggregation in platelets. (A) The topological arrangement of the membrane-spanning segments (TM1–7), extracellular loops (e1-e4), and intracellular loops (i1-i4) of PAR1 is based on the x-ray structure of rhodopsin (1) and is illustrated on the left. Thrombin cleaves the extracellular domain (e1) at the R41—S42 bond creating a new N terminus, S42FLLRN, which functions as a tethered PAR1 agonist. The composition of the peptides used in this study is shown on the right, and their corresponding effects on platelet Ca2+ are shown immediately below. (B) Platelets from healthy volunteer donors were isolated by gel filtration chromatography, and Ca2+ measurements were performed as described (11). Intracellular Ca2+ concentration was monitored as the ratio of fluorescence excitation intensity at 340/380 nm. (C) PAR1 i3 loop peptides cause full platelet aggregation. Individual aggregation traces of platelets stimulated with 10 μM of indicated peptides or 200 μM palmitic acid are shown. Platelet aggregation was monitored as percent of light transmittance of stirred platelets at 37°C, as described (19).
A series of progressively truncated versions of P1-i3–40 were then made to determine whether the N-terminal hydrophobic region was required for stimulation of Ca2+. The P1-i3–19 peptide, which completely lacks hydrophobic N-terminal residues, causes no stimulation of Ca2+ fluxes (Fig. 1B). In contrast, the P1-i3–33 peptide has similar potency to the P1-i3–40 peptide, demonstrating that 14 hydrophobic amino acid residues confer full in vivo activity to the i3 intracellular loop. Studies with short membrane-translocating sequences have shown that 11 to 12 hydrophobic amino acid residues are sufficient to transfer proteins up to 120 kDa into intact cells (12) and tissues of mice (13). The P1-i3–26 peptide has only seven N-terminal hydrophobic residues and would be expected to partition to only the outside leaflet of the lipid bilayer. Indeed, P1-i3–26 gives no Ca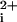 response (Fig. 1B). Hence, membrane tethering alone is not sufficient for activity—the hydrophobic tether must be of sufficient length or hydrophobicity to transfer its cargo across the membrane.
response (Fig. 1B). Hence, membrane tethering alone is not sufficient for activity—the hydrophobic tether must be of sufficient length or hydrophobicity to transfer its cargo across the membrane.
We then replaced the N-terminal hydrophobic residues from the TM5 helix with a palmitate lipid (C16H31O) to drastically reduce the size of the i3 peptides. Indeed, lipidated peptides have been used as cell-penetrating antiprotease alkylating agents (14) and as αIIbβ3-based activators of platelets (15). As shown in Fig. 1B, the palmitoylated i3 loop peptide, P1pal-19, causes a rapid Ca2+ transient that is identical in profile to that caused by the extracellular PAR1 ligand, SFLLRN. In addition, P1pal-19 fully activates platelet aggregation (Fig. 1C) with an EC50 of 8 ± 3 μM. Palmitic acid (≤200 μM) by itself has no effect on platelet aggregation or Ca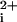 (Fig. 1C and data not shown). Next, we wanted to determine which regions of the PAR1 i3 loop were necessary for activity. Deletion of the N-terminal six residues of P1pal-19 created P1pal-13, which retained nearly full activity for both platelet Ca2+ fluxes and aggregation (Fig. 1 B and C). Further N-terminal deletion to create P1pal-7 resulted in neither Ca
(Fig. 1C and data not shown). Next, we wanted to determine which regions of the PAR1 i3 loop were necessary for activity. Deletion of the N-terminal six residues of P1pal-19 created P1pal-13, which retained nearly full activity for both platelet Ca2+ fluxes and aggregation (Fig. 1 B and C). Further N-terminal deletion to create P1pal-7 resulted in neither Ca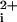 nor stimulation of platelet aggregation. The corresponding N-terminal fragment, P1pal-12, also lacked agonist activity (Fig. 1 B and C).
nor stimulation of platelet aggregation. The corresponding N-terminal fragment, P1pal-12, also lacked agonist activity (Fig. 1 B and C).
Membrane-Tethered i3 Peptides Require the Presence of Receptor for G Protein Signaling.
A major question to be addressed was whether the PAR1 i3 loop peptides required the presence of the PAR1 receptor to activate signaling or were directly stimulating G proteins in the absence of receptor. To address this question, we tested Rat1 fibroblasts expressing PAR1 versus untransfected Rat1 fibroblasts. PAR1 couples to both Gq and Gi(βγ) to stimulate PLC-β. Quite strikingly, P1pal-19 and P1pal-13 stimulated PLC-β-dependent inositol triphosphate (InsP) production only in the presence of PAR1 with EC50 values of 180 ± 20 nM (Fig. 2A) and 700 ± 50 nM (Fig. 2B), respectively, and with similar efficacies as the natural agonist thrombin. The activation curves are biphasic with a steep activating phase followed by a steep inhibitory phase. The two smaller i3 loop fragments, P1pal-12 and P1pal-7, did not stimulate PLC-β (Fig. 2B). Significantly, neither P1pal-19 nor P1pal-13 stimulates InsP (11%) in the absence of the PAR1 receptor in Rat1 (Figs. 2A and 5A) or COS7 cells (Fig. 5 A and B). These results demonstrate that activation of G protein signaling by the membrane-tethered peptides requires the presence of receptor.
Figure 2.

Membrane-tethered PAR1 i3 loop peptides require the presence of their cognate receptor to activate signaling. (A and B) PAR1-Rat1 cells (24) were challenged with 1 nM to 50–100 μM i3 peptide. PLC-β activity was determined by measuring total [3H]-InsP formation (17). PLC-β activity was converted to percent of the full response relative to 0.1 nM thrombin (100%) and plotted as a function of peptide concentration by using a two-site equation that fits the biphasic activation and inhibition profile. The full PAR1 thrombin responses for individual experiments were 12.4-fold for P1pal-19 and P1pal-19/untransfected Rat1 cells, 18-fold for P1pal-19Q (Pal-RCLSSSAVANQSQQSQALF), 12.4-fold for P1pal-19E (Pal-RCESSSAEANRSKKERELF), 7.6-fold for P1pal-13, and 9.4-fold for P1pal-12 and P1pal-7.
Figure 5.

Receptor selectivity profiles of cell-penetrating PAR1 i3 loop agonists. COS7 cells were transiently transfected with the human receptors PAR1, PAR2, PAR4, CCKA, CCKB, SubP, or rat SSTR2. Transfected cells were challenged with 0.1–10 μM P1pal-19 (A) or P1pal-13 (B), and the highest stimulation of the individual receptors is shown as a black column. The extracellular agonists used to define maximum stimulation for each receptor (open column) were 10 nM thrombin for PAR1, 100 μM SLIGKV for PAR2, 100 nM thrombin for PAR4, 300 nM CCK-8 for CCKA and CCKB, 1 μM AGCKNFFWKTFTSC for SSTR2, and 1.5 μM RPKPQQFFGLM for SubP.
Previous studies using soluble i3 loop peptides with cell-free systems have shown that positively charged residues in the C-terminal region of i3 loop peptides (BBXB motif) are essential for the activation of G proteins (7). We tested whether these exogenous membrane-tethered i3 loop peptides would mimic the i3 loop of the intact receptor and therefore require this BBXB motif to activate G proteins. Surprisingly, mutation of the positively charged residues in the C terminus of the lipidated i3 loop peptide (P1pal-19Q) gave only a 2-fold loss in efficacy of InsP production in PAR1-Rat1 cells (Fig. 2A) or in platelet aggregation (Fig. 1C). In contrast, mutation of the conserved more hydrophobic residues in the P1pal-19E peptide results in ≥90% loss of agonist activity (Figs. 1C and 2A). Thus, these membrane-tethered peptides are not simply acting as positively charged amphipathic helixes, like mastaparan, to activate G protein signaling in a receptor-independent manner.
Another important issue to resolve was whether these membrane-tethered peptide agonists nonspecifically block a downstream component in the Ca2+ flux pathway. This was readily addressed by determining whether P1pal-19 blocks the Ca2+ flux evoked by another GPCR. For instance, human platelets contain PAR1 and PAR4, both of which couple to Gq to turn on PLC-β and evoke IP3-dependent Ca2+ fluxes. If Plpal-19 nonspecifically targets a downstream component, then it should also block the PAR4 response. As shown in Fig. 3A, P1pal-19 does not block the PAR4 response to GYPGKF and hence does not target downstream signaling components.
Figure 3.

Lipidated PAR1 i3 loop peptides are cell-penetrating and do not activate receptor at the extracellular ligand-binding site. Human platelet Ca2+ responses were determined as in Fig. 1B. (A) Palmitoylated i3 loop peptides do not inhibit Ca2+ flux pathways downstream of PAR4. Platelets were stimulated with either 3 μM SFLLRN or 2 μM P1pal-19 alone, followed by 2 mM GYPGKF. (B) Palmitoylated i3 loop peptides penetrate intact cells. Flow cytometry was conducted on platelets or Rat1 fibroblasts stably transfected with PAR1 (24) that were treated with Fluor-labeled peptides, Fluor-Pal-i3 (Fluor-P1pal-19), or Fluor-i3 (Fluor-P1-i3–19), as indicated. Cells were incubated with 10 μM Fluor-Pal-i3 or Fluor-i3 for 2 min in PBS/0.1% FCS and then treated with 2 units of pronase for 15 min at 37°C and washed before flow cytometry. (C) Platelets were stimulated with 3 μM SFLLRN or 3 μM P1pal-19 alone. In the right trace, platelets were pretreated with the anti-PAR1 small molecule, 1 μM BMS-200661, then sequentially challenged with 3 μM SFLLRN and 3 μM P1pal-19.
Lipidated PAR1 i3 Loop Peptides Are Cell-Penetrating and Do Not Activate the Receptor at the Extracellular Ligand Site.
To directly determine whether palmitoylation conferred cell-penetrating abilities, P1-i3–19 and P1pal-19 were tagged with Fluor and incubated with intact platelets or Rat1 fibroblasts. The cells were then treated with pronase to completely digest extracellularly bound peptides and analyzed by flow cytometry. As shown in Fig. 3B, both platelets and fibroblasts retained 5-fold higher fluorescence when incubated with Fluor-Pal-i3, as compared with the nonpalmitoylated Fluor-i3. These results indicate that palmitoylation confers cell-penetrating ability as has been seen with other lipidated peptides (14, 15).
Next, we wanted to exclude the possibility that these membrane-tethered i3 loop peptides were acting as receptor agonists by binding to the extracellular ligand-binding site. We took advantage of the anti-PAR1 small molecule, BMS-200661, which inhibits SFLLRN in a competitive manner by binding at the extracellular ligand binding site (ref. 10; S. Seeley, L.C., J. Baleja, and A.K., unpublished work). As shown in Fig. 3C, BMS-200661 completely inhibits SFLLRN activity with no effect on activation of platelet Ca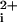 fluxes by the P1pal-19 agonist. These data rule out the possibility that the P1pal-19 peptide acts as a receptor agonist by binding to the extracellular ligand binding site. Together, these results indicate that the lipidated i3 loop peptides are localizing to the intracellular membrane surface to exert their effects on PAR1.
fluxes by the P1pal-19 agonist. These data rule out the possibility that the P1pal-19 peptide acts as a receptor agonist by binding to the extracellular ligand binding site. Together, these results indicate that the lipidated i3 loop peptides are localizing to the intracellular membrane surface to exert their effects on PAR1.
Cell-Penetrating i3 Loop Peptides Do Not Correct a Signaling Defect in a PAR1 i3 Mutant Receptor.
As we have shown in Fig. 2, these cell-penetrating i3 loop peptides require the presence of receptor for G proteins to be activated. There are two simple models of activation of the receptor: (i) the exogenous membrane-tethered i3 loop replaces its cognate i3 loop in the intact receptor to directly activate G protein, or (ii) the exogenous i3 loop peptide binds the receptor at an allosteric site to indirectly activate G protein. To distinguish between direct versus indirect activation of the G protein by the lipidated i3 loop peptides, a point mutation was introduced at position S309 located in the C terminus of the i3 loop/N terminus of TM6 of intact PAR1. This perimembranous region has been shown to be important for the fidelity of G protein coupling for many receptors (3–5) and comes into direct contact with the critical DRY residues of TM3 (1). A S309P mutant was constructed and transiently expressed in COS7 cells to the same level as wild-type PAR1 (11). The S309P mutant is deficient in thrombin- and SFLLRN-dependent stimulation of InsP with 17- and 28-fold loss of potency, and 1.6- and 3.3-fold loss of efficacy, respectively (Fig. 4 A and B). Interestingly, P1pal-19 also stimulates the S309P mutant with parallel losses in potency (13-fold) and efficacy (4.3-fold) relative to its effects on wild-type PAR1 (Fig. 4C). Because P1pal-19 did not correct the signaling defect of the S309P mutation, this indicates that the crucial C-terminal portion of the i3 region in the intact receptor exerts dominant effects in coupling to G protein over that of the exogenous peptide. Therefore, the exogenous membrane-tethered i3 loop peptide must indirectly activate the receptor by an allosteric mechanism.
Figure 4.

P1pal-19 does not activate C-tail-deleted PAR1 but activates a PAR1 i3-mutant. COS7 cells were transiently transfected with wild-type (WT), S309P, or Δ377 PAR1 receptors. Cells were challenged with (A) thrombin, (B) SFLLRN, or (C) P1pal-19, and PLC-β activity determined by measuring total [3H]-inositol phosphate formation relative to 100% stimulation (9.6-fold) of WT PAR1 with 0.1 nM thrombin.
Cell-Penetrating i3 Loop Peptides Require the Cytoplasmic Tail of the Receptor for Activity.
To define the region(s) of the receptor that might directly contact the exogenous i3-peptide, we deleted the entire C-terminal i4 domain of PAR1 (Δ377). The x-ray structure of rhodopsin (1) indicates that the i3 loop may contact the N-terminal region of α-helix 8 and residues to the C-terminal side of the Cys-palmitate moieties within the i4 C-terminal domain. As shown in Fig. 4C, the P1pal-19 peptide gives effectively no stimulation of PLC-β in the presence of the Δ377 mutant. In contrast, the Δ377 PAR1 mutant can still be stimulated by the extracellular agonists thrombin and SFLLRN, although efficacy is reduced by 2- to 3-fold, and potency is reduced by 22- to 30-fold (Fig. 4 A and B). Therefore, these data demonstrate that the C-tail of PAR1 is required for P1pal-19 to activate G protein and that the C-tail may provide a binding surface for these cell-penetrating agonists.
Receptor Selectivity.
Having established that the PAR1-derived i3 peptides require PAR1 for activation of G protein signaling, we then asked whether homologous receptors such as PAR2 or nonhomologous receptors such as cholecystokinin A (CCKA) can also be activated by PAR1-derived i3 peptides. We tested P1pal-19 for agonist activity against PAR2, PAR4, CCKA, cholecystokinin B (CCKB), somatostatin (SSTR2), and substance P (SubP). Of these, PAR2 (16) is a trypsin/tryptase-activated receptor that is important in inflammation, pain, and cancer, and PAR4 (17, 18) is a second thrombin receptor that controls irreversible platelet aggregation (19). COS7 cells were transiently transfected with each receptor and InsP production measured. P1pal-19 can fully activate the highly homologous PAR2 receptor and stimulates CCKB to about 30% of its maximal activity but does not activate the less homologous PAR4, or the nonhomologous CCKA, SSTR2, and SubP receptors (Fig. 5A). We then tested the shorter P1pal-13 peptide, which lacks the N-terminal 6-aa residues of P1pal-19, for its selectivity against the panel of seven GPCRs. As shown in Fig. 5B, P1pal-13 was completely selective for PAR1. Together, these data indicate that both P1pal-19 and P1pal-13 exhibit complementarity of binding to PAR1. Therefore, the C-terminal 13 amino acids of P1pal-19 are sufficient to activate PAR1. It is possible that the N-terminal six residues of P1pal-19 may bind to receptor at an additional site that is more generally shared by other receptors such as PAR1/PAR2/CCKB.
Extension of Approach to Other Receptor–G Protein Systems.
Next, we wanted to determine whether we could construct intracellular agonists for GPCRs other than PAR1. Because PAR2 was also activated by the P1pal-19 i3 loop, we synthesized lipidated i3 peptides based on PAR2. We found that the full-length wild-type PAR2 i3 peptide, P2pal-21 was a partial agonist with an efficacy of 18% for PAR2 (Fig. 6A). We hypothesized that the reduced efficacy of P2pal-21 for PAR2 as compared with P1pal-19 was due to a single positively charged residue located at the extreme C terminus of the PAR2 i3 loop in an otherwise hydrophobic region. We mutated the C-terminal Lys to Phe, which is the homologous residue in the PAR1 sequence. Quite strikingly, mutation of the C-terminal Lys to Phe converts the PAR2 peptide, the P2pal-21F, into a potent (EC50 = 25 nM) full agonist of PAR2 with biphasic properties (Fig. 6A). P2pal-21F is the most potent agonist tested to date for PAR2, which indicates the potential importance of this C-terminal hydrophobic region in activation of GPCRs by this class of agonists. Not surprisingly, P2pal-21F also activated the closely homologous PAR1 but not PAR4, CCKA, CCKB, or SSTR2 (Fig. 6B). These data suggest that the i3 loop peptides must be tethered or embedded in a lipophilic environment at both termini to be efficacious as agonists for PAR1 and PAR2.
Figure 6.

Cell-penetrating PAR2 and MC4 i3 loop peptides activate their cognate receptors. (A) A single point mutation of the C-terminal residue of the PAR2 i3 loop peptide results in full activation of PAR2. COS7 cells were transiently transfected with PAR2 and challenged with 0.1–10 μM P2pal-21 (Pal-MLRSSAMDENSEKKRKRAIK) or P2pal-21F (Pal-MLRSSAMDENSEKKRKRAIF). PLC-β activity was converted to percent of the full response relative to 100 μM SLIGKV (100%) and plotted as in Fig. 2. In these experiments, 100 μM SLIGKV gave 3.9- and 3.1-fold maximal stimulation of PAR2 for the P2pal-21 and P2pal-21F, respectively. (B) Receptor selectivity profile of P2pal-21F. COS7 cells were transiently transfected with the indicated receptors and challenged with 0.1–10 μM of P2pal-21F; the highest stimulation of the individual receptors is shown as a black column. The extracellular agonists used to define maximum stimulation for each receptor (open column) are the same as described in Fig. 5. (C) The lipidated i3 loop peptide of the MC4 obesity receptor stimulates Gs/adenylate cyclase in COS7 cells transiently transfected with human MC4 receptors. Transfected cells were challenged with 3 nM–10 μM MC4pal-14 (Pal-TGAIRQGANMKGAI), and adenylate cyclase activity was converted to percent of the full response relative to 30 nM α-melanocyte-stimulating hormone. Adenylate cyclase activity was measured by using the RIA kit for cAMP (NEN, Lifescience Products).
To test the generality of this approach with non-Gi/Gq-coupled GPCRs, we synthesized lipidated peptides based on the wild-type i3 sequence of the melancortin-4 (MC4) obesity receptor. Because the MC4 receptor couples solely to Gs to stimulate adenylate cyclase (20), we measured cAMP production in COS7 fibroblasts expressing the human MC4 receptor. We found that the MC4pal-14 peptide is a potent (EC50 = 100 nM) agonist with biphasic properties (Fig. 6C). Its efficacy of activation is 40% compared with its native agonist α-melanocyte-stimulating hormone. To reflect their properties as GPCR-specific regulators of signal transduction, we have termed these cell-penetrating peptides “pepducins.” To our knowledge, this is the first report of intracellular reagents that exhibit receptor-specific and receptor-dependent effects on G protein signaling.
Pepducin Antagonists Block Receptor–G Protein Signaling.
We also tested whether lipidated i3 peptides that lacked agonist activity could block receptor–G protein signaling. We hypothesized that pepducins might bind at a second lower-affinity inhibitory site, perhaps situated at the receptor–G protein interface, to block signal transference from receptor to G protein. Human platelets were useful to test the efficacy and selectivity of the anti-PAR1 pepducins, because they possess PAR1 and PAR4 thrombin receptors, both of which couple to Gq-PLC-β to activate Ca2+ fluxes. As shown in Fig. 7A, stimulation of PAR1 and PAR4 with their respective ligands gives rise to easily distinguishable (19) Ca2+ transients. We found that the PAR1 i3 peptide, P1pal-12, can block the PAR1 Ca2+ transient without significantly affecting the PAR4 Ca2+ transient (Fig. 7A). As shown in Fig. 7B, 3 μM P1pal-12 effectively inhibits PAR1-dependent platelet aggregation by SFLLRN with IC50 of 1 μM (Fig. 7C) but does not block PAR4 aggregation by AYPGKF (Fig. 7B). This clearly demonstrates that the P1pal-12 pepducin can selectively block PAR1 without affecting PAR4 or its immediate downstream signaling partners that include Gq and PLC-β. The P1pal-12 anti-PAR1 pepducin was also able to completely block PAR1 signaling to PLC-β in fibroblasts (Fig. 7D).
Figure 7.

Inhibitory pepducins derived from the i3 loop sequence of PAR1 and PAR2 are antagonists of their cognate receptors. (A) The cell-penetrating PAR1 i3 loop peptide, P1pal-12, selectively inhibits the Ca2+ signal from PAR1. Platelet Ca2+ measurements were performed as in Fig. 1. Platelets were pretreated with 3 μM P1pal-12 (open arrowhead) and then stimulated with 3 μM SFLLRN or 200 μM AYPGKF, as indicated. (B) Platelets were preincubated with either vehicle (−) or 3 μM P1pal-12 for 1 min and then sequentially challenged with 3 μM SFLLRN and 200 μM AYPGKF and aggregation monitored. (C) Platelets were pretreated for 1 min with 0.01–5 μM P1pal-12 and challenged with 3 μM SFLLRN. (D) PAR1 and PAR2-expressing COS7 fibroblasts were pretreated with 0.03–100 μM P1pal-12 or P2pal-21 for 5 min, and then challenged with extracellular agonists 0.1 nM thrombin or 100 μM SLIGKV, respectively. Percent InsP inhibition was calculated relative to the full extracellular agonist-stimulated response: 5.2-fold for P1pal-12 and 3.1-fold for P2pal-21. (E) Proposed mechanism of activation and inhibition of receptor–G protein complexes by pepducins. The receptor is shown in its off-state (R) and on-state (R*) bound to G protein (G). Pepducin agonists are shown as ellipses, pepducin antagonists as rectangles, and the extracellular ligand as a triangle.
Next, we wanted to determine whether inhibitory pepducins might be used against other receptors besides PAR1 to inhibit activation by their naturally occurring extracellular agonists. We had already shown (Fig. 6A) that concentrations of P2pal-21 above 100 nM exhibited the typical inhibition of pepducins against their own agonist activity. Thus, higher concentrations of P2pal-21 lose agonist activity on occupancy of the inhibitory site and should also be able to block PAR2 signaling evoked by the extracellular agonist, SLIGKV. Indeed, as shown in Fig. 7D, P2pal-21 inhibits stimulation of PLC-β by the extracellular PAR2 ligand, SLIGKV, with an IC50 of 1 μM. These data are again consistent with a two-site model, that at higher concentrations P2pal-21 binds to PAR2 at a second lower-affinity site and blocks signaling from the SLIGKV-liganded receptor.
Mechanism of Pepducin Activation and Inhibition.
To explain the ability of the pepducins to both activate and inhibit receptor–G protein signaling, we propose the two-site mechanism shown in Fig. 7E. First, the pepducin agonist occupies a high-affinity site at the intracellular surface of the GPCR. The bound pepducin agonist either stabilizes or induces the activated state of the receptor to turn on the associated G protein(s). After this first site becomes saturated, higher concentrations of pepducin begin to occupy a second lower-affinity inhibitory site that blocks signal transference to G protein, perhaps by mimicking or stabilizing receptor i3 loop ground-state interactions with the G protein. The inhibition by the pepducin antagonists is coincident with the inhibitory phase of the agonists, thus the antagonists may also bind at this lower-affinity site. Exogenous activation or inhibition of receptors by pepducins could reflect a potential dimerization mode whereby one receptor donates its intracellular loops to an adjacent receptor. We have previously shown that PAR1 forms homodimers (21); however, the functional significance of these complexes is currently under investigation. Moreover, there are now many examples of receptor homo- and heterodimers that display distinct signaling properties including allosteric activation (22) and inhibition (23).
In conclusion, we have shown that pepducins rapidly transduce the plasma membrane and achieve high effective molarity at the perimembranous target interface. Having surmounted the membrane obstacle, the rich diversity of intracellular receptor structures may be exploited for both generation of new therapeutic agents and delineation of the mechanism of receptor–G protein coupling under in vivo conditions. Thus, in this postgenomic era, the pepducin approach may be widely applicable to the targeting of diverse membrane proteins and may open up new experimental avenues in systems previously not amenable to traditional molecular techniques.
Acknowledgments
We thank Martin Beinborn, Dan Cox, Matt Waldor, and Maria Diverse for critically reviewing the manuscript and for many thoughtful discussions. We especially thank Ci Chen for her generous help in setting up the phospholipase C assays. This work was supported by National Institutes of Health Grants R01HL64701, R01HL57905, and DK39428, and by a Scholar Award from the Pew Charitable Trusts (to A.K.).
Abbreviations
- GPCR
G protein-coupled receptor
- TM
transmembrane helix
- i3
third intracellular loop
- PAR
protease-activated receptor
- PLC-β
phospholipase C-β
- InsP
inositol triphosphate
- Fluor
fluorescein
- CCKA
cholecystokinin A
- SSTR2
somatostatin
- SubP
substance P
- MC4
melancortin-4
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Palczewski K, Kumasaka T, Hori T, Behnke C A, Motoshima H, Fox B A, Le Trong I, Teller D C, Okada T, Stenkamp R E, et al. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 2.Gether U, Kolbilka B K. J Biol Chem. 1998;273:17979–17982. doi: 10.1074/jbc.273.29.17979. [DOI] [PubMed] [Google Scholar]
- 3.Cotecchia S, Ostrowski J, Kjelsberg M A, Caron M G, Lefkowitz R J. J Biol Chem. 1992;267:1633–1639. [PubMed] [Google Scholar]
- 4.Kostenis E, Conklin B R, Wess J. Biochemistry. 1997;36:1487–1495. doi: 10.1021/bi962554d. [DOI] [PubMed] [Google Scholar]
- 5.Kjelsberg M A, Cotecchia S, Ostrowski J, Caron M G, Lefkowitz R J. J Biol Chem. 1992;267:1430–1433. [PubMed] [Google Scholar]
- 6.Luttrell L M, Ostrowski J, Cotecchia S, Kendal H, Lefkowitz R J. Science. 1993;259:1453–1457. doi: 10.1126/science.8383880. [DOI] [PubMed] [Google Scholar]
- 7.Okamoto T, Murayama Y, Hayashi Y, Inagaki M, Ogata E, Nishimoto I. Cell. 1991;67:723–730. doi: 10.1016/0092-8674(91)90067-9. [DOI] [PubMed] [Google Scholar]
- 8.Gilman A G. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 9.Higashijima T, Uzu S, Nakajima T, Ross E M. J Biol Chem. 1988;263:6491–6494. [PubMed] [Google Scholar]
- 10.Bernatowicz M S, Klimas C E, Hartl K S, Peluso M, Allegretto N J, Seiler S M. J Med Chem. 1996;39:4879–4887. doi: 10.1021/jm960455s. [DOI] [PubMed] [Google Scholar]
- 11.Kuliopulos A, Covic L, Seeley S K, Sheridan P J, Helin J, Costello C E. Biochemistry. 1999;38:4572–4585. doi: 10.1021/bi9824792. [DOI] [PubMed] [Google Scholar]
- 12.Rojas M, Donahue J P, Tan Z, Lin Y-Z. Nat Biotechnol. 1998;16:370–375. doi: 10.1038/nbt0498-370. [DOI] [PubMed] [Google Scholar]
- 13.Schwarze S R, Ho A, Vocero-Akbani A, Dowdy S F. Science. 1999;285:156–159. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 14.Wikstrom P, Kirschke H, Stone S, Shaw E. Arch Biochem Biophys. 1989;270:286–293. doi: 10.1016/0003-9861(89)90030-1. [DOI] [PubMed] [Google Scholar]
- 15.Stephans G, O'Luanaigh M, Reilly D, Harriott P, Walker B, Fitzgerald D, Moran N. J Biol Chem. 1998;273:20317–20322. doi: 10.1074/jbc.273.32.20317. [DOI] [PubMed] [Google Scholar]
- 16.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Proc Natl Acad Sci USA. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu W-F, Andersen H, Whitmore T E, Presnell S R, Yee D P, Ching A, Gilbert T, Davie E W, Foster D C. Proc Natl Acad Sci USA. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahn M L, Zheng Y-W, Huang W, Bigornia V, Zheng D, Moff S, Farese R V, Tam C, Coughlin S R. Nature (London) 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 19.Covic L, Gresser A L, Kuliopulos A. Biochemistry. 2000;39:5458–5467. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- 20.Oosterom J, Garner K M, Dekker W K d, Nijenhuis W A, Gispen W H, Burbach J P, Barsh G S, Adan R A. J Biol Chem. 2001;276:931–936. doi: 10.1074/jbc.M007261200. [DOI] [PubMed] [Google Scholar]
- 21.Swift S, Sheridan P J, Covic L, Kuliopulos A. J Biol Chem. 2000;275:2627–2635. doi: 10.1074/jbc.275.4.2627. [DOI] [PubMed] [Google Scholar]
- 22.Milligan G. Science. 2000;288:65–67. doi: 10.1126/science.288.5463.65. [DOI] [PubMed] [Google Scholar]
- 23.Pfeiffer M, Koch T, Schroder H, Klutzny M, Kirscht S, Kreienkamp H J, Hollt V, Schulz S. J Biol Chem. 2001;276:14027–14036. doi: 10.1074/jbc.M006084200. [DOI] [PubMed] [Google Scholar]
- 24.Ishii K, Chen J, Ishii M, Koch W J, Freedman N J, Lefkowitz R J, Coughlin S R. J Biol Chem. 1994;269:1125–1130. [PubMed] [Google Scholar]