

Abstract
RING domains act in a variety of essential cellular processes but have no general function ascribed to them. Here, we observe that purified arenaviral protein Z, constituted almost entirely by its RING domain, self-assembles in vitro into spherical structures that resemble functional bodies formed by Z in infected cells. By using a variety of biophysical methods we provide a thermodynamic and kinetic framework for the RING-dependent self-assembly of Z. Assembly appears coupled to substantial conformational reorganization and changes in zinc coordination of site II of the RING. Thus, the rate-limiting nature of conformational reorganization observed in the folding of monomeric proteins can also apply to the assembly of macromolecular scaffolds. These studies describe a unique mechanism of nonfibrillar homogeneous self-assembly and suggest a general function of RINGs in the formation of macromolecular scaffolds that are positioned to integrate biochemical processes in cells.
Keywords: protein association|oligomerization|macromolecular scaffold| arenavirus|zinc
Physiological processes are comprised of a network of discrete biochemical activities such as acetylation in chromatin remodeling and thioester transfer in ubiquitin-mediated protein degradation. Their biological specificity and functional plasticity depend on their spatial and temporal coupling in cells. The framework for this coupling often is provided by macromolecular assemblages, which are typically observed by confocal microscopy as punctate structures, variably termed dots, speckles, and bodies. They are diverse in function, composition, and subcellular location. Although much is known about the biochemical activities that characterize biological function, the physical basis of their macromolecular organization remains poorly understood.
Intriguingly, a number of these macromolecular assemblages contain proteins with RING domains (Fig. 1a). RING domains are of great interest because of their widespread occurrence and functional diversity as well as their involvement in human disease (1). Establishing a general function for RINGs has been problematic, because their functions extend to a wide range of biological processes including regulation of cell growth, apoptosis, antiviral response, and organelle biogenesis (1). Only a few discrete biochemical actions have been ascribed to RINGs, namely transcriptional repression by KAP-1/TIF1β (2), translational repression by promyelocytic leukemia protein (PML) and Z (3), and ubiquitin conjugating (E3) activity by RINGs such as Cbl (4) and BRCA1 (5).
Figure 1.

Purified LCMV and LFV Z self-assemble. (a) Alignment of lymphocytic choriomeningitis virus (LCMV) and Lassa fever virus (LFV) Z amino acid sequences and schematic of zinc binding by their RING domains using the cross-brace topology (6). Site I is in blue and site II is in red. Arrows delineate N- and C-terminal regions deleted in the RING construct. (b) Z is present in punctate nuclear and cytoplasmic bodies (indicated by arrow heads) and in diffuse pattern throughout the cytoplasm of NIH 3T3 cells 90 h postinfection with LCMV, stained with an antibody for Z, and visualized by using confocal laser scanning microscopy. Micrograph represents a single confocal section using an ×100 objective lens. (c) Electron micrograph of purified and negatively stained Z showing a morphologically homogeneous population of 300–500-Å spherical particles. Nominal magnification is ×100,000.
However, over 50 of the 200-member family of RING proteins are found in discrete subcellular structures that are visible by confocal microscopy in vivo and/or isolated from cells as high molecular weight assemblies (1, 6). For instance, the RING domain of the PML is required for the formation of discrete spherical multiprotein structures, 0.1–1 μm in diameter, referred to as PML nuclear bodies (7).
To investigate properties specific to RING domains that may underlie the macromolecular organization of the multidomain proteins that contain them, we focused our attention on the smallest known RING protein, the arenaviral protein Z, in which 60 of its 90 residues comprise the RING (8). Arenaviruses include LCMV and LFV and carry a substantial health toll (9). During infection, Z is required for viral replication, and its RING domain is thought to repress translation of host cell mRNAs (3, 10, 11). Z and PML form spherical cytoplasmic and nuclear structures that are similar in size and morphology to those formed by PML in uninfected cells (12). The structural integrity of these RING domains is causally linked to the integrity and function of PML bodies in growth control (7) and of Z bodies in viral replication (3, 10, 11, 13). The integrity of many RING-containing assemblages requires an intact RING (6), suggesting that this domain may act in the assembly of these structures in cells.
Here we demonstrate that purified RING protein Z forms spherical structures in vitro that are similar in dimensions and morphology to structures formed by Z in infected cells. By using electron microscopy (EM), analytical ultracentrifugation, light scattering, and circular dichroism (CD) spectroscopy, we provide a thermodynamic and kinetic framework for the self-assembly properties of Z and its RING domain. In all, these studies reveal a unique mechanism of nonfibrillar self-assembly. The deduced determinants of efficient self-assembly of Z likely will apply to other RING-containing proteins that form macromolecular assemblages in cells.
We use the following strategy to examine the self-assembly of Z. First, we demonstrate that the observed self-association of Z represents ordered assembly and not aggregation. Second, analyses of both guanidine denaturation and analytical ultracentrifugation data discriminate between two possible thermodynamic models of assembly. Finally, analysis of the kinetics of assembly and disassembly identifies the principal features of the assembly pathway and the uniqueness of the nonfibrillar self-assembly of RINGs.
Methods
LCMV (Armstrong strain) and LFV Z were expressed and purified as described in ref. 3. LCMV infection and confocal immunofluorescence microscopy were performed as described in ref. 11. All reagents were of ACS grade from Sigma–Aldrich except for ultrapure guanidine hydrochloride (ICN) and Tris-carboxyethyl phosphine (Pierce). Solution conditions were 0.3 M NaCl, 10 mM phosphate, 10 μM Tris-carboxyethyl phosphine, and 1 μM Zn2+, pH 7.5, at 25°C, degassed, and under argon unless stated otherwise.
Analysis.
For detailed descriptions of data processing and analysis, please see Methods and Analysis, which is published as supporting information on the PNAS web site, www.pnas.org. All energies are referenced to 100 μM monomer unless noted otherwise.
Analytical Gel Filtration.
Measurements were done using an ÄKTAexplorer chromatograph and Superose 6 HR column (Amersham Pharmacia). Void volume was measured by using Dextran blue 2000. Samples (100 μl) were injected directly onto the column and separated at 0.2–0.5 ml/min in under an hour while recording UV absorbance. The relationship between partition coefficient and globular molecular mass was determined by linear regression of Kp as a function of log M.
Analytical Ultracentrifugation.
All measurements were performed by using a Beckman XL-I ultracentrifuge equipped with the An-60 Ti rotor, double-sector cell, 12-mm Epon centerpiece, and quartz windows. Samples were centrifuged at 3,000–50,000 rpm in continuous mode and monitored by using absorbance optics. Partial protein specific volume was calculated from those of component amino acids (14). Sedimentation coefficients were corrected to standard temperature and pressure (15).
Dynamic Light Scattering.
Measurements were performed by using a DynaPro 801-TC laser photometer. Correlation spectra were recorded at 90° using a 1 × 1-mm2 fluorescence cuvette (Hellma, Forest Hills, NY) and 20-channel correlator tuned to a 1–1,000-μs delay (Protein Solutions, Lakewood, NJ). Measured intensity autocorrelation spectra were converted to field correlation spectra and analyzed by single-value decomposition (16).
CD.
Measurements were performed by using a Jasco J-810 spectropolarimeter with either 0.1- or 1-cm quartz cuvettes (Hellma). The bandwidth was 1 nm for spectral measurements and 2–4 nm for single-wavelength measurements.
EM.
Formvar-coated nickel grids (Electron Microscopy Sciences, Fort Washington, PA) were glow-discharged, floated on sample drops, blotted, and air-dried. They were then negatively stained with 1% aqueous uranyl acetate. Grids were photographed by using a JEOL JEM 100-CX electron microscope under low-dose conditions at 80 kV.
Inductively Coupled Plasma Optical Emission Spectroscopy.
Inductively coupled plasma optical emission spectroscopy was conducted at Galbraith Laboratories (Knoxville, TN) by using a Perkin–Elmer P2000 instrument. Samples were prepared as described (17).
Results and Discussion
Z Reversibly Self-Assembles into Macromolecular Structures in Vivo and in Vitro.
In LCMV-infected cells, Z localizes to discrete structures in the nucleus and cytoplasm (12). These structures appear round and are ≈0.1–0.5 μm in diameter (Fig. 1b). Thus, when bacterially expressed LCMV Z appeared to form high molecular weight species during purification, we were interested in whether this behavior results from ordered assembly or aggregation.
Several features distinguish amorphous aggregation and structured self-assembly. Aggregation is characterized by a polydisperse collection of heterogeneously aggregated species both in terms of size and morphology, is independent of native folding, and is usually irreversible. In contrast, self-assembly is defined by dependence on native structures and presence of distinct oligomeric states and is frequently reversible. As can be seen from Fig. 1c, bacterially expressed and purified Z high molecular weight state consists of a morphologically homogeneous population of round particles with variable diameters of up to 500 Å. It is unknown whether the apparent variability in size reflects physical heterogeneity or differential grid adsorption, because hydrodynamic measurements indicate a uniform size (see below). These particles possess significant secondary structure, corresponding to mean α-helical content of >70% (Fig. 2a). Because RING domains require Zn2+ coordination for folding and stability (refs. 18 and 19; Fig. 1a), we examined the zinc dependence of assembly. The addition of a 10-fold excess of EDTA eliminates all secondary structure (Fig. 2a). Furthermore, EDTA chelation of Zn2+ eliminates the presence of bodies in EM (Fig. 2b Lower), indicating that native zinc binding and folding by the RING domain are required for assembly. In agreement with this, Z1 that contains Cys → Phe and Cys → Gly substitutions of the first two ligands in the first zinc-binding site (Fig. 1a), which is only partially structured (Fig. 2a), does not form bodies (Fig. 2b Lower), indicating that their formation depends on a stable and structured RING domain. Moreover, deletion of N- and C-terminal sequences flanking the RING domain abolishes body formation (Figs. 1a and 2b Lower), while preserving wild-type per residue secondary structure content (Fig. 2a). In contrast, Z-GST fusion forms a heterogeneous population of amorphous aggregates (Fig. 2b Lower), suggesting that the GST fusion interferes with self-assembly, possibly by sterically inhibiting the formation of intermediates on the assembly pathway (see below).
Figure 2.

Assembly behavior of Z and its mutants and their secondary structure content. (a) Far-UV CD spectra of wild-type Z (solid black), first zinc-binding site mutant (Z1, red), second zinc-binding site mutant (Z2, blue), Z deletion mutant containing only the minimal RING domain (RING, orange), Z at 40°C (green), wild-type Z in the presence of 10-fold excess of EDTA (dashed black), and Z in the presence of 4 M guanidine hydrochloride (dotted black). (b Upper) Fractional sedimentation boundary plateau values as derived from sedimentation velocity (SV) data for assembly species for wild-type Z (Z), RING, fusion of Z with glutathione S-transferase (Z-GST), Z with 10-fold excess of EDTA, Z1, and Z2. Note that destabilization of site I in the Z1 leads to precipitation of most of the protein. SV profiles of the fusion of Z with GST and Z2 were fit to two sedimenting species, with error bars representing the uncertainty in estimating the plateau concentration caused by broad boundary widths. Asterisks represent inconsistencies between calculated sedimentation and diffusion coefficients (see Methods and Analysis and Table 2). (b Lower) Single-particle electron micrographs of negatively stained preparations showing fully assembled bodies of Z and Z2, lack of assembly of the minimal RING domain, Z1, and EDTA-treated Z as well as amorphous aggregation of the fusion of Z with GST. (Bar, 0.1 μm.) (c) Gel filtration profile as a function of elution volume (Ve) for Z (solid) and Z2 (dashed) at 6°C. Z elutes with three well separated peaks corresponding to molecular masses for monomer (M), tetramer (T), and body (B), whereas the Z2 exhibits broad overlapping tetramer and body peaks, suggesting fast assembly and disassembly kinetics. Note that peak heights, normalized to total eluted absorbance, correspond to SV assembly species' fractions and are in good agreement with relative species' concentrations as measured by SV at 4°C (Figs. 3b and 4c). Elution of globular molecular mass standards are represented by solid triangles (from left to right: thyroglobulin, 667 kDa; catalase, 232 kDa; albumin, 67 kDa; RNase A, 14 kDa). (d) SV profile of Z consisting of radial scans of absorbance as a function of sedimentation time and showing three sedimentation boundaries that move outward to the edge of the cell at 7.2 cm and become broader because of diffusion in the course of the experiment. Absorbance values of the plateaus of sedimentation boundaries are related directly to the concentration of the sedimenting species. The meniscus is located at 6.2 cm.
The distribution of Z oligomeric states in solution was examined by using gel filtration and SV ultracentrifugation. Analytical gel filtration of Z reveals three species of molecular masses of 12 ± 2.4, 44 ± 5.1, and 250 ± 12 kDa (Fig. 2c). Moreover, Z exhibits three discrete sedimentation boundaries (Fig. 2d and Table 2, which is published as supporting information on the PNAS web site), with velocity sedimentation and diffusion coefficients consistent with the presence of three discrete species with molecular masses of 8.6 ± 1.9, 39 ± 4.8, and 240 ± 9.3 kDa, indicating a population of monomers, tetramers, and 24-mers, heretofore referred to as bodies, with the expected monomer molecular mass of 10.2 kDa. We were surprised that an assembly composed of 24 subunits, 10 kDa each, should have dimensions on the order of 500 Å by EM (Fig. 1c). However, dehydration and negative staining are known to distort physical dimensions as compared with those measured in solution (20). Fig. 2b presents fractional SV boundary plateau values in histogram form, confirming CD and EM observations.
The importance of the RING domain is underscored by the self-assembly of LFV Z, which is indistinguishable by SV from that of LCMV Z, despite substantial differences in the sequences flanking their RINGs (Fig. 1d). Examination of the pH dependence of self-assembly indicates that assembly is favored under acidic conditions, where all of Z exists in body form at pH 5 (Fig. 4a, which is published as supporting information on the PNAS web site). This acidic stability is interesting, because the arenaviral life cycle involves endosomal acidification during viral entry with the endosomal pH of ≈5 (21, 22).
We assessed the reversibility of Z self-assembly by using guanidine denaturation and renaturation. In the presence of increasing concentrations of guanidine, Z undergoes a cooperative transition with the disappearance of bodies and appearance of monomers, which are unfolded (Figs. 2a and 3a). Incubation of these unfolded monomers in the presence of decreasing concentrations of guanidine reproduces the denaturation/disassembly transition without any significant hysteresis (Fig. 3a), indicating that assembly and disassembly processes are thermodynamically reversible.
Figure 3.

Thermodynamics and kinetics of Z assembly. (a) Guanidine denaturation profile of Z bodies (○), Z tetramers (◊), and Z monomers (□) as ascertained from SV boundary plateau concentrations. Reversibility is indicated by the formation of bodies (●) from monomers (■). Denaturation (★) and renaturation (⋆) profile of Z as monitored by CD at 222 nm. (b) van't Hoff plot of the apparent equilibrium constants (K) of assembly as derived from the plateau concentration analysis for the monomer-tetramer (dashed line, blue ◊) and tetramer-body (solid line, green ○) assembly steps. (c) Kinetics of Z assembly as monitored by CD at 222 nm (lines) and SV boundary plateau analysis (blue dots). Assembly was initiated by manual dilution of fully unfolded and reduced protein into native conditions in the presence of stoichiometric Zn2+ and reductant, upon which the protein folded and tetramerized within the experimental 30-s dead time (supporting Fig. 4e). Assembly kinetics of T → B for wild-type Z in 0.1 M guanidine (black) and 0.1 M guanidine in the presence of 1% fully assembled bodies (red, with the arrow indicating time of addition of seed) and Z2 in 0.1 M guanidine (green). Nucleation kinetics were tested by using both bodies assembled at pH 5 and 0.5 M guanidine hydrochloride and produced identical results (data not shown). Horizontal and vertical error bars reflect the uncertainty in estimating the time point of observation by SV and the error in estimating the fraction body from sedimentation boundary plateau concentrations, respectively. (d) Guanidine dependence of observed assembly (left limb) and disassembly rates (right limb) for Z (solid line, ●) and Z2 (dashed line, ○). Assembly rates scale linearly with guanidine concentration even in <0.5 M guanidinium, where rollover of rates would be expected because of the presence of the tetrameric intermediate. The lack of observation of such a rollover is likely caused by the experimental dead time of 30 s and subsecond interconversion of monomers and tetramers (Methods and Analysis). The apparent two-state nature of the kinetic pathway thus is due to preequilibration of monomer and tetramer as a result of current experimental limitations.
In all, an intact and stable RING domain is necessary but insufficient for ordered and reversible self-assembly, and macromolecular organization of Z involves a structural element shared by the RING domain as well as its immediate contextual sequence.
Bodies Assemble from Tetramers.
Understanding of the assembly of the RING domain of Z into functional structures is incomplete without a detailed understanding of the energetic means by which the assembled body is stable and the kinetic mechanism by which it achieves this stability efficiently. At neutral pH, Z exists as a population of monomers (M), tetramers (T), and bodies (B). Two limiting assembly mechanisms are possible, being primarily distinguished by whether the tetrameric intermediate is on (Eq. 1) or off (Eq. 2) the pathway of productive assembly:
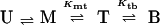 |
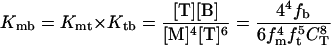 |
1 |
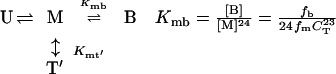 |
2 |
where fm, ft, and fb are fractions of monomer, tetramer, and body, respectively. Concerted assembly (Eq. 2) is characterized by a much steeper dependence of Kmb on monomer concentration, [M], as well as total concentration, CT.
To distinguish between the two models, we use guanidine denaturation, which differentially affects the stability of assembly species, thereby simplifying their partitioning. Guanidine denaturation studies indicate that the addition of low concentrations of guanidine abolishes the presence of monomers and tetramers, with only bodies being present at 0.5 M guanidine (Fig. 2a). Thus, these guanidine denaturation measurements report on the equilibrium between fully assembled bodies and fully unfolded monomers (U). Fits of the CD and SV guanidine denaturation profiles are in agreement with each other: ΔG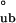 = −12.3 ± 0.65 kcal/mol and ΔG
= −12.3 ± 0.65 kcal/mol and ΔG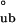 = −12.2 ± 0.86 kcal/mol, respectively. Because the free energy requirement for assembly from monomers into bodies (ΔG
= −12.2 ± 0.86 kcal/mol, respectively. Because the free energy requirement for assembly from monomers into bodies (ΔG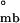 ) cannot exceed that of monomeric folding coupled with assembly (ΔG
) cannot exceed that of monomeric folding coupled with assembly (ΔG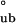 ), we use this as the energetic upper limit to discriminate between hierarchical and concerted mechanisms. Sedimentation boundary plateau values contain information about the relative population of each assembly species and therefore can be used to calculate the apparent equilibrium constants and free energy changes of the assembly process (Methods and Analysis). Because concerted assembly is highly cooperative, it involves a large nonlinear change in free energy upon assembly (24th power in Eq. 2). Indeed, application of this model to SV boundary plateau concentration data yields ΔG
), we use this as the energetic upper limit to discriminate between hierarchical and concerted mechanisms. Sedimentation boundary plateau values contain information about the relative population of each assembly species and therefore can be used to calculate the apparent equilibrium constants and free energy changes of the assembly process (Methods and Analysis). Because concerted assembly is highly cooperative, it involves a large nonlinear change in free energy upon assembly (24th power in Eq. 2). Indeed, application of this model to SV boundary plateau concentration data yields ΔG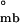 = −18.2 ± 0.9 kcal/mol, a value that greatly exceeds the energetic upper limit established by ΔG
= −18.2 ± 0.9 kcal/mol, a value that greatly exceeds the energetic upper limit established by ΔG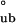 . In contrast, application of the hierarchical model results in ΔG
. In contrast, application of the hierarchical model results in ΔG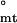 = −2.57 ± 0.9 and ΔG
= −2.57 ± 0.9 and ΔG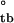 = −4.67 ± 0.9 kcal/mol, with the consequent ΔG
= −4.67 ± 0.9 kcal/mol, with the consequent ΔG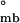 = −7.24 ± 1.3 kcal/mol, which is consistent with ΔG
= −7.24 ± 1.3 kcal/mol, which is consistent with ΔG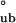 . Moreover, dependence of the observed partitioning among assembly species on protein concentration is consistent only with the hierarchical mode of assembly, as could be anticipated from Eq. 2 (data not shown). Thus, thermodynamically, self-assembly is best described hierarchically, where monomers form tetramers that oligomerize into bodies, presumably hexamers of tetramers.
. Moreover, dependence of the observed partitioning among assembly species on protein concentration is consistent only with the hierarchical mode of assembly, as could be anticipated from Eq. 2 (data not shown). Thus, thermodynamically, self-assembly is best described hierarchically, where monomers form tetramers that oligomerize into bodies, presumably hexamers of tetramers.
Assembly Is Entropically Driven and Its Steps Are Thermodynamically Unique.
To characterize the differences between the two steps of the assembly process and understand the thermodynamic origin of its stability, we investigated its temperature dependence. Assembly and polymerization are distinguished from other biological interactions in that they must overcome extremely large changes in translational entropy, whereby an ensemble of multiple particles comes together into an ordered collective (23). Thus, the relative entropic and enthalpic dependencies of assembly thermodynamics contain information about the driving force for assembly, because the principal opposing force is known. Consequently, we estimated the relative entropic and enthalpic dependencies of the two assembly equilibria by using van't Hoff analysis (Figs. 3b and 4c and Methods and Analysis).
Strikingly, the two steps of the assembly have different dependencies on temperature (Fig. 3b). Because the slope of the van't Hoff plot is directly proportional to changes in enthalpy, it can be seen that tetramer-body equilibrium is largely entropically driven, whereas that of the monomer-tetramer equilibrium depends on changes in enthalpy: ΔH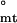 = −2.5 ± 0.8 and ΔH
= −2.5 ± 0.8 and ΔH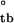 = −0.3 ± 0.2, whereas ΔG
= −0.3 ± 0.2, whereas ΔG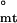 = −2.57 ± 0.9 and ΔG
= −2.57 ± 0.9 and ΔG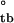 = −4.67 ± 0.9 kcal/mol. Because both assembly steps involve unfavorable changes in translational entropy, this suggests that the tetrameric intermediate overcomes them by accumulating favorable enthalpic interactions, whereas the fully assembled body, consisting of six tetramers, does so by directly offsetting the entropic cost, likely as a result of conformational reorganization (see below). Examination of the ionic strength dependence of assembly confirms that stability of the tetramer is considerably enthalpic in nature (Fig. 4b). Notably, no substantial changes in heat capacity are observed in the 4–40°C range, as reflected by lack of significant deviations from linearity in van't Hoff behavior (Fig. 3b). Estimation of ΔCp for the tetramer-body step reveals that it cannot exceed 0.13 ± 0.24 kcal/mol/K (Methods and Analysis). The lack of significant changes in heat capacity upon assembly is surprising, because actin polymerization, for example, exhibits >16 kcal/mol/K change in heat capacity in the 4–20°C range (24). Such behavior may reflect the distinctiveness of RING self-assembly as compared with fibrillar assembly reactions.
= −4.67 ± 0.9 kcal/mol. Because both assembly steps involve unfavorable changes in translational entropy, this suggests that the tetrameric intermediate overcomes them by accumulating favorable enthalpic interactions, whereas the fully assembled body, consisting of six tetramers, does so by directly offsetting the entropic cost, likely as a result of conformational reorganization (see below). Examination of the ionic strength dependence of assembly confirms that stability of the tetramer is considerably enthalpic in nature (Fig. 4b). Notably, no substantial changes in heat capacity are observed in the 4–40°C range, as reflected by lack of significant deviations from linearity in van't Hoff behavior (Fig. 3b). Estimation of ΔCp for the tetramer-body step reveals that it cannot exceed 0.13 ± 0.24 kcal/mol/K (Methods and Analysis). The lack of significant changes in heat capacity upon assembly is surprising, because actin polymerization, for example, exhibits >16 kcal/mol/K change in heat capacity in the 4–20°C range (24). Such behavior may reflect the distinctiveness of RING self-assembly as compared with fibrillar assembly reactions.
The Kinetic Mechanism of Z Self-Assembly.
To define the kinetic mechanism of self-assembly, we monitor assembly and disassembly kinetics as a function of denaturant perturbation. Thus, we have extended the Hammond analysis (25) as it has been applied to protein folding (26) to the study of self-assembly of macromolecular complexes (Methods and Analysis). Hammond analysis involves the measurement of forward and back reaction rates as a function of perturbation of the system, thereby allowing characterization of the transition state of the reaction, and in this case the defining features of the assembly mechanism.
Assembly and disassembly kinetics were monitored by SV, EM, CD, and dynamic light scattering. As can be seen from Fig. 3c, after dilution from denaturing conditions where Z is monomeric and unfolded into native conditions where folding is favored, assembly proceeds on the time scale of hours with an apparent rate constant of 0.0037 ± 12 × 10−4 min−1 at 0.1 M guanidinium, as quantified by SV. Monitoring secondary structure rearrangements by using CD reveals that Z acquires a native-like CD profile within the experimental dead time of 30 s and subsequently loses ≈10% of ellipticity at 222 nm at a rate of 0.0032 ± 5 × 10−4 min−1 (Fig. 3c), which is in agreement with the formation of bodies as monitored by SV. Examination of the same process by dynamic light scattering, which monitors the population-weighted average molecular mass, reveals that the tetramer (40 ± 7 kDa) forms within the dead time (Fig. 4e and Methods and Analysis). Comparison of SV, CD, and dynamic light-scattering kinetic profiles as well as time points in assembly and disassembly examined by EM reveals that folding and tetramerization occur on the time scale of seconds, whereas assembly of tetramers into bodies takes hours (Methods and Analysis). Thus, the observed loss of ellipticity at 222 nm that accompanies assembly coincides with the conformational reorganization of tetramers as they self-associate into bodies.
By definition, the transition state captures the essential features of the reaction pathway, and its characterization for the assembly of tetramers into bodies exposes the cardinal features of the self-assembly process. To apply Hammond analysis to self-assembly, as it has been applied to simple chemical reactions and protein folding (25, 26), the uniqueness of the barrier for forward and reverse processes must be validated. Operationally, this can be achieved by recovering the equilibrium free energy of assembly from the activation free energies of assembly and disassembly. Because monomeric folding and tetramerization occur on relatively fast time scales within the experimental dead time, we treat them as fast preequilibria (Methods and Analysis) and examine the dependence of the observed rates of T → B and B → T transitions on guanidine concentration (Fig. 3d), with the left limb of the chevron showing the decrease in the assembly rate with increasing guanidine concentrations and the right limb showing the increase in the disassembly rate with increasing guanidine concentrations. Extrapolation of the two limbs to 0 M guanidine yields values for activation energies that are comparable directly with energies derived from equilibrium methods (Methods and Analysis). Table 1 lists the thermodynamic parameters of Z assembly derived from the SV plateau concentration analysis and kinetic parameters derived from Hammond analysis of guanidine equilibration measurements. As can be seen from Table 1, ΔG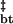 − ΔG
− ΔG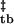 = −4.59 ± 0.41 kcal/mol, which agrees well with the equilibrium value of ΔG
= −4.59 ± 0.41 kcal/mol, which agrees well with the equilibrium value of ΔG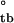 = −4.67 ± 0.13 kcal/mol. Thus, the assembly kinetics apparently are two-state and involve a unique transition state, because the equilibrium free energy of assembly can be recovered fully from the activation free energies of assembly and disassembly.
= −4.67 ± 0.13 kcal/mol. Thus, the assembly kinetics apparently are two-state and involve a unique transition state, because the equilibrium free energy of assembly can be recovered fully from the activation free energies of assembly and disassembly.
Table 1.
Thermodynamic and kinetic parameters of Z assembly from tetramers to bodies
| Thermodynamics
|
Kinetics
|
||||
|---|---|---|---|---|---|
ΔG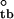
|
ΔG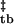
|
−mtb | ΔG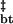
|
mbt | |
| Z | −4.67 ± 0.13 | −17.42 ± 0.27 | 0.47 ± 0.05 | −22.01 ± 0.31 | 1.25 ± 0.03 |
| Z2 | −15.94 ± 0.16 | 0.41 ± 0.07 | −22.80 ± 0.26 | 1.40 ± 0.07 | |
All values are extrapolated to 0 M guanidine hydrochloride and 100 μM protein. Energies are in kcal/mol and m values are in kcal/mol/M; both are ±1σ.
The dependence of apparent free energy on guanidine concentration is proportional to the extent of hydrophobic surface desolvation and is described by the m value (ref. 27, Table 1). Thus, −mtb/(−mtb + mbt) describes the fraction of hydrophobic surface area buried in the assembly transition state relative to that in the body, where −mtb is the difference in surface area buried in the tetramer and the transition state (0.47 ± 0.07 kcal/mol/M), and −mtb + mbt is the total extent of hydrophobic desolvation between tetramers and bodies (1.72 ± 0.11 kcal/mol/M). In this fashion, the assembly transition state is relatively solvent-exposed, with (1.72–0.47)/1.72 or ≈70% of available guanidine-sensitive surface area being buried once in the assembled body (Table 1).
Many self-assembling systems such as actin (28), tobacco mosaic virus protein (29), and prions (30) involve nucleation as the rate-limiting step of assembly. The assembly rate of Z is not affected significantly by the presence of 1% (wt/wt) seed of assembled bodies, indicating that the assembly process does not involve template- or nucleus-mediated oligomerization (Fig. 3c).
A Structural Rearrangement Around Site II Is Coupled to Self-Assembly.
Self-assembly of Z depends on an intact RING domain. However, the two zinc-binding sites in RINGs are not identical, both with respect to ligands as well as thermodynamics. For example, in the RINGs of BRCA1 and PML, the two sites are anticooperatively coupled, whereby zinc binding by the first site reduces the affinity for Zn2+ of the second site (18, 19, 31). Thus, we tested the assembly of a mutant Z (Z2) containing a double cysteine to alanine substitution of the last two ligands in the second zinc-binding cluster (Fig. 1a). In contrast to Z1, which is monomeric, partially folded, and does not assemble, Z2 is fully folded (Fig. 2a). Furthermore, Z2 assembles into bodies with size and morphology similar to those of wild-type Z as observed by EM (Fig. 2b). Moreover, gel filtration of Z2 reveals that its hydrodynamic behavior is similar to that of Z (Fig. 2c). Thus, Z2's self-assembly mimics that of wild-type Z. However, Z2 exhibits sedimenting boundaries that are fast and broad (Methods and Analysis and Table 2), suggesting fast assembly kinetics that repopulate sedimenting boundaries caused by the transport of quickly assembling species in the course of sedimentation (32).
The kinetics of assembly of Z2 were measured as they were for wild-type Z. Destabilization of the second zinc-binding site accelerates the assembly rate of tetramers into bodies by more than an order of magnitude (Fig. 3 c and d). No significant effects on the disassembly rate can be observed across a wide range of guanidine concentrations (Fig. 3d). Because this mutation only affects the T → B assembly rate, this perturbation only affects ΔG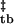 without significant effects on ΔG
without significant effects on ΔG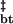 (Table 1). Results of equilibrium sedimentation of Z2 support the notion that Z2 bodies are more stable (data not shown), as predicted from the increased assembly rate of Z2 as compared with Z. In Z2, it is expected that zinc binding in site II is either extremely weak or absent altogether (33). These data suggest that efficient self-assembly of Z involves a rearrangement of the second zinc-binding site, whereby this rearranged cluster is present in the assembly transition state and in fully assembled bodies but not in the tetrameric intermediate or unassembled monomers. Consistent with this reasoning, Z2 that exists largely in body form (Fig. 2b, data not shown) binds 1.1 ± 0.2 zinc ions per RING, presumably in site I, as measured by using inductively coupled plasma optical emission spectroscopy. In contrast, the RING construct, which does not assemble (Fig. 2b), binds 2.0 ± 0.1 zinc ions per RING, as measured by inductively coupled plasma optical emission spectroscopy. These observations are consistent with the structural biochemistry of BRCA1 RING and its site II mutants (34, 35). Thus, efficient self-assembly of Z and its RING domain is accomplished by the favorable geometry of the otherwise poorly stable tetrameric intermediate, which allows for the rearrangement of regions surrounding the second zinc-binding site, presumably facilitating assembly of tetramers into bodies. The high translational entropy cost associated with self-assembly is offset by the structural rearrangement of the RING subunits involving the second zinc-binding site and consequent desolvation of more than 70% of available surface area.
(Table 1). Results of equilibrium sedimentation of Z2 support the notion that Z2 bodies are more stable (data not shown), as predicted from the increased assembly rate of Z2 as compared with Z. In Z2, it is expected that zinc binding in site II is either extremely weak or absent altogether (33). These data suggest that efficient self-assembly of Z involves a rearrangement of the second zinc-binding site, whereby this rearranged cluster is present in the assembly transition state and in fully assembled bodies but not in the tetrameric intermediate or unassembled monomers. Consistent with this reasoning, Z2 that exists largely in body form (Fig. 2b, data not shown) binds 1.1 ± 0.2 zinc ions per RING, presumably in site I, as measured by using inductively coupled plasma optical emission spectroscopy. In contrast, the RING construct, which does not assemble (Fig. 2b), binds 2.0 ± 0.1 zinc ions per RING, as measured by inductively coupled plasma optical emission spectroscopy. These observations are consistent with the structural biochemistry of BRCA1 RING and its site II mutants (34, 35). Thus, efficient self-assembly of Z and its RING domain is accomplished by the favorable geometry of the otherwise poorly stable tetrameric intermediate, which allows for the rearrangement of regions surrounding the second zinc-binding site, presumably facilitating assembly of tetramers into bodies. The high translational entropy cost associated with self-assembly is offset by the structural rearrangement of the RING subunits involving the second zinc-binding site and consequent desolvation of more than 70% of available surface area.
Unique Properties of Self-Assembly Behavior of RING Domains.
Classically, protein assembly has been described by two types of microscopic models. Isodesmic polymerization (36), as observed with tubulin (37), involves noncooperative sequential addition of monomers to an infinitely growing assembling structure with equilibrium accumulation of dimers, trimers, tetramers, pentamers, etc. In contrast, nucleated polymerization (29), as observed with actin (38), is based on strongly cooperative association involving a thermodynamically unstable nucleus. In this work, we observe that macroscopic behavior of self-assembling systems can exhibit substantial deviations from limits established by the above models. The rate-limiting step of self-assembly of Z and its RING is not nucleated (Fig. 3c). Moreover, assembly is not isodesmic, being finite and without accumulation of {n, n + 1} intermediates (Fig. 2 b–d). Instead, slow self-assembly kinetics of Z seem to be caused by conformational restructuring and changes in zinc coordination of the RING domain near the second zinc-binding site, which are coupled to the assembly of tetramers into bodies. Because regions surrounding the RING of Z contain prolines and the RING domain coordinates zinc in a cross-brace topology, the slow step in the assembly process could be due to prolyl peptide bond isomerization (39) or loop threading (40).
Furthermore, the hierarchical mode of assembly and its dependence on the tetrameric intermediate create a self-assembly process that is accessible to biological energies on the order of 10 kcal/mol (41), allowing for dynamic remodeling of the assembled structure, which is in contrast to other systems such as amyloid fibrils (30). It is noteworthy that such remodeling by partner proteins in vivo may occur through interactions with the tetrameric intermediate, because its reorganization constitutes the rate-limiting step of the assembly process. Understanding the spatial encoding of assembly by an intermediate as well as structural characterization of the macromolecular assemblages formed by RING domains are important directions of future work.
Macromolecular Assemblages Constituted by RING Domain Proteins in Vivo.
Because of the diverse functional distribution of RING domains, definition of their general function has been difficult. Three discrete biochemical activities have been described for RINGs: (i) RING of KAP-1/TIF1β is required for the formation of a homohexameric complex, which associates with the KRAB domains of the Krüppel-associated family of proteins, with this association being required for DNA binding and transcriptional repression (2); (ii) RINGs of PML and Z repress translation by directly binding translation initiation factor eIF4E and reducing its affinity for the 5′ 7-methyl guanosine cap of mRNA (3); and (iii) RINGs from a number of proteins including Cbl and BRCA1 indirectly support polymerization of ubiquitin and its conjugation to substrates for ubiquitin-mediated proteolysis (4, 5). At the biochemical level, the three processes are apparently disparate, involving divergent mechanisms and substrates. We propose that self-assembly is a general property of RING domains and that their consequent organization of macromolecular assemblages underlies their function in diverse biochemical and physiological processes by coupling discrete biochemical activities. Such scaffolding behavior would be analogous to activation of receptor tyrosine kinases, where clustering of monomers leads to potentiation of inherent kinase activity (42).
To date, over 50 of the 200 RING proteins described are observed in and/or isolated from cells as parts of macromolecular assemblies (1, 6). Here we observe that arenaviral Z constituted almost entirely by its RING domain reversibly self-assembles into ordered structures that approximate in size and morphology those observed in infected cells. This is a striking finding considering that Z is a 10-kDa protein and can self-assemble into structures that are readily visible by EM. A compelling question is whether these structures participate in Z function in vivo. The RING of Z is required to selectively repress mRNA translation of host cell growth regulatory factors, likely inhibiting cellular responses and facilitating viral replication (11). Destabilization of the first zinc-binding site abolishes the capacity to self-assemble in vitro (this work) to form discrete structures and repress translation both in vitro and in vivo (3, 10, 11). In contrast, Z2 self-assembles into bodies indistinguishable from those formed by Z and has biological activities similar to those of wild-type Z (3, 10, 11). This suggests that RING-dependent self-assembly of Z into bodies underlies its translational repression in arenavirus-infected cells. In this way, the scaffolding properties of Z are correlated to its physiological functions in vivo.
Perhaps the most exciting promise of this work is the elucidation of the general function of RING domains as templates for macromolecular assemblages in cells. Only future studies will tell whether other RING-containing proteins depend on self-assembly properties of their RINGs for the organization of functional macromolecular scaffolds.
Supplementary Material
Acknowledgments
We are grateful to Roman Osman, Maria Salvato, and Allan Capili for many fruitful discussions, Hilal Lashuel and Carlos Escalante for help with ultracentrifugation, Larry Shapiro for reading the manuscript, and reviewers for many insightful comments. A.K. is supported by the National Institutes of Health Medical Scientist Training Program and the Louis L. Seaman Medical Student Microbiology grant. K.L.B.B. is a scholar of the Leukemia and Lymphoma Society. Funding was provided by National Institutes of Health Grant CA80728.
Abbreviations
- LCMV
lymphocytic choriomeningitis virus
- LFV
Lassa fever virus
- PML
promyelocytic leukemia protein
- EM
electron microscopy
- CD
circular dichroism
- SV
sedimentation velocity
- GST
glutathione S-transferase
- Z1 and Z2
first and second zinc-binding site mutants, respectively
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Kentsis A, Borden K L B. Curr Protein Peptide Sci. 2000;1:49–73. doi: 10.2174/1389203003381478. [DOI] [PubMed] [Google Scholar]
- 2.Peng H, Begg G E, Schultz D C, Friedman J R, Jensen D E, Speicher D W, Rauscher F J., III J Mol Biol. 2000;295:1139–1162. doi: 10.1006/jmbi.1999.3402. [DOI] [PubMed] [Google Scholar]
- 3.Kentsis A, Dwyer E C, Perez J M, Sharma M, Chen A, Pan Z Q, Borden K L. J Mol Biol. 2001;312:609–623. doi: 10.1006/jmbi.2001.5003. [DOI] [PubMed] [Google Scholar]
- 4.Miyake S, Lupher M L, Jr, Druker B, Band H. Proc Natl Acad Sci USA. 1998;95:7927–7932. doi: 10.1073/pnas.95.14.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruffner H, Joazeiro C A, Hemmati D, Hunter T, Verma I M. Proc Natl Acad Sci USA. 2001;98:5134–5139. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saurin A J, Borden K L, Boddy M N, Freemont P S. Trends Biochem Sci. 1996;21:208–214. [PubMed] [Google Scholar]
- 7.Melnick A, Licht J D. Blood. 1999;93:3167–3215. [PubMed] [Google Scholar]
- 8.Riviere Y. Curr Top Microbiol Immunol. 1987;133:59–65. doi: 10.1007/978-3-642-71683-6_5. [DOI] [PubMed] [Google Scholar]
- 9.Peters C J. Clinical Virology. New York: Churchill Livingstone; 1997. [Google Scholar]
- 10.Borden K L, Campbelldwyer E J, Carlile G W, Djavani M, Salvato M S. J Virol. 1998;72:3819–3826. doi: 10.1128/jvi.72.5.3819-3826.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell Dwyer E J, Lai H, MacDonald R C, Salvato M S, Borden K L. J Virol. 2000;74:3293–3300. doi: 10.1128/jvi.74.7.3293-3300.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borden K L, Campbell Dwyer E J, Salvato M S. J Virol. 1998;72:758–766. doi: 10.1128/jvi.72.1.758-766.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salvato M S, Rai K S. In: Topley and Wilson's Microbiology and Microbial Infections. Collier L H, Mahy B W J, editors. London: Arnold; 1996. [Google Scholar]
- 14.Perkins S J. Eur J Biochem. 1986;157:169–180. doi: 10.1111/j.1432-1033.1986.tb09653.x. [DOI] [PubMed] [Google Scholar]
- 15.Tanford C. Physical Chemistry of Macromolecules. New York: Wiley; 1961. [Google Scholar]
- 16.Pecora R. Dynamic Light Scattering: Applications of Photon Correlation Spectroscopy. New York: Plenum; 1985. [Google Scholar]
- 17.Capili A D, Schultz D C, Rauscher I F, Borden K L. EMBO J. 2001;20:165–177. doi: 10.1093/emboj/20.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roehm P C, Berg J M. Biochemistry. 1997;36:10240–10245. doi: 10.1021/bi970863d. [DOI] [PubMed] [Google Scholar]
- 19.Brzovic P S, Meza J, King M C, Klevit R E. J Biol Chem. 1998;273:7795–7799. doi: 10.1074/jbc.273.14.7795. [DOI] [PubMed] [Google Scholar]
- 20.Hayat M A. Principles and Techniques of Electron Microscopy. New York: Nostrand Reinhold; 1976. [Google Scholar]
- 21.Di Simone C, Zandonatti M A, Buchmeier M J. Virology. 1994;198:455–465. doi: 10.1006/viro.1994.1057. [DOI] [PubMed] [Google Scholar]
- 22.Di Simone C, Buchmeier M J. Virology. 1995;209:3–9. doi: 10.1006/viro.1995.1225. [DOI] [PubMed] [Google Scholar]
- 23.Hill T. An Introduction to Statistical Thermodynamics. New York: Dover; 1986. [Google Scholar]
- 24.Quirion F, Gicquaud C. Biochem J. 1993;295:671–672. doi: 10.1042/bj2950671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hammond G S. J Am Chem Soc. 1955;77:334–338. [Google Scholar]
- 26.Matouschek A, Fersht A R. Proc Natl Acad Sci USA. 1993;90:7814–7818. doi: 10.1073/pnas.90.16.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santoro M M, Bolen D W. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]
- 28.Frieden C. Annu Rev Biophys Biophys Chem. 1985;14:189–210. doi: 10.1146/annurev.bb.14.060185.001201. [DOI] [PubMed] [Google Scholar]
- 29.Schuster T M, Scheele R B, Adams M L, Shire S J, Steckert J J, Potschka M. Biophys J. 1980;32:313–329. doi: 10.1016/S0006-3495(80)84959-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly J W. Structure (London) 1997;5:595–600. doi: 10.1016/s0969-2126(97)00215-3. [DOI] [PubMed] [Google Scholar]
- 31.Borden K L, Boddy M N, Lally J, O'Reilly N J, Martin S, Howe K, Solomon E, Freemont P S. EMBO J. 1995;14:1532–1541. doi: 10.1002/j.1460-2075.1995.tb07139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demeler B, Saber H, Hansen J C. Biophys J. 1997;72:397–407. doi: 10.1016/S0006-3495(97)78680-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berg J M, Godwin H A. Annu Rev Biophys Biomol Struct. 1997;26:357–371. doi: 10.1146/annurev.biophys.26.1.357. [DOI] [PubMed] [Google Scholar]
- 34.Brzovic P S, Rajagopal P, Hoyt D W, King M C, Klevit R E. Nat Struct Biol. 2001;8:833–837. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 35.Brzovic P S, Meza J E, King M C, Klevit R E. J Biol Chem. 2001;276:41399–41406. doi: 10.1074/jbc.M106551200. [DOI] [PubMed] [Google Scholar]
- 36.Vickers L P, Ackers G K. Arch Biochem Biophys. 1976;174:747–749. doi: 10.1016/0003-9861(76)90406-9. [DOI] [PubMed] [Google Scholar]
- 37.Lobert S, Vulevic B, Correia J J. Biochemistry. 1996;35:6806–6814. doi: 10.1021/bi953037i. [DOI] [PubMed] [Google Scholar]
- 38.Grazi E, Ferri A, Cino S. Biochem J. 1983;213:727–732. doi: 10.1042/bj2130727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mann C J, Shao X, Matthews C R. Biochemistry. 1995;34:14573–14580. doi: 10.1021/bi00044a036. [DOI] [PubMed] [Google Scholar]
- 40.De Young L R, Schmelzer C H, Burton L E. Protein Sci. 1999;8:2513–2518. doi: 10.1110/ps.8.11.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makhatadze G I, Privalov P L. Adv Protein Chem. 1995;47:307–425. doi: 10.1016/s0065-3233(08)60548-3. [DOI] [PubMed] [Google Scholar]
- 42.Schlessinger J. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.