

Abstract
The problem of global optimization is pivotal in a variety of scientific fields. Here, we present a robust stochastic search method that is able to find the global minimum for a given cost function, as well as, in most cases, any number of best solutions for very large combinatorial “explosive” systems. The algorithm iteratively eliminates variable values that contribute consistently to the highest end of a cost function's spectrum of values for the full system. Values that have not been eliminated are retained for a full, exhaustive search, allowing the creation of an ordered population of best solutions, which includes the global minimum. We demonstrate the ability of the algorithm to explore the conformational space of side chains in eight proteins, with 54 to 263 residues, to reproduce a population of their low energy conformations. The 1,000 lowest energy solutions are identical in the stochastic (with two different seed numbers) and full, exhaustive searches for six of eight proteins. The others retain the lowest 141 and 213 (of 1,000) conformations, depending on the seed number, and the maximal difference between stochastic and exhaustive is only about 0.15 Kcal/mol. The energy gap between the lowest and highest of the 1,000 low-energy conformers in eight proteins is between 0.55 and 3.64 Kcal/mol. This algorithm offers real opportunities for solving problems of high complexity in structural biology and in other fields of science and technology.
Many problems in life sciences and in other fields of science and technology are of high complexity, thus requiring sophisticated methods of searching and scoring to achieve the ability to study and to simulate them by means of a computer simulation. An excellent search method coupled with a highly reliable scoring method should allow comparisons to some natural phenomena. In this article, we have taken the approach of comparing best populations found by a stochastic search method to a full, exhaustive search, as the crucial test of this method. However, comparisons to experimental results also are included. The problem chosen to exemplify this method is the positions of side chains in proteins, which is essential for both theoretical and experimental purposes. On the theoretical side, it is a subproblem in de novo protein structure prediction. It is essential for structure-based drug design (1), for inverse folding and threading algorithms (2), for predicting the effect of mutations on structure (3), for ab initio predictions of tertiary structure (4), for homology-based modeling (5), and others. From the x-ray crystallographer's point of view, it could speed the placement of side chains using the electron density maps of the main chain before refinement calculations. The main limitation is the large amount of possible conformations that each side chain may adopt (6). An exhaustive search of all possible conformations is beyond the scope of state of the art computers.
Current strategies for side chain addition to a given backbone differ in three categories. The first is the conformational space of each side chain. In continuous space methods (7, 8), any side chain torsion angle may be sampled. Discrete space methods are based on the assumption that side chains exist in energetically preferred conformations called rotamers, which are local minima conformers that have been sampled by statistical analysis of known structures (9–14). Discrete space methods cannot predict conformations that are not present in the rotamer database. There is no agreement regarding the optimal size of a rotamer library. Several groups showed that large rotamer databases that contain very rare conformations do not necessarily yield better predictions than smaller databases (15–18). On the other hand, Xiang & Honig (19) have recently extended the accuracy of predictions with an extensive rotamer library. Rotamer databases also can be classified as backbone dependent and backbone independent. The former are based on a relationship between the side chain conformation and the local backbone conformation (20–21), whereas the latter are not (7, 16, 22).
The second category is the scoring or cost function for evaluating solutions. Energy-based methods rely on nonbonding terms (6, 15, 16, 18, 23–25). The assumption is that the lower the energy, the more accurate the prediction. Knowledge-based methods also were proposed: Sutcliffe et al. (26) suggested a procedure for building side chains using spatial information from side chains in topologically equivalent positions—as far as such a correlation may be observed—and most probable conformations of the side chains in the respective secondary structure type. Sali & Blundell (27) described a comparative protein-modeling method designed to find the most probable structure for a sequence, given its alignment with related structures. Bower et al. (28) located residues in their most favorable backbone-dependent rotamers and systematically resolved the conflicts that arise from that structure.
Accurate computer location of protein side chains is a complicated task because of the large number of minimum energy conformers on the potential energy surface, even with a rigid backbone. Conventional methods for side chain addition usually result in a single structure of the protein, which is then compared with an experimental structure, if available. The conformational space is disregarded, although protein function and molecular recognition depend on structural plasticity (29), and conformational flexibility of receptor proteins is considered to be one of the major factors that affect ligand docking (30).
Our algorithm focuses on the third category, the search strategy, and not on the energy function or the rotamer library. There are numerous examples of search strategies for highly complex problems. Metropolis Monte Carlo methods (15), Gibbs sampling Monte Carlo (18), Neural networks (31), Conformational Space Annealing (32), Genetic and Evolutionary Algorithms (33–35), Simulated Annealing (6), Mean Field Optimization (23), and Locally Enhanced Sampling (8). Combinatorial Searches (21, 24, 33) are used on discrete conformers and may be followed by a continuous minimization in the final stage of refinement. It should be noted that there is no guarantee that any of the above will converge to a valid solution. Another widely used method is Dead End Elimination (DEE). It is based on the identification of rotamers that are absolutely incompatible with the global minimum energy conformation, eliminating rotamers that cannot contribute to local energy minima of a certain or higher order. Conformations composing such rotamers can be qualified as dead ending (30, 36, 37). If enough rotamers can be eliminated by recursive applications, the global minimum can be found (38, 39). If no conditions can be established to eliminate further rotamers during the calculation, DEE might not converge. The global minimum can be found by an exhaustive search of the remaining rotamers (38), provided the remaining search space is not prohibitively large. Because of the mechanism of elimination of rotamers in DEE, there is little chance of forming an optimal population of solutions.
However, a combination of DEE with the A* algorithm (40) has been suggested for constructing a population of low-energy side chain conformations in proteins, and was used for constructing partition functions. The A* algorithm approach may find the best N solutions, but it is restricted to relatively small proteins. The largest protein solved by this algorithm so far contained 68 amino acids, which comprise about 1043 combinations—depending on the complexity of the rotamer library–whereas proteins with a much larger number of combinations are common. As a “stand alone” algorithm (without the DEE preprocessing stage) the A* algorithm reaches a maximum of 1021 combinations. An effective search by the A* algorithm must have a good estimate of the cost to reach a goal node. Estimation is problematic because of interactions between residues that have not yet been assigned. Those limitations raise the need for a robust algorithm that finds the global minimum and the lowest energy conformations in larger systems. Such a search algorithm is presented here.
Methods
The Search Technique.
The code uses a backbone-dependent rotamer library (13, 21, 28, 41). We used the August 1997 update of the rotamer library of Dunbrack & Karplus, with united atoms (42). Energy is computed by Eq. 1 with the AMBER nonbonding 12–6 Lennard–Jones and electrostatic energy terms (43), where Ai,j is the repulsion parameter for the two (i, j) atoms, Bi,j is their attractive polarizability parameter, qi is the partial charge, ri,j is the distance between atoms, and ɛ is the dielectric constant. A distance-dependent dielectric constant of ɛ = r has been used. The nonbonded energy is calculated for interactions with the backbone and with other residues' rotamers. If the nonbonded energy term exceeds the value of 10 Kcal/mol for a given pair of atoms, it is truncated at 10 Kcal/mol.
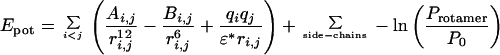 |
1 |
As suggested by Bower et al. (28) and implemented in the SCWRL (Side Chains With a Rotamer Library) algorithm, every rotamer is given a local energy based on its probability in the backbone-dependent rotamer library. Energies are taken from the probabilities of the backbone-dependent rotamer library, as −ln(protamer/p0), where p0 is the probability of the most probable rotamer, and protamer is the probability of a given rotamer (assuming kT = 1). The search strategy includes several steps:
(I) Steric clashes elimination stage and preliminary rotamer location.
The input for the calculation are the backbone (N, Cα, C, O) coordinates of a protein with a known, high-resolution structure. Those, together with standard bond lengths and bond angles from AMBER 4.1 (43) and with ϕ and ψ angles of the backbone are used to create the initial placement of possible rotamers for each residue. Possible disulfide bonds between cysteine residues are calculated by the distance between sulfur atoms. All rotamers that clash with the backbone are excluded by a threshold value of 18 Kcal/mol. If all rotamers of a residue clash with the backbone, the rotamer with the lowest “clash energy” remains. The algorithm treats single rotamers as part of the backbone; i.e., other rotamers that clash with those residues also will be excluded. The algorithm also searches for all side chain clashes between rotamer i of amino acid j and rotamer k of amino acid l. The algorithm excludes such pairs from being part of the solution, and, therefore, they are not sampled in the stochastic stage (see below).
(II) Stochastic stage.
It is obvious that in the case of a large biological system such as a protein, a very large combinatorial problem results. In Hydrolase (1arb; ref. 44), for example, there are 2.4 × 10105 alternative positioning options after step I. A stochastic algorithm is used to reduce the size of the problem. In the protein, the side chain rotamers in d0 amino acids are unknown. For each amino acid there is usually more than one rotamer, but only one would give the lowest energy. Let Xj = (xj1, xj2 … xjd0) be a conformation of the protein that includes randomly picked rotamers for d0 amino acids. For each conformation Xj, the energy Ej = E(Xj) may be calculated according to the energy function described above. The objective is to find the conformation that minimizes E. Because it is impossible to evaluate all of the alternative conformations because of the large number of combinations, the following steps are taken: (i) Sample at random n conformations of the large population of combinations X1 = (x11, x12, … x1d0), … , Xn = (xn1, xn2, … , xnd0), where x11 is a randomly picked rotamer for the first amino acid in the first conformation, and xn1 is a randomly picked rotamer for the same amino acid in the nth conformation. We use n = 1,000 to create a large enough number of protein conformations and compute the corresponding energy values: E1 = E(X1) to En = E(Xn).
(ii) Construct the distribution F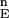 (n = 103). F
(n = 103). F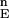 is the set of energies of all of the N-sampled conformations for the full protein. Define cutoff points H and L in F
is the set of energies of all of the N-sampled conformations for the full protein. Define cutoff points H and L in F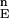 . H contains all variable values satisfying Ei ≥ F
. H contains all variable values satisfying Ei ≥ F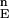 (1 − α), where F
(1 − α), where F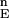 (α) is the α−th percentile of F
(α) is the α−th percentile of F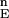 , and L contains all variable values satisfying Ei ≤ F
, and L contains all variable values satisfying Ei ≤ F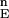 (α). The number of conformations in each of H and L is n0 = n × α. When n = 1,000 conformations and α = 0.01 (1%) for highest and lowest energy conformations, n0 = α × n = 0.01 × 1,000 = 10, so L = 10 and H = 10. In other words, H stands for the 10 highest energy conformations, and L stands for the 10 conformations with the lowest energy. (iii) Construct the vector h for all rotamer variables corresponding to the conformations in H. The vector h is the element-wise intersection of all of the rotameric states in H, in the following manner: if all rotameric states in H share the same rotamer at component j (corresponding to xnj of conformation Xn), then hj = rotamer_number; otherwise, hj = 0 (no common rotamer for j in all high-energy conformations.) (iv) Construct the vector l for rotamer variables corresponding to the conformations in L. Unlike vector h, more than one rotamer may appear for each amino acid j up to a maximum of n0 values in lj. It is the union of all rotamers of component j that appear in the low-energy conformations of L. (v) Compare h and l. If both hj and lj have a similar rotamer, it will remain as a viable rotameric state, because it contributes also to low-energy values. However, if hj does not correspond to any element of lj, then the corresponding rotamer hj will be evicted from subsequent iterations. If an amino acid has only one rotamer, it will not be evicted from subsequent iterations because it is the only remaining solution. (vi) Repeat steps i to iv for the reduced set of variables' values until the number of possible combinations of all variables is smaller than a user-defined “end of stochastic stage criteria”.
(α). The number of conformations in each of H and L is n0 = n × α. When n = 1,000 conformations and α = 0.01 (1%) for highest and lowest energy conformations, n0 = α × n = 0.01 × 1,000 = 10, so L = 10 and H = 10. In other words, H stands for the 10 highest energy conformations, and L stands for the 10 conformations with the lowest energy. (iii) Construct the vector h for all rotamer variables corresponding to the conformations in H. The vector h is the element-wise intersection of all of the rotameric states in H, in the following manner: if all rotameric states in H share the same rotamer at component j (corresponding to xnj of conformation Xn), then hj = rotamer_number; otherwise, hj = 0 (no common rotamer for j in all high-energy conformations.) (iv) Construct the vector l for rotamer variables corresponding to the conformations in L. Unlike vector h, more than one rotamer may appear for each amino acid j up to a maximum of n0 values in lj. It is the union of all rotamers of component j that appear in the low-energy conformations of L. (v) Compare h and l. If both hj and lj have a similar rotamer, it will remain as a viable rotameric state, because it contributes also to low-energy values. However, if hj does not correspond to any element of lj, then the corresponding rotamer hj will be evicted from subsequent iterations. If an amino acid has only one rotamer, it will not be evicted from subsequent iterations because it is the only remaining solution. (vi) Repeat steps i to iv for the reduced set of variables' values until the number of possible combinations of all variables is smaller than a user-defined “end of stochastic stage criteria”.
The value of α that is used to determine n0 should be selected with care. If n0 is too large, no rotamers will be eliminated. If n0 is too small, an unjustified elimination of rotamers might occur. At best, n0 should be adjusted by the number of possible rotamers of each amino acid, to allow an equal probability for the elimination of rotamers. To explain the determination of α, let us assume that each rotamer is not affected by interactions with any other amino acid in its environment. The n0 values for 2 to 29 possible rotamers of a single residue that would lead to the correct rotamer elimination with a certainty >99.983% are presented (Fig. 1). Those values were calculated in the following manner. Given a residue with three rotamers, if we want to remove one rotamer with a certainty (Pcorrect) higher than 99.99%, the error probability (Perror) must be smaller than 0.01% (0.0001). For erroneously evicting a rotamer, it must first appear in all of the high-energy conformations. In this case, the probability is (1/3)n0. In addition, this rotamer must not appear in any low-energy conformation. In this case the probability is (2/3)n0. The total error probability is Perror = (1/3)n0(2/3)n0. Thus, one may tune the calculation to nearly 100% confidence by employing the general formula in Eq. 2, where m is the number of variable values (rotamers).
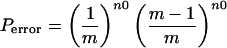 |
2 |
When m = 1 (there is one rotamer) Perror = 0. Assigning a value of Perror = 0.0001 and solving the equation for m = 3 leads to a value of n0 = 6.12. When n0 is very large, Perror = 0, but the odds of evicting any variable value are very low. Thus, we employ the n0 values from Fig. 1, which allow eviction of variable values, with Pcorrect = 99.983–99.9988%.
Figure 1.

Values of n0 for 2 to 29 possible rotamers of a single residue that lead to elimination with high certainty. Each number of rotamers has an associated value of n0 (▵). The larger the number of rotamers, the smaller is n0. For each given number of rotamers and n0, the percentage of certainty is calculated (□).
(III) End of search.
Once there are less than M combinations remaining (M ≈ 105), an exhaustive search is conducted to yield the N lowest energy conformers of the protein.
Results
A Test of the Search Method's Validity.
To test the accuracy and efficiency of our method, we impose our stochastic algorithm to find the lowest energy combinations—given the constraints of the energy function and the rotamer library—and compare them to the results of an exhaustive search. We applied the stochastic algorithm to eight high-quality x-ray structures (resolution < 1.5 Å, R factor < 0.17) of proteins taken from the Protein Data Bank (45) with various sizes (54 to 263 residues) that were chosen to cover a range of protein-fold families as shown (Table 1). These proteins are: rubredoxin (5rxn) (46), ovomucoid third domain (2ovo) (47), erabutoxin B (3ebx) (48), ribosomal protein (1ctf) (49), ribosomal protein (1whi) (50), lysozyme (2ihl) (unpublished work), endonuclease (2end) (51) and hydrolase (1arb) (44). We limited the number of rotamers each residue could adopt by employing the most probable rotamers from the SCWRL backbone-dependent rotamer library (28), so that the exhaustive, full search calculation may end in a reasonable computer (CPU) time.
Table 1.
Systems selected for comparison
| Name | PDB code | Size | Number of combinations* | Number of combinations for comparing exhaustive and stochastic* | Average RMSD† for 1,000 lowest energy conformers, Å | Energy gap† in Kcal/mol between the 1,000th conformer and the global minimum | Residues‡ with different χ1 among lowest 1,000 energy conformers, % |
|---|---|---|---|---|---|---|---|
| Rubredoxin | 5rxn | 54 | 3.90 × 1027 | 1.26 × 109 | 2.20 | 1.64 | 14.6 |
| Ovomucoid third domain | 2ovo | 56 | 1.06 × 1025 | 8.49 × 107 | 2.03 | 3.64 | 16.7 |
| Erabutoxin B | 3ebx | 62 | 1.50 × 1031 | 6.37 × 108 | 2.50 | 1.35 | 12.3 |
| Ribosomal protein | 1ctf | 68 | 3.23 × 1034 | 3.58 × 108 | 2.33 | 3.28 | 6.4 |
| Ribosomal protein | 1whi | 122 | 4.97 × 1073 | 8.49 × 107 | 2.48 | 3.33 | 5.9 |
| Lysozyme | 2ihl | 129 | 2.17 × 1061 | 5.66 × 107 | 2.26 | 1.98 | 5.7 |
| Endonuclease | 2end | 137 | 1.31 × 1082 | 2.01 × 109 | 2.68 | 3.03 | 5.9 |
| Hydrolase | 1arb | 263 | 2.40 × 10105 | 1.61 × 109 | 2.24 | 0.55 | 3.0 |
After backbone clashes are relieved.
For a calculation with all the rotamers.
Except Gly and Ala.
Two stochastic searches were conducted for each test protein. A seed number of 100,000 was used for the first search and was replaced by 8,242,117 in the second. In Fig. 2 A and B, we compare the energies resulting from the two stochastic searches to the exhaustive one for the 1,000 low-energy conformations. When employing a seed number of 100,000, the low-energy conformations were identical by the stochastic and the exhaustive searches in all of the proteins except 1ctf. In 1ctf (Fig. 2A), the first 213 solutions were identical, and the 1,000th solution differed by 0.08 Kcal/mol. In the second stochastic search with a seed number of 8,242,117, the low energy conformations were identical in the stochastic and the exhaustive searches in all of the proteins except 2ovo (Fig. 2B), where the first 141 solutions were the same and the 1,000th solution differed by 0.15 Kcal/mol.
Figure 2.

Comparison of stochastic searches when employing two different seed numbers to an exhaustive search on two test proteins. Lowest energy conformations (1,000) are presented. Error is calculated as the energy difference between the given conformation in the stochastic and exhaustive searches. (A) Ribosomal protein (1ctf). (B) Ovomucoid third domain (2ovo).
The Search Method's Efficiency.
By applying our algorithm for rotamer prediction, computing time grows linearly and not exponentially, with an increase in the number of residues. The algorithm was applied to the eight proteins with an initial number of rotamer combinations that range from 1.06 × 1025 to 2.4 × 10105 as shown (Table 1). The ln(number of combinations) vs. the number of iterations is depicted (Fig. 3). The number of iterations to convergence ranged between 516 for 2ovo (1.06 × 1025 combinations) to 4,441 for 1arb (2.4 × 10105). The ratio between the combinations for these two proteins is 2.26 × 1080, whereas the ratio between the iterations was 8.6. The 129 residues of 2ihl required 1,894 iterations to end the stochastic stage, whereas the 263 residues of 1arb needed 4,440 iterations. The number of starting combinations for these two was 2.17 × 1061 vs. 2.40 × 10105, respectively.
Figure 3.

ln(number of combinations) vs. the number of iterations for eight proteins.
Comparison of the Algorithm to Experimental Results.
Results (Table 1) are given for the average RMSD (root mean square deviation) of the lowest energy populations of 1,000 conformations for each protein. The values are between 2.03 to 2.68. The lowest energy conformations (data not shown) did not have, in most proteins, the lowest RMSD to the x-ray structure. The energy gap between the lowest and highest energies among the low-energy populations ranges between 0.55 for 1arb to 3.64 for 2ovo. Among the thousand results, we find (last column in Table 1) that a relatively small number of residues (expressed as percentage) deviate from the crucial χ1, which is the angle closest to the backbone and affects most strongly the conformation of the side chain. We evaluated the number of side chains in each protein that adopted multiple positions by calculating the percentage of residues (except Ala and Gly) with different χ1 among the 1,000 lowest energy conformations found by our search method. An angle χ1 that deviates by 30° or more from the rest was considered as different. For example, rubredoxin contains (Table 1, line a) 48 residues that are neither Ala nor Gly. We found that seven residues exhibited χ1 deviations, thus 14.6%.
Discussion
We present a stochastic search technique and an example of its possible applications, exploring a given conformational space of proteins' side chains. The algorithm successfully explores the conformational space of various sizes of proteins and can deal with a large number of combinations after eliminating rotamers that clash with the backbone. The robustness of the stochastic algorithm in handling complex combinatorial searches is clearly demonstrated (Fig. 2 A and B and Fig. 3). Comparing it to an exhaustive search proves the reliability of the stochastic algorithm in reproducing most of the population of lowest energy conformations. In all proteins, the global minimum has been consistently detected. The 1,000 low-energy conformations were identical in the stochastic and the exhaustive searches in 14 of 16 comparisons, whereas 2 cases had a smaller set of lowest energy conformations that were identical in the two searches. Even in these cases, with 141/1,000 and 213/1,000 identical lowest conformations, the prevailing contributors to the molecular partition function are included, and may subsequently be used to estimate the conformational entropy. Indeed, Leach & Lemon (40) used low-energy rotamer combinations to evaluate the partition function and, thus, calculated the side chain contribution to the conformational entropy of the folded protein. One must bear in mind that both our method and Leach & Lemon's method are conducted in discrete space. A numeric comparison to entropy values obtained from continuous searches may give further insight into the reliability of a discrete search. Full conformational freedom of the backbone is required to extract real entropy values for proteins.
Table 1 presents the energy gaps between the 1,000th conformer and the global minimum of each protein. These energy differences indicate that the rotamers have a considerable degree of conformational flexibility that varies between the different proteins: it is 0.55 Kcal/mol for 1arb and 3.64 Kcal/mol for 2ovo. The energy gap variations between proteins may reflect the relative flexibilities of their side chains and should be studied further in connection with other indices of flexibility. Also, the lack of relations between protein size and energy gap warrants further examination.
The algorithm presented here belongs to the class of heuristic solutions. One of the tools used to assess the quality of our results is changing the seed number. Like other stochastic heuristic methods, our algorithm is not immune to such an effect. Nevertheless, we demonstrated that the algorithm found the global minimum in all of the proteins, when employing different seed numbers. Thus, the global minimum has been retained and not evicted in any of the large number of iterations for each of the proteins. In these test cases we have demonstrated that by combining two different seed numbers we succeeded in finding all of the required low-energy populations. Also, it should be noted that no accidental eviction of values is possible: each such eviction is a result of a systematic test. Those values that are not evicted remain for the final exhaustive step, in which all their combinations are evaluated. Thus, each one of the total of initial values must be probed and either evicted or retained for the final full search.
The hub of this work is a search methodology and neither a rotamer library nor a cost function. Most deviations from maximal accuracy may be caused by the size limitation of the rotamer library and the deficiencies of the energy function. Indeed, the current rotamer library's best possible RMSD for the tested proteins (found by positioning the rotamer that is closest to the x-ray structure) is between 0.94 Å to 1.52 Å. A way to overcome the search-space limitation was suggested by Mendes et al. (52); it presents a rotamer as a continuous ensemble of conformations that cluster around the classic rigid rotamer. A different approach to expanding the search space was recently devised by Honig and coworkers (19), which achieved accurate predictions by using an extensive rotamer library containing over 7,560 members, in which bond lengths and bond angles were taken from the database rather than simply assuming idealized values. Further, the performance of CHARMM (53) was better than that of AMBER in that work. The limitations of the force field are noticeable mostly in the fact that, in most proteins, the lowest energy conformations did not have the lowest RMSD from the x-ray structure.
Currently, there are four main methods to study the conformational space of a given protein: x-ray crystallography, NMR, molecular dynamics (MD), and rotamer library-based methods. Experimental information of biomolecular structure and conformations has its own limitations. X-ray crystallography usually supplies a single structure which reflects the biomolecule in the highly ordered crystal lattice, as opposed to the more physiologically relevant solution environment of an NMR structure. The former might be biased toward specific conformational substates in the crystal, which may not be among the ensemble of conformations in solution (54). Observation of alternate rotamers is beyond the detection limits of conventional x-ray crystallographic techniques, except at the very highest resolution. At least 10% of all side chains in proteins adopt multiple, discrete conformations in carefully refined crystal structures (55). MacArthur & Thornton (56) found a significant and unexpected correlation between χ1 mean values and resolution, mainly for small flexible side chains. All of the data support the hypothesis that this observation reflects local conformational flexibility and disorder, which at low resolution might be interpreted as a single distorted conformer.
We used the algorithm to explore the side chain conformational space of E. coli ribonuclease HI (57) and compared the results to experimental and theoretical methods that offer an insight into the multiple conformations that each side chain may adopt under different conditions: x-ray crystallography, NMR, and MD. Our algorithm found 82% of the multiple side chain conformers in this case (data not shown). The advantage of our algorithm is straightforward: it extends the single conformation into a population of viable conformations.
Unlike x-ray crystallography, NMR suggests alternative conformations by deciphering the two-dimensional and three-dimensional coupling maps (57, 58). NMR does not teach us about the shape of the energy minima on the potential energy surface. NMR of proteins is a long and tedious experiment limited by the time scale of conformational variations, especially in large proteins. In this case, our algorithm may be an additional tool for suggesting alternative conformations. When NMR structures are available, our algorithm may be used to extend this information by allowing the determination of the conformations' energy weights, thus enabling an assessment of their contribution to the overall population at equilibrium.
Classical MD simulations suggest conformations that may not be detected by NMR or by x-ray crystallography. With current technology, MD simulations of systems consisting of tens of thousands of atoms for a few nanoseconds are becoming more common (59). However, relevant time scales for biomolecular functions range from nanoseconds to more than seconds. The time required to reach an equilibrium between different conformers of a protein by MD is prohibitive for such simulations, and we may acquire only a glimpse of the protein's behavior in its surrounding. As a result, the ability of MD to detect the global minimum or the population of lowest-energy conformations in large biomolecules is limited. The reliability of our stochastic algorithm in finding both has been demonstrated in this article. Whereas MD trajectories imply a mechanism of conformational interconversions, our stochastic approach, like Monte Carlo, concentrates on products and not pathways, because of the employment of discrete values and its nondeterministic nature.
Dill and Chan (60, 61) suggested that the native state of a given protein corresponds to the global minimum in free energy, which is not necessarily the computed global minimum potential energy, even with a reliable function. The missing entropy evaluation may be contributed partially by our algorithm, as it yields most of the low-energy conformers. Our search offers, in addition to finding the global minimum, the next N best solutions for rotamers in large proteins without any mean field approximation and is unique in that sense. Thus, it may be used for studying thermodynamic properties of complex molecular systems. The stochastic algorithm can treat more than 250 residues (the maximum at this stage has been 2.29 × 10105 combinations, with no optimization of the CPU time), which is more than any algorithm known to us that is able to generate side chain populations and not single minima. Another advantage is in its ability to form populations by employing the stochastic algorithm in a stand-alone mode without any preprocessing algorithm (such as DEE, in the case of the A* algorithm). Also, one should note that the numbers of combinations presented (Table 1) for the stochastic algorithm refer to possible numbers of combinations that remain after evicting rotamers that clash with the backbone. Hence, the real number of possible combinations is much higher. This algorithm can be applied to other issues (62) of complex optimization.
It may be possible to simplify the combinatorial nature of the side chain problem and reduce it to pairwise (36, 37, 38) or to self-consistency (19) methods. However, such approaches cannot produce an accurate or close approximation to the ensemble of structures, the “best population” that may be crucial for the physical and biological characteristics of a protein. Our method, however, transcends the side chain issue that was used here as a test case. We regard our comparison of these heuristic search results to full exhaustive results as the most significant test of this method's performance and suggest it as a yardstick for future comparisons of methodologies in this field and others.
Our approach for finding low-energy minima of a complex biomolecular system is not necessarily limited to the life sciences. After adjusting the number of sampled solutions in each iteration (n) and cutoff points H and L in F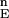 to the specific nature and complexity of the problem (i.e., the number of variable values, which is the number of rotamers in this example), this strategy may be used in other problems as long as the search space is discrete and a reliable or reasonable cost function may be used. This algorithm thus may evolve to be useful for other fields such as telecommunications (to design efficient networks), transportation, and economics.
to the specific nature and complexity of the problem (i.e., the number of variable values, which is the number of rotamers in this example), this strategy may be used in other problems as long as the search space is discrete and a reliable or reasonable cost function may be used. This algorithm thus may evolve to be useful for other fields such as telecommunications (to design efficient networks), transportation, and economics.
Acknowledgments
We thank Dr. Andrew Leach from GlaxoSmithKline for his prompt response and advice. This project was supported in part by the Israel Ministry of Trade and Industry, in the framework of the Daat (“knowledge”) consortium (Magnet project) and by a grant of the Israel Science Foundation established by the Israel Academy of Sciences and Humanities. Equipment was supplied by the Alex Grass Center for Drug Design and Synthesis of Novel Therapeutics at the School of Pharmacy, Hebrew University of Jerusalem.
Abbreviations
- DEE
Dead End Elimination
- MD
molecular dynamics
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Defay T, Cohen F E. Proteins Struct Funct Genet. 1995;23:431–445. doi: 10.1002/prot.340230317. [DOI] [PubMed] [Google Scholar]
- 2.Bahar I, Jernigan R. J Mol Biol. 1997;266:195–214. doi: 10.1006/jmbi.1996.0758. [DOI] [PubMed] [Google Scholar]
- 3.Wong K B, DeDecker B S, Freund S M V, Proctor M R, Bycroft M, Fersht A R. Proc Natl Acad Sci USA. 1999;96:8438–8442. doi: 10.1073/pnas.96.15.8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang E S, Koehl P, Levitt M, Pappu R V, Ponder J W. Proteins Struct Funct Genet. 1998;33:204–217. doi: 10.1002/(sici)1097-0134(19981101)33:2<204::aid-prot5>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 5.Blundell T L, Sibanda B L, Sternberg M J E, Thornton J M. Nature (London) 1987;326:347–352. doi: 10.1038/326347a0. [DOI] [PubMed] [Google Scholar]
- 6.Lee C, Subbiah S. J Mol Biol. 1991;217:373–388. doi: 10.1016/0022-2836(91)90550-p. [DOI] [PubMed] [Google Scholar]
- 7.Eisenmenger F, Argos P, Abagyan R. J Mol Biol. 1993;231:849–860. doi: 10.1006/jmbi.1993.1331. [DOI] [PubMed] [Google Scholar]
- 8.Roitberg A, Elber R. J Chem Phys. 1991;95:9277–9287. [Google Scholar]
- 9.Chandrasekaran R, Ramachandran G N. Int J Protein Res. 1970;2:223–233. [PubMed] [Google Scholar]
- 10.Lovell S C, Word J M, Richardson J S, Richardson D C. Proteins Struct Funct Genet. 2000;40:389–408. [PubMed] [Google Scholar]
- 11.Ponder J W, Richards F M. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 12.Gelin B R, Karplus M. Biochemistry. 1979;18:1256–1268. doi: 10.1021/bi00574a022. [DOI] [PubMed] [Google Scholar]
- 13.Dunbrack R L, Jr, Karplus M. Nat Struct Biol. 1994;1:334–340. doi: 10.1038/nsb0594-334. [DOI] [PubMed] [Google Scholar]
- 14.Cheng B, Nayeem A, Scheraga H A. J Comput Chem. 1996;17:1453–1480. [Google Scholar]
- 15.Holm L, Sander C. Proteins Struct Funct Genet. 1992;14:213–223. doi: 10.1002/prot.340140208. [DOI] [PubMed] [Google Scholar]
- 16.Laughton C A. J Mol Biol. 1994;235:1088–1097. doi: 10.1006/jmbi.1994.1059. [DOI] [PubMed] [Google Scholar]
- 17.Tanimura R, Kidera A, Nakamura H. Protein Sci. 1994;3:2358–2365. doi: 10.1002/pro.5560031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vasquez M. Biopolymers. 1995;36:53–70. [Google Scholar]
- 19.Xiang Z, Honig B. J Mol Biol. 2001;311:421–430. doi: 10.1006/jmbi.2001.4865. [DOI] [PubMed] [Google Scholar]
- 20.McGregor M J, Islam S A, Sternberg M J E. J Mol Biol. 1987;198:295–310. doi: 10.1016/0022-2836(87)90314-7. [DOI] [PubMed] [Google Scholar]
- 21.Dunbrack R L, Jr, Karplus M. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 22.Levitt M. J Mol Biol. 1992;226:507–533. doi: 10.1016/0022-2836(92)90964-l. [DOI] [PubMed] [Google Scholar]
- 23.Koehl P, Delarue M. J Mol Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 24.Wilson C, Gregoret L M, Agard D A. J Mol Biol. 1993;229:996–1006. doi: 10.1006/jmbi.1993.1100. [DOI] [PubMed] [Google Scholar]
- 25.Vasquez M. Curr Opin Struct Biol. 1996;6:217–221. doi: 10.1016/s0959-440x(96)80077-7. [DOI] [PubMed] [Google Scholar]
- 26.Sutcliffe M J, Hayes F R, Blundell T L. Protein Eng. 1987;1:385–392. doi: 10.1093/protein/1.5.385. [DOI] [PubMed] [Google Scholar]
- 27.Sali A, Blundell T L. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 28.Bower M J, Cohen F E, Dunbrack R L., Jr J Mol Biol. 1997;267:1268–1282. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- 29.Garcia K C, Degano M, Pease L R, Huang M, Peterson P A, Teyton L, Wilson I A. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- 30.Desmet J, Wilson I A, Joniau M, De Maeyer M, Lasters I. FASEB J. 1997;11:164–172. doi: 10.1096/fasebj.11.2.9039959. [DOI] [PubMed] [Google Scholar]
- 31.Hwang J K, Liao W F. Protein Eng. 1995;8:363–370. doi: 10.1093/protein/8.4.363. [DOI] [PubMed] [Google Scholar]
- 32.Pillardy A, Czaplewski C, Liwo A, Lee J, Ripoll D R, Kazmierkiewicz R, Oldziej S, Wedemeyer W J, Gibson K D, Arnautova Y A, et al. Proc Natl Acad Sci USA. 2001;98:2329–2333. doi: 10.1073/pnas.041609598. . (First Published February 20, 2001; 10.1073/pnas.041609598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuffery P, Etchebest C, Hazout S, Lavery R. J Biomol Struct Dyn. 1991;8:1267–1289. doi: 10.1080/07391102.1991.10507882. [DOI] [PubMed] [Google Scholar]
- 34.Bowie J U, Eisenberg D. Proc Natl Acad Sci USA. 1994;91:4436–4440. doi: 10.1073/pnas.91.10.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forrest S. Science. 1993;261:872–878. doi: 10.1126/science.8346439. [DOI] [PubMed] [Google Scholar]
- 36.Desmet J, De Maeyer M, Hazes B, Lasters I. Nature (London) 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 37.Lasters I, Desmet J. Protein Eng. 1993;6:717–722. doi: 10.1093/protein/6.7.717. [DOI] [PubMed] [Google Scholar]
- 38.Looger L L, Hellinga H W. J Mol Biol. 2001;307:429–445. doi: 10.1006/jmbi.2000.4424. [DOI] [PubMed] [Google Scholar]
- 39.Voigt C A, Gordon D B, Mayo S L. J Mol Biol. 2000;299:789–803. doi: 10.1006/jmbi.2000.3758. [DOI] [PubMed] [Google Scholar]
- 40.Leach A R, Lemon A P. Proteins Struct Funct Genet. 1998;33:227–239. doi: 10.1002/(sici)1097-0134(19981101)33:2<227::aid-prot7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 41.Dunbrack R L, Jr, Cohen F E. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiner S J, Kollman P A, Case D A, Singh U C, Ghio C, Alagona G, Profeta S, Jr, Weiner P. J Amer Chem Soc. 1984;106:765–784. [Google Scholar]
- 43.Pearlman D A, Case D A, Caldwell J W, Ross W S, Cheatham T E, III, Ferguson D M, Seibel G L, Singh U C, Weiner P K, Kollman P A. amber. San Francisco: Univ. of California; 1995. , version 4.1. [Google Scholar]
- 44.Tsunasawa S, Masaki T, Hirose M, Soejima M, Sakiyama F. J Biol Chem. 1989;264:3832–3839. [PubMed] [Google Scholar]
- 45.Berman H M, Westbrook J, Feng Z, Gilliland G, Bhat T N, Weissig H, Shindyalov I N, Bourne P E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watenpaugh K D, Sieker L C, Jensen L H. J Mol Biol. 1980;138:615–633. doi: 10.1016/s0022-2836(80)80020-9. [DOI] [PubMed] [Google Scholar]
- 47.Empie M W, Laskowski M., Jr Biochemistry. 1982;21:2274–2284. doi: 10.1021/bi00539a002. [DOI] [PubMed] [Google Scholar]
- 48.Smith J L, Corfield P W, Hendrickson W A, Low B W. Acta Crystallogr A. 1988;44:357–368. doi: 10.1107/s0108767388000303. [DOI] [PubMed] [Google Scholar]
- 49.Leijonmarck M, Liljas A. J Mol Biol. 1987;195:555–579. doi: 10.1016/0022-2836(87)90183-5. [DOI] [PubMed] [Google Scholar]
- 50.Davies C, White S W, Ramakrishnan V. Structure (London) 1996;4:55–66. doi: 10.1016/s0969-2126(96)00009-3. [DOI] [PubMed] [Google Scholar]
- 51.Morikawa K, Matsumoto O, Tsujimoto M, Katayanagi K, Ariyoshi M, Doi T, Ikehara M, Inaoka T, Ohtsuka E. Science. 1992;256:523–526. doi: 10.1126/science.1575827. [DOI] [PubMed] [Google Scholar]
- 52.Mendes J, Baptista A M, Carrondo M A, Soares C M. Proteins Struct Funct Genet. 1999;37:530–543. doi: 10.1002/(sici)1097-0134(19991201)37:4<530::aid-prot4>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 53.Brooks B R, Bruccoleri R E, Olafson B D, States D J, Swamirathan S, Karplus M. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 54.Brunger A T. Nat Struct Biol. 1997;4,Suppl.:862–865. [PubMed] [Google Scholar]
- 55.Smith J L, Hendrickson W A, Honzatko R B, Sheriff S. Biochemistry. 1986;25:5018–5027. doi: 10.1021/bi00366a008. [DOI] [PubMed] [Google Scholar]
- 56.MacArthur M W, Thornton J M. Acta Crystallogr D. 1999;55:994–1004. doi: 10.1107/s0907444999002231. [DOI] [PubMed] [Google Scholar]
- 57.Philippopoulos M, Lim C. Proteins Struct Funct Genet. 1999;36:87–110. doi: 10.1002/(sici)1097-0134(19990701)36:1<87::aid-prot8>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 58.Ellgaard L, Riek R, Herrman T, Guntert P, Braun D, Helenius A, Wutrich K. Proc Natl Acad Sci USA. 2001;98:3133–3138. doi: 10.1073/pnas.051630098. . (First Published March 6, 2001; 10.1073/pnas.051630098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sagui C, Darden T A. Annu Rev Biophys Biomol Struct. 1999;28:155–179. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 60.Chan H S, Dill K A. Proteins Struct Funct Genet. 1998;30:2–33. doi: 10.1002/(sici)1097-0134(19980101)30:1<2::aid-prot2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 61.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 62.Wales D J, Scheraga H A. Science. 1999;285:1368–1372. doi: 10.1126/science.285.5432.1368. [DOI] [PubMed] [Google Scholar]