

Abstract
PapD is a periplasmic chaperone essential for P pilus formation in pyelonephritic strains of E. coli. It is composed of two domains, each of which contains a tryptophan residue (Trp-36 and Trp-128, in the N- and C-terminal domains, respectively). To explore the role of domain–domain interactions during folding, the protein was labeled with 6-fluorotryptophan for use in 19F-NMR experiments. 19F-NMR data collected as a function of urea concentration revealed the presence of a resonance caused by Trp-128 that was distinct from either the folded or unfolded resonances. The time course of refolding from urea was monitored by stopped-flow fluorescence, CD, and 19F-NMR, each method showing multiple kinetic phases. The 19F-NMR stopped-flow spectra, collected at 70 μM of protein with a fluorine cryoprobe, demonstrated that the intermediate was populated early in the folding process (<5 s). The slow disappearance of the intermediate and unfolded resonance occurred at the same rate as the appearance of the native resonances of both domains. The data are consistent with a model in which the C-terminal domain collapses rapidly to an intermediate, whereas the stabilization of the final structure is slow and requires folding of the N-terminal domain with concomitant readjustment of the C-terminal domain structure.
Keywords: fluorescence‖circular dichroism‖folding intermediates‖P pili
The ability of pyelonephritic strains of E. coli to adhere to and to infect the kidney depends on the presence of long, hair-like structures called P pili on the surface of the bacterium (1). P pili are composite fibers consisting of an adhesive tip fibrillum joined to a thicker pilus rod. These composite fibers are assembled from subunits that are Ig-like domains without their C-terminal G β-strands. The folding of pilus subunits requires the presence of the periplasmic chaperone PapD (2). PapD is the prototype member of a superfamily of chaperones that are required for the assembly of diverse adhesive structures in a multitude of different Gram-negative pathogens. PapD-like chaperones facilitate the folding of pilus subunits by transiently donating their G1 β-strand from the N-terminal domain to complete the Ig fold of the subunit in a process called “donor strand complementation” (3, 4, 5). Because this G1 β-strand is necessary for both binding subunits and for acquiring the proper fold of the subunit (5), it becomes important to examine the folding of PapD itself.
19F-NMR is a powerful technique to study protein folding, as are other methods such as CD and fluorescence (6). Depending on the number and environments of the fluorine nuclei, the resonances are typically well resolved, facilitating the use of one-dimensional NMR to follow equilibrium or kinetic processes. Although highly electronegative, the fluorine atom is small (slightly larger than a hydrogen) and, thus, is expected to have little impact on the native structure. Furthermore, the high sensitivity of the fluorine nucleus to its local environment is advantageous for protein-folding studies, allowing the changes in local environment that can occur during the folding process to be monitored. Finally, the recent development of a 19F-NMR cryoprobe has facilitated the use of low concentrations (μM) of protein that resemble concentrations used in fluorescence and CD experiments.
PapD is a two-domain boomerang-shaped protein (Fig. 1A and ref. 3). The N-terminal domain exhibits the s-type Ig-like fold (7), whereas the C-terminal domain is structurally similar to the small α-amylase inhibitor tendamistat (8). PapD has one Trp residue in each of the two domains. Trp-36, a conserved residue among the pilin family of chaperones, is located along the C1 β-strand on the surface of the N-terminal domain, whereas Trp-128 in the C-terminal domain is mostly buried and is located near the domain–domain interface (2). Here, we describe the equilibrium and kinetic folding properties of 6-fluorotryptophan (6-19F-Trp)-labeled PapD measured by CD, fluorescence, and 19F-NMR. The folding properties as observed by these three techniques are similar. However, the 19F-NMR kinetic and equilibrium spectra show the presence of an intermediate resonance that is due to Trp-128. The intermediate is identified by a distinct chemical shift from both the folded and unfolded resonances, and stopped-flow 19F-NMR data indicate that this intermediate forms early in the folding process. The disappearance of the intermediate and unfolded resonance, and the appearance of both native Trp-36 and Trp-128 resonances, occurs slowly and at identical rates, indicating that final domain folding and association are coupled events. The presence of an intermediate at early times during refolding and at concentrations of urea that are above the folding transition suggests that the area around this residue may serve as a nucleation site for the folding of the protein. This nucleation is followed then by the slow folding of the N-terminal domain that, once folded, allows the cooperative interactions between both domains to form that stabilize the final folded structure.
Figure 1.

(A) The crystal structure of PapD showing the location of the two tryptophan residues in the N- and C-terminal domains (3, 27). (B) Far-UV CD spectra of PapD 19F-labeled (---) and unlabeled (—) in 30 mM Mops/HCl, pH 7.0. (C) Fluorescence (●) and CD (□) changes of PapD wt (Upper) and 19F-labeled (Lower) as a function of urea. The data are normalized to the fraction folded by using the standard equation: F = ([θ/F] − [θ/F]u)/([θ/F]n − [θ/F]u), where [θ/F] represents ellipticity at 233 nm/fluorescence emission at 350 nm for fully folded (n) or unfolded (u) states.
Materials and Methods
Chemicals.
Ultrapure urea was obtained from United States Biochemical. The concentration of urea was determined refractometrically at 25°C (9). 6-19F-Trp was obtained from Sigma. All other chemicals were reagent grade.
Site-Directed Mutagenesis.
Mutant proteins were made by using the splicing-by-overlap-extension-PCR method (10). The following primers were used to introduce mutations into PapD: W36F, coding strand, 5′GCTCAGGCATTTATAGAAAAT3′; noncoding, 5′ATTTTCTATAAATGCCTGAGC3′. W128F, coding strand, 5′AATGAAGTATTTCAGGACCAG3′; noncoding, 5′ CTGGTCCTGAAATACTTCATT3′. Mutations in PapD were confirmed by Big-Dye sequencing. All mutant PapD genes were cloned into vector pMMB91 under control of the IPTG-inducible pTac promoter, as described (11).
Production of Labeled and Unlabeled PapD.
E. coli strain W3110 trpA33 (ref. 12; auxotrophic for tryptophan) was used to produce 6-19F-Trp-labeled and unlabeled PapD, respectively. Labeling of PapD was done according to Hoeltzli and Frieden (13). PapD and the mutant proteins were purified from the periplasm as described (14). Labeling efficiency was determined by electrospray mass spectroscopy (Protein and Nucleic Acid Chemistry Laboratory, Washington Univ.). Concentrations were determined by using an extinction coefficient of 24,977 M−1 cm−1 as determined by the method of Pace and Schmid (15).
Fluorescence.
Equilibrium fluorescence data were acquired on an Alpha Scan PTI fluorometer (Photon Technologies, Ontario, Canada) using an excitation wavelength of 290 nm and an emission wavelength of 350 nm. The buffer used was 30 mM Mops/HCl at pH 7.0. For the kinetic experiments, an Applied Photophysics SX.18 MV (Applied Photophysics, Surrey, U.K.) was used. Excitation was at 290 nm, and fluorescence above 305 nm was observed by using a cutoff filter. Refolding was monitored by a 1:1 dilution of protein at 2 μM in 4.5 M urea into buffer. All experiments were performed in 30 mM Mops/HCl, pH 7.0, and the temperature for all experiments was kept at 20.0°C.
CD.
Equilibrium measurements were made with a Jasco-J715 spectropolarimeter with a cell of 1-mm pathlength. The temperature was controlled to 20.0°C. Spectra were recorded from 260 to 206 nm or lower, depending on the urea concentration, and were recorded by an average of eight scans. Response times were 2 s, and scan rates were 20 nm per m−1. A buffer baseline (30 mM Mops/HCl, pH 7) was subtracted from each spectrum. Final protein concentrations were ≈0.2 mg/ml. Kinetic experiments were performed by using an Applied Photophysics RX1000 rapid kinetics accessory with a 0.2 cm pathlength stopped-flow cell. Measurements were recorded at 233 nm using a 1-nm bandwidth. For data to 60 s, a response time of 0.125 s and a resolution of 2 s were used. The data represent an average of seven injections. For data to 4,000 s, a response time of 2 s and a resolution of 10 s were used.
19F-NMR Spectroscopy.
NMR spectra were recorded on a Varian Unity-Plus 500 MHz spectrometer operating at 470.3 MHz with either a Nalorac Proton/Fluorine 5-mm probe or a Varian Cryo-Q dedicated 5-mm 19F probe. The cryoprobe was maintained at 20°K with the Varian Cryo-Q Open Cycle Cryogenic system. The temperature of the sample was maintained at 20°C with the XRII851 Air-Jet Crystal Cooler heat exchanger (FTS Systems, Stone Ridge, NY). The 90o pulse width was 13 ms for the room temperature probe and 6.3 ms for the cryoprobe, and all spectra processed with 5 Hz line broadening. The T1 relaxation times were determined by the inversion recovery method. All spectra were referenced to an internal standard of 4-fluorophenylalanine (0.3 mM), and all samples contained 10% (vol/vol) D2O and 30 mM Mops/HCl, pH 7.0. No correction to pH with D2O in the sample was made. Stopped-flow NMR data were acquired with an Applied Photophysics RX1000 rapid kinetics accessory, as described (16). Folding was initiated by a 1:1 mixing of PapD denatured in 4.5 M urea/30 mM Mops/HCl, pH 7.0/10% (vol/vol) D2O/0.3 mM 4-fluorophenylalanine and buffer containing 30 mM Mops/HCl, (pH 7.0), 10% (vol/vol) D2O and 0.3 mM 4-fluorophenylalanine at 20°C. The stop syringe was set at 0.8 ml, which displaces >90% of the old solution (16). Data were recorded with a modification of the VNMR s2pul pulse sequence, as described (16). After injection and an initial delay of 1.5 s, 30 transients were recorded at 1.24 s apart, and 20 transients were recorded at 3.3 s apart. Data for each time point were recorded for 0.2 s. Sixty-four injections were made, and the data at each time point were summed. Kinetic data recorded at less than 4 × T1 (0.8 s) were corrected for T1, as described (17). For the long time course experiment, 16 transients were recorded for each data point, with a recycle time of 3.3 s. Four injections were made, and the resulting transients were summed as indicated above. The amplitudes of the resonances observed were determined with BAYESANALYZE (L. Bretthorst, Washington University; ref. 18).
Data Analysis.
Equilibrium transitions were fit to a two-state transition, as described (19). Data were fit by using the program KALEIDAGRAPH (Synergy Software, Reading, PA).
Results
Stability of Wild-Type and 6-19F-Tryptophan-Labeled PapD.
PapD was labeled biosynthetically with 6-19F-Trp, with a labeling efficiency that was greater than 90%, as determined by electrospray mass-spectrometry. Fig. 1B compares the far-UV CD spectrum of PapD wild-type (WT) and 6-19F-Trp-labeled proteins. The shape of the curves can be superimposed, suggesting little alteration in native secondary structure. Fig. 1C compares the equilibrium folding properties of the WT unlabeled to that of the labeled protein as a function of urea concentration by using fluorescence or CD at 233 nm. CD changes at 233 nm were used because the change at 216 nm (which reflects β-sheet structure) between folded and unfolded forms is too small to be accurately measured. The coincidence of the curves in Fig. 1C, fit to a two-state transition model (19), suggests that the CD measured at 233 nm is an accurate reflection of structural changes associated with denaturation and, thus, can be used for the stopped-flow experiments shown below.
19F-NMR Spectra as a Function of Urea Concentration.
Equilibrium unfolding of PapD was monitored by 19F-NMR as a function of urea. Fig. 2A shows representative spectra recorded at various urea concentrations. At 0 M urea, two resonances are observed that correspond to the completely folded forms of Trp-36 (−46.7 ppm) and Trp-128 (−43.66 ppm). The resonances were assigned by independently changing each Trp to a Phe. At 5 M urea, two resonances again were observed corresponding to Trp-36 (−46.37 ppm) and Trp-128 (−46.33 ppm). The close proximity of the folded Trp-36 resonance to its unfolded resonance (Δppm = 0.33) suggested that this resonance was likely caused by the solvent-exposed Trp-36. At 2.2 M urea, a resonance is observed at −45.5 ppm that is distinct from all of the folded and unfolded resonances. This resonance disappears only at 5 M urea, well beyond the midpoint of denaturation as determined by fluorescence or CD (Fig. 1C)
Figure 2.

(A) 19F-NMR spectra of PapD as a function of urea. Spectra were recorded at 20°C with 0.7 mM PapD in 30 mM Mops/HCl, pH 7.0/10% (vol/vol) D2O/0.3 mM 4-fluorophenylalanine (as an internal reference). The data represent 1,024 transients processed with 5-Hz line broadening. 19F-NMR spectra of PapD wild-type (WT) (B), mutants W128F (W128F) (C), and W36F (W36F) (D) at 0, 3, and 5 M urea. The data for the W36F mutant were obtained under the same conditions as in A, by using 0.15 mM PapD. The data for W128F were obtained as in A, by using 0.34 mM PapD, and represent 2,048 transients processed with 5-Hz line broadening.
To assign the −45.5-ppm resonance as well as the folded and unfolded resonances, site-directed mutants of PapD were made in which Trp-36 or Trp-128 was changed to phenylalanine. Then, these mutants were labeled with 6-19F-Trp, and the spectra were recorded at 0, 3, and 5 M urea, as shown in Fig. 2 B–D. At 3 M urea, the midpoint of the transition, the resonance at −45.5 ppm is observed in both the WT (Fig. 2B) and W36F (Fig. 2D) mutant but is not observed at 3 M urea in the W128F (Fig. 2C) mutant. Therefore, we have assigned this resonance to an intermediate of Trp-128.
The spectra obtained for the WT protein as a function of urea were deconvoluted by using Bayesian analysis to obtain the relative amplitudes of the resonances (18). Fig. 3 A and B show the amplitudes obtained by using Bayesian analysis for the various resonances as a function of urea. As with CD and fluorescence measurements, the transitions are highly cooperative, and the folding parameters also are summarized in Table 1. The midpoints of both Trp-36 and Trp-128 folded resonances are coincident, suggesting that the equilibrium unfolding of the two domains occurs concomitantly. However, the unfolded resonances, which are also coincident, have a higher transition midpoint than the folded resonances (Table 1). The peak amplitude of the resonance at −45.5 ppm, although small, is in between these two midpoints (≈3.2 M urea).
Figure 3.
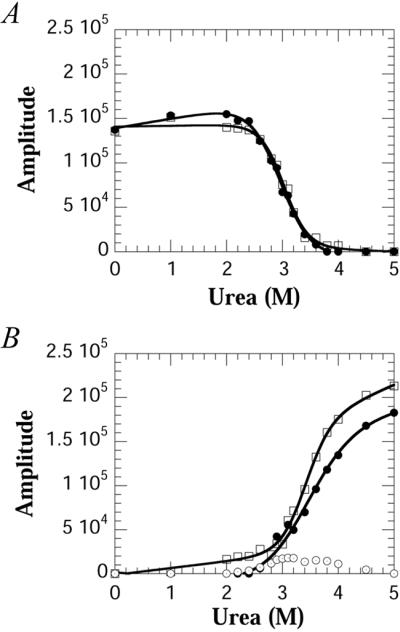
Amplitudes obtained by using Bayesian analysis of the data in Fig. 2A as a function of urea. (A) Amplitudes of the folded resonances of Trp-36 (□) and Trp-128 (●) (B) Amplitudes of the unfolded resonances of Trp-36 (□), Trp-128 (●), and the intermediate resonance of Trp-128 (⊙). Note in B the amplitude of the intermediate resonance which is present above the denaturation midpoint.
Table 1.
Fluorescence, CD, and 19F-NMR equilibrium and kinetic parameters of WT and 6-19F-tryptophan labeled PapD*
| Experiment | Equilibrium data†
|
Kinetic data‡
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| ΔG0,§ kcal⋅mol−1 | m, kcal⋅mol−1 M−1 | midpoint, M | k1 s−1 | k2 s−1 | k3 s−1 | ||||
| Fluorescence | |||||||||
| WT | 8.95 | 2.7 | 3.31 | 2.2 | 0.05 | 0.0012 | |||
| 6-19F-tryptophan | 9.3 | 2.98 | 3.12 | 2 | 0.03 | 0.0012 | |||
| Circular Dichroism | |||||||||
| WT | 8.06 | 2.43 | 3.31 | 1.24 | 0.03 | 0.00064 | |||
| 6-19F-tryptophan | 8.14 | 2.59 | 3.14 | 0.94 | 0.03 | 0.00063 | |||
ΔG ¶, kcal⋅mol−1 ¶, kcal⋅mol−1
|
mf, kcal⋅mol−1 M−1 | midpoint, M | ΔG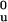 ,‖ kcal⋅mol−1 ,‖ kcal⋅mol−1
|
mu, kcal⋅mol−1 M−1 | midpoint, M | ||||
| 19F-NMR | |||||||||
| W128 resonances | 6.18 | 2.08 | 2.97 | 7.67 | 2.28 | 3.36 | nd** | 0.03‡‡ | 0.001†† |
| W36 resonances | 8.45 | 2.8 | 3.02 | 8.73 | 2.57 | 3.4 | nd** | 0.022§§ | 0.0011†† |
All experiments were performed in 30 mM Mops/HCl, pH 7.0 at 20°C.
ΔG0, the cooperativity index (m) value and midpoint were obtained from fitting the data to a two-state model (ref. 9).
Errors are less than 5% for all kinetic values.
Errors are <1.5 kcal mol−1, and <0.5 kcal mol−1 for the ΔG0 and m value, respectively.
Equilibrium data obtained from a two-state model fit to the folded (f) resonances.
Equilibrium data obtained from a two-state model fit to the unfolded (u) resonances.
nd, too fast to determine.
Value determined from the amplitude change in the intermediate resonance at −45.5 ppm.
Values for the intermediate, unfolded, and folded resonances of W128 and W36.
Value determined from the amplitude change in the unfolded resonance at −43.6 ppm.
Real-Time Refolding of PapD.
Because of the highly cooperative nature of the folding transition, intermediates in protein-folding studies are detected only transiently with kinetic techniques (20). To determine whether the intermediate observed at equilibrium also forms during the kinetic folding of PapD, the refolding of PapD was measured by using stopped-flow 19F-NMR in combination with stopped-flow fluorescence and CD. The kinetics of PapD folding, as measured by stopped-flow fluorescence and CD, are complex and exhibit at least four phases (J.G.B., G. Aronsson, and C.F., unpublished work). A shallow (1:1 mixing) urea jump from 4.5 to 2.25 M urea was chosen to slow the refolding process such that the third and fourth phases could be resolved with stopped-flow 19F-NMR (Fig. 4). The stopped-flow 19F-NMR data were obtained with 64 transients at a final protein concentration of 70 μM, a concentration made possible by the use of a fluorine cryoprobe. Final concentrations higher than 70 μM could not be used because a visible precipitate was observed upon dilution of the denatured protein in control experiments. At 70 μM, the sum of the intensities of all peaks was constant throughout the time course of the experiment, and thus all forms of the protein can be accounted for as part of the folding pathway (data not shown).
Figure 4.
Stopped-flow 19F-NMR, CD and fluorescence kinetic data of PapD on changing the urea concentration from 4.5 to 2.25 M urea. (A) Initial 19F-NMR spectrum of PapD obtained 1.24 s after mixing. The data represent 64 transients collected from 64 separate injections at a final concentration of 70 μM, 20°C. (B) Amplitudes of the kinetic data for the folded resonances of Trp-36 (□) and Trp-128 (●), the intermediate resonance of Trp-128 (⊙), and the unfolded resonance (♦), obtained by Bayesian analysis. The line through each curve represents the fit to a sum of two exponentials. (C) Time dependence of the CD change at 233 nm for WT (---) and 19F-labeled (—). (D) Time dependence of the fluorescence change WT (---) and 19F-labeled (—). The fluorescence data were normalized to the fraction folded by using the equation F = ([Fl] − [Fl]u)/([Fl]n − [Fl]u) where Fl represents the fluorescence.
At the earliest time point measured, 1.24 s, only two resonances are observed: the intermediate at −45.5 ppm and an unfolded resonance (Fig. 4A). Fig. 4B shows the relative amplitudes obtained by fitting the spectra at each time point by using Bayesian analysis. The unfolded resonance observed at the earliest time point was fitted as one peak, and the chemical shift (−46.4 ppm) matches that of Trp-36. Although there is high spectral overlap between both of the unfolded resonances, the analysis suggests that the majority of this resonance is caused by Trp-36. The 19F-NMR data at short times shows that the intermediate is highly populated within 5 s but increases in concentration up to 50 s with a rate constant of 0.03 s−1 (Fig. 4B and Table 1). This rate constant correlates with the rate constant observed on the same time scale for the decrease in amplitude of the unfolded resonance. It also correlates with the third phase observed by CD and fluorescence, in both the labeled and unlabeled protein (Table 1). The CD changes at 233 nm (Fig. 4C) also show that about 40% of the total amplitude occurs before the phases observed by NMR, suggesting that considerable structure is formed within 5 s.
A slow phase (k = 0.001 s−1) is observed for the disappearance of the intermediate, the disappearance of the unfolded resonances, and appearance of the two folded resonances. These changes also correlate with the slowest phase of folding as observed by CD or fluorescence (Fig. 4 C and D). Other experiments (double-jump, rate constants as a function of temperature, and experiments with the E. coli periplasmic proline isomerase PpiA) show that the final two phases are caused by the cis-trans isomerization of proline residues (J.G.B., G. Aronsson, and C.F., unpublished work). The rate constants obtained by using the three techniques are summarized in Table 1. The data show that the rate constants observed for both the labeled and unlabeled proteins as measured for a dilution of 4.5 to 2.25 M urea are the same, irrespective of the method used to measure folding. However, the data obtained with stopped-flow 19F-NMR strongly suggests that the area around Trp-128 has a substantial amount of secondary structure early on, presumably representing a partially collapsed form of the C-terminal domain.
Discussion
In this study, the equilibrium and kinetic folding properties of a two-domain protein, the E. coli periplasmic chaperone PapD, were elucidated by using 19F-NMR combined with CD and fluorescence methods. Although these three methods are complementary, only the 19F-NMR data revealed the existence of an intermediate resonance arising from Trp-128 in the C-terminal domain. This intermediate is present in both equilibrium and kinetic experiments. Because of the proximity of Trp-128 to the domain–domain interface, this tryptophan could potentially provide information on three distinct environments. These environments include the folded state, where the interdomain contacts of PapD are intact, an intermediate, where the C-terminal domain is still partially folded but the interdomain contacts are lost, and the unfolded state. Indeed, three distinct resonances are observed for Trp-128, indicating that three distinct environments are accessible to this residue.
Because of the high degree of overlap between the folded and unfolded resonances of Trp-36, an intermediate form for this residue cannot be ruled out. In contrast to the equilibrium data, however, the NMR kinetic data suggest that at the initial time point (1.24 s), only two resonances are represented, Trp-128 intermediate and the unfolded forms of Trp-36/128. The population of the intermediate at this time point is substantial, accounting for >40% of the total amplitude of Trp-128 observed. Although an intermediate form of Trp-36 may be present at early times as well, its population must be small compared with that of the intermediate of Trp-128.
From both the equilibrium and kinetic data presented, we propose a model in which the folding of PapD is initiated by a collapse of structure around Trp-128 in the C-terminal domain, giving rise to the intermediate resonance. This collapse corresponds to the large change observed by CD at early times, suggesting that a substantial amount (approximately half) of the secondary structure is formed. Subsequent to this step, the appearance of the folded resonances and disappearance of the intermediate and unfolded resonances occur concomitantly and at the same rate. This rate is likely due to the cis-trans isomerization of proline residues. PapD has a total of 16 proline residues, two of which are in the cis conformation in the native protein and are located in the N-terminal domain. This finding may explain why, at early times, we are unable to observe a resonance for Trp-36 other than its unfolded resonance, because the folding of the N-terminal domain is likely due to the trans->cis isomerization of one or both of these two cis proline residues. As this domain folds, the interface between the domains form, and thus the final native structure is stabilized.
These results also may explain why the N-terminal domain is unable to fold when expressed as an independent moiety (21). During secretion across the cytoplasmic membrane, the N-terminal domain is presumably first to emerge into the periplasm. The folding of the N-terminal domain may be prevented transiently because of its requirement for the C-terminal domain to fold. This mechanism differs from that of intramolecular chaperones in which an N-terminal propeptide, also required for folding, is cleaved off after the protein has folded (22, 23).
To our knowledge, this observation is the first direct report of an intermediate of a two-domain protein, either kinetically or at equilibrium. An intermediate involving a tryptophan residue, Trp-82, in equilibrium with the folded and unfolded forms of the intestinal fatty acid-binding protein, was implied through complete line-shape analysis (24). In a manner similar to PapD, this intermediate persisted at concentrations of denaturant well above the folding transition as monitored by fluorescence and CD, and it suggested that the region around this tryptophan is a likely nucleus for folding. Recent studies with 1H-NMR (25), or site-directed mutagenesis (26, 27), confirm that the area around this tryptophan is involved in an early nucleation event. Thus, the intermediate observed for PapD also could represent a nucleus for folding. Further studies similar to these on PapD will be required to understand the nature of the intermediate, as well as the forces that govern the cooperative interactions between the two domains. Undoubtedly, 19F-NMR has provided a starting point toward achieving these goals.
Acknowledgments
We thank Dr. Sydney Hoeltzli for advice and help in collecting the NMR stopped-flow data. We are grateful to Chia and Danielle Hung for help in constructing the mutants. We thank Göran Aronsson for the fluorescence data on WT PapD used in Fig. 1. This work was supported by National Institutes of Health Grant DK13332 and a Keck Fellowship Award (to J.G.B.).
Abbreviation
- WT
wild type
References
- 1.Hultgren S J, Jones S N, Normark S N. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, Curtis R III, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 2730–2756. [Google Scholar]
- 2.Hung D L, Knight S D, Woods R M, Pinkner J S, Hultgren S J. EMBO J. 1996;15:3792–3805. [PMC free article] [PubMed] [Google Scholar]
- 3.Sauer F G, Futterer K, Pinkner J S, Dodson K W, Hultgren S J, Waksman G. Science. 1999;285:1058–1061. doi: 10.1126/science.285.5430.1058. [DOI] [PubMed] [Google Scholar]
- 4.Choudhury D, Thompson A, Stojanoff V, Langermann S, Pinkner J, Hultgren S J, Knight S D. Science. 1999;285:1061–1066. doi: 10.1126/science.285.5430.1061. [DOI] [PubMed] [Google Scholar]
- 5.Barnhart M M, Pinkner J S, Soto G E, Sauer F G, Langermann S, Waksman G, Frieden C, Hultgren S J. Proc Natl Acad Sci USA. 2000;97:7709–7714. doi: 10.1073/pnas.130183897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerig J T. Prog Nucl Magn Reson Spectrosc. 1994;26:293–370. [Google Scholar]
- 7.Bork P, Holm L, Sander C. J Mol Biol. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- 8.Wiegand G, Epp O, Huber R. J Mol Biol. 1995;247:99–110. doi: 10.1006/jmbi.1994.0125. [DOI] [PubMed] [Google Scholar]
- 9.Pace C N, Scholtz J M. In: Protein Structure: A Practical Approach. Creighton T E, editor. Oxford: Oxford Univ. Press; 1997. pp. 299–321. [Google Scholar]
- 10.Morrison H G, Dessovires R C. Biotechniques. 1993;14:454–457. [PubMed] [Google Scholar]
- 11.Slonim L N, Pinkner J S, Branden C I, Hultgren S J. EMBO J. 1992;11:4747–4756. doi: 10.1002/j.1460-2075.1992.tb05580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drapeau G R, Brammar W J, Yanofsky C. J Mol Biol. 1968;35:357–367. doi: 10.1016/s0022-2836(68)80030-0. [DOI] [PubMed] [Google Scholar]
- 13.Hoeltzli S D, Frieden C. Biochemistry. 1996;35:16843–16851. doi: 10.1021/bi961896g. [DOI] [PubMed] [Google Scholar]
- 14.Lindberg F, Tennent J M, Hultgren S J, Lund B, Normark S. J Bacteriol. 1989;171:6052–6058. doi: 10.1128/jb.171.11.6052-6058.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pace C N, Schmid F X. In: Protein Structure: A Practical Approach. Creighton T E, editor. Oxford: Oxford Univ. Press; 1997. pp. 253–259. [Google Scholar]
- 16.Hoeltzli S D, Ropson I J, Frieden C. Tech Prot Chem. 1994;V:455–465. [Google Scholar]
- 17.Becker E D. Anal Chem. 1979;51:1413–1420. [Google Scholar]
- 18.Bretthorst G L. J Magn Reson. 1990;88:533–551. [Google Scholar]
- 19.Santoro M M, Bolen D W. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin R L. Curr Opin Struct Biol. 1993;3:84–91. [Google Scholar]
- 21.Hermanns U, Sebbel P, Eggli V, Glockshuber R. Biochemistry. 2000;39:11564–11570. doi: 10.1021/bi000549a. [DOI] [PubMed] [Google Scholar]
- 22.Silen J L, Frank D, Fujishige A, Bone R, Agard D A. J Bacteriol. 1989;171:1320–1325. doi: 10.1128/jb.171.3.1320-1325.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shinde U P, Liu J J, Inouye M. Nature (London) 1997;389:520–522. doi: 10.1038/39097. [DOI] [PubMed] [Google Scholar]
- 24.Ropson I J, Frieden C. Proc Natl Acad Sci USA. 1992;89:7222–7226. doi: 10.1073/pnas.89.15.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodsdon M E, Frieden C. Biochemistry. 2001;40:732–742. doi: 10.1021/bi001518i. [DOI] [PubMed] [Google Scholar]
- 26.Kim K, Ramanathan R, Frieden C. Protein Sci. 1997;6:364–372. doi: 10.1002/pro.5560060212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim K, Frieden C. Protein Sci. 1998;7:1821–1828. doi: 10.1002/pro.5560070818. [DOI] [PMC free article] [PubMed] [Google Scholar]