

Abstract
P‐glycoprotein (P‐gp, ABCB1) is a well‐researched ATP‐binding cassette (ABC) drug efflux transporter linked to the development of cancer multidrug resistance (MDR). Despite extensive studies, approved therapies to safely inhibit P‐gp in clinical settings are lacking, necessitating innovative strategies beyond conventional inhibitors or antibodies to reverse MDR. Photodynamic therapy is a globally approved cancer treatment that uses targeted, harmless red light to activate non‐toxic photosensitizers, confining its cytotoxic photochemical effects to disease sites while sparing healthy tissues. This study demonstrates that photodynamic priming (PDP), a sub‐cytotoxic photodynamic therapy process, can inhibit P‐gp function by modulating cellular respiration and ATP levels in light accessible regions. Using chemoresistant (VBL‐MDA‐MB‐231) and chemosensitive (MDA‐MB‐231) triple‐negative breast cancer cell lines, we showed that PDP decreases mitochondrial membrane potential by 54.4% ± 30.4 and reduces mitochondrial ATP production rates by 94.9% ± 3.46. Flow cytometry studies showed PDP can effectively improve the retention of P‐gp substrates (calcein) by up to 228.4% ± 156.3 in chemoresistant VBL‐MDA‐MB‐231 cells, but not in chemosensitive MDA‐MB‐231 cells. Further analysis revealed that PDP did not alter the cell surface expression level of P‐gp in VBL‐MDA‐MB‐231 cells. These findings indicate that PDP can reduce cellular ATP below the levels that is required for the function of P‐gp and improve intracellular substrate retention. We propose that PDP in combination with chemotherapy drugs, might improve the efficacy of chemotherapy and overcome cancer MDR.
Keywords: ABC transporter, cellular ATP levels, drug delivery, multidrug resistance, P‐glycoprotein, photodynamic priming
P‐glycoprotein (P‐gp) is a well‐researched ATP‐binding cassette (ABC) drug efflux transporter linked to the development of cancer multidrug resistance (MDR). Despite extensive studies, therapies to inhibit P‐gp in clinical settings have largely failed due to high toxicity and low selectivity, necessitating alternative strategies beyond conventional inhibitors or antibodies to reverse MDR. Photodynamic priming (PDP) provides an innovative strategy, harnessing sub‐therapeutic photochemistry to induce sub‐lethal effects in cancer cells with minimal toxicity and with high selectivity. Here, we show that PDP results in a decrease in mitochondrial and glycolytic ATP production, thereby decreasing P‐gp efflux function in chemoresistant cancer cells.
Abbreviations
- BPD
benzoporphyrin derivative
- BSA
bovine serum albumin
- DMEM
Dulbecco's Modified Eagle Medium
- FBS
fetal bovine serum
- IMDM
Iscove's Modified Dulbecco's Medium
- MDR
multidrug resistance
- OXPHOS
oxidative phosphorylation
- PDP
photodynamic priming
- PDT
photodynamic therapy
- P‐gp
P‐glycoprotein
- TMRE
tetramethyl rhodamine, ethyl ester
INTRODUCTION
ATP‐binding cassette (ABC) transporters comprise a group of membrane proteins found in both prokaryotic and eukaryotic organisms. 1 These transporters harness the energy generated from ATP binding and hydrolysis to transport a range of chemically diverse compounds across cellular membranes, defying concentration gradients. In mammals, ABC transporters are naturally present in excretory organs, serving as the initial defense against foreign substances in many tissues, including the intestine, liver, kidney, blood‐testis barrier, and placental barrier. 2 , 3 Consequently, they exert significant influence over the pharmacokinetic pathways of numerous drugs and govern systemic drug levels. Malignant cells, such as those in cancer, can inherently possess or acquire this detoxification mechanism, effectively effluxing a wide array of drugs from cells and rendering treatment ineffective. Among the 48 human ABC proteins, P‐glycoprotein (P‐gp, ABCB1) is known to be one of the crucial contributors to the development of multidrug resistance (MDR) in cancer. 4 , 5 , 6 , 7 Inhibiting P‐gp has become a pivotal strategy in combating cancer MDR. Despite more than four decades of research, the targeted inhibition of ABC transporters at specific sites of interest without causing harm to healthy cells has remained elusive with respect to clinical practice. 8
Efforts aimed at developing effective inhibitors to block the activity of ABC drug transporters have been underway for several decades. 1 , 9 The most extensively explored approach involves the use of small molecule inhibitors, including tyrosine kinase inhibitors. 10 , 11 , 12 , 13 Notably, some of these inhibitors, such as verapamil and cyclosporine A, also function as substrates for P‐gp. Certain inhibitors, such as cyclosporine A, have been observed to reduce the ATPase activity of P‐gp. 14 Despite three generations of inhibitors developed over a span of 30 years, most have proved to be either insufficiently potent or too toxic to non‐cancer tissues when administered in conjunction with chemotherapy. Consequently, their success in treating cancer patients has been limited. 1 An emerging approach involves the utilization of monoclonal antibodies, exemplified by the monoclonal anti‐P‐gp antibody UIC2, to inhibit P‐gp function. 15 , 16 , 17 , 18 This strategy involves antibody binding to the external loops of ABC transporters, effectively constraining them and preventing the efflux of substrates. However, it is important to note that antibodies typically bind to only 10–40% of the ABC transporters on the cell surface, resulting in partial inhibition. 15
Previous research has highlighted pronounced metabolic distinctions between chemoresistant and chemosensitive cancer cells, predominantly attributed to the overexpression of drug transporters. 19 This revelation opens a compelling avenue for the development of inhibitors. In line with the Warburg effect, cancer cells exhibit a distinct preference for glycolysis over oxidative phosphorylation (OXPHOS) as their primary means of ATP generation, driving the progression of the disease. 20 Recent insights have underscored the pivotal role of mitochondrial respiration in numerous cellular functions of cancer cells. Notably, Cavalli et al. 21 have observed that inhibiting OXPHOS heightens the sensitivity of cancer cells to treatment. Given that transporters are reliant on the availability of ATP and OXPHOS represents the most efficient ATP production pathway, exploring strategies to inhibit mitochondrial ATP production becomes an appealing avenue for disrupting transporter function and thereby preventing drug efflux.
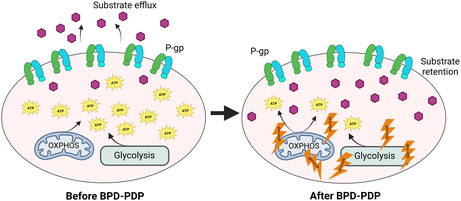
Understanding the importance of an approach that is locally selective with minimal systemic toxicity, photochemistry‐based approaches stand out as a clear first choice. Photodynamic therapy (PDT) employs the excitation of a photosensitizer at a certain wavelength of light, resulting in the generation of reactive oxygen species (ROS) to locally destroy tumors. Since light intensity diminishes exponentially in tissues and PDT‐induced damage is a threshold‐based phenomenon, cancer cells distant from the light source might not receive a sufficient PDT dose (a function of photosensitizer concentration and light dose) to undergo ablation. Depending on the tissue depth, the lethal PDT effects may not be observed. The non‐uniform distribution of photosensitizer and light, however, remains active, albeit at a sub‐lethal threshold. This leads to a “disturbance” of tissues remote from the light source, known as photodynamic priming (PDP). Instead of outright cell and tissue destruction, this disturbance primes them to be sensitized to respond better to future treatments, alter the tumor microenvironment or stimulate the immune system. Examples of resultant PDP reactions include decreasing stromal density for solid tumors, permeabilization of tumor vasculature causing significant improvements in drug delivery, and immune system stimulation and mobilization for immunogenic cell death. 22 , 23 , 24 , 25 These processes collectively enhance drug delivery to the target tissue. Examples of improving drug delivery using PDP demonstrated synergistic effects sensitizing drug‐resistant cancer cells to chemotherapy by directly modulating P‐gp and other ABC drug transporters. 18 , 26 , 27 , 28
Employing the photosensitizer benzoporphyrin derivative (BPD) with photochemistry has demonstrated its capability to depolarize the mitochondrial membrane potential. 29 This intriguing observation suggests a potential alternative approach to indirectly disrupt the function of P‐gp by influencing ATP production. In this study, we present the evidence supporting the feasibility of employing PDP to reduce cellular ATP levels, a critical factor required for the functionality of P‐gp, and to enhance the intracellular retention of substrates in chemoresistant cancer cells. Our investigation revealed that chemoresistant cancer cells exhibit a heightened baseline metabolic demand in comparison to their chemosensitive counterparts. Furthermore, we observed that PDP‐induced depolarization of the mitochondrial membrane leads to a reduction in mitochondrial ATP production. This alteration in cellular energetics correlates with an augmented retention of model drugs, which are recognized substrates of P‐gp. Collectively, these findings underscore the potential utility of PDP as a valuable tool that could be used to circumvent multidrug resistance in cancer.
MATERIALS AND METHODS
Chemicals
The fluorescent compounds calcein‐AM and rhodamine 123 were purchased from Invitrogen (Carlsbad, CA) and Sigma Aldrich (St. Louis, MO), respectively. The photosensitizer benzoporphyrin derivative (BPD) was purchased from U.S. Pharmacopeia (Rockville, MD) and was dissolved in dimethyl sulfoxide (DMSO), purchased from Sigma Aldrich (St. Louis, MO). The P‐gp‐specific monoclonal antibody MRK16 and IgG2a were obtained from Kyowa Medex Company (Tokyo, Japan). FITC‐labeled anti‐mouse secondary antibody IgG2a was obtained from BD Biosciences (San Jose, CA). All other chemicals and reagents were purchased from Thermo Fisher Scientific (Waltham, MA) or Sigma‐Aldrich (St. Louis, MO).
Cell culture
The human triple‐negative breast cancer (TNBC) cell line, MDA‐MB‐231, was obtained from the National Cancer Institute. 30 The subline VBL‐MDA‐MB‐231 was established by continuous culture of MDA‐MB‐231 cells in 100 ng/mL vinblastine. 31 MDA‐MB‐231 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) growth medium (Gibco), and VBL‐MDA‐MB‐231 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium (Gibco). Both cell culture media were supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. All cell lines were confirmed to be free of mycoplasma and cultured in 5% CO2 at 37°C.
Photodynamic priming, cytotoxicity, and mitochondrial membrane potential
Cells were seeded in a monolayer and allowed to incubate overnight in 5% CO2 at 37°C. The next day, PDP was performed using BPD as a photosensitizer. Cells were incubated with BPD (prepared in DMSO) in media at 37°C for 90 min. The cells were then washed twice with phosphate buffered saline (PBS) containing calcium and magnesium and fresh media was added to each well. PDP was performed at 690 nm and an irradiance of 10 mW/cm2 and a radiant exposure of 1 J/cm2 using ML6600 (Modulight Corp., Tampere, Finland). 32 Cells were then incubated in 5% CO2 at 37°C for 2 h postillumination.
The 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay (Invitrogen) was used to measure cytotoxicity after PDT, measured by decreases in enzyme activity. In brief, 20,000 cells/well were plated in a 96‐well plate. Cells were incubated with BPD photosensitizer (0–20 μM) for 90 minutes in 5% CO2 at 37°C. Cells were then washed twice with PBS containing calcium and magnesium, and fresh media was added to each well. To evaluate phototoxicity, cells were incubated with MTT solution (0.25 mg/mL) for 1 h. The solution was aspirated, and the formazan crystals were solubilized with DMSO. The absorbance was read at 570 nm using a plate reader (BioTek Synergy Neo2). The sublethal PDP dose was determined to be the concentration of BPD at the fixed light dose of 1 J/cm2 that resulted in decreases in enzyme activity of less than approximately 30% for the chemoresistant cells, based thresholds agreed upon by previous studies. 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42
Mitochondrial membrane potential depolarization was assessed using a tetramethylrhodamine, ethyl ester (TMRE) assay (Abcam). TMRE (300 nM) and the P‐gp inhibitor tariquidar (2.5 μM) were incubated with the cells for 2 h. The cells were washed with PBS containing 0.2% bovine serum albumin (BSA) twice and fresh PBS + 0.2% BSA solution was added to each well. TMRE fluorescence was measured using a plate reader (BioTek Synergy Neo2) and imaged on BioTek Lionheart Imager.
Seahorse assay
The Agilent Seahorse XF Real Time ATP Assay kit was used to measure glycolysis and oxidative phosphorylation as well as to calculate ATP production rates from each process over time and after stimulation from ETC inhibitors, oligomycin and rotenone/antimycin A. Twenty thousand cells were plated per well in a Seahorse XF96 cell culture microplate and were allowed to incubate overnight in 5% CO2 at 37°C. The Seahorse utility plate was hydrated overnight using Ultrapure water (Invitrogen) in 0% CO2 at 37°C and the XF calibrant was left overnight in 0% CO2 at 37°C. The following day, the XF calibrant was added to the utility plate and was left to incubate in 0% CO2 at 37°C until ready to load with inhibitors. Seahorse DMEM was supplemented with glutamate (2 mM), pyruvate (1 mM), and glucose (10 mM). BPD was incubated with cells (0–2 μM) in Seahorse DMEM for 90 min in 5% CO2 at 37°C. Cells were then washed twice with PBS containing calcium and magnesium, and fresh Seahorse DMEM media was added to each well. The cells were then incubated at 37°C for 2 h. Electron transport chain inhibitors, oligomycin and rotenone/antimycin A, were prepared at concentrations 15 μM and 5 μM, respectively, and were loaded onto the utility plate. After 2 h of incubation, the cells were then washed once with PBS containing calcium and magnesium, and fresh Seahorse DMEM was added to each well. The seahorse analyzer was calibrated with the utility plate, and then the cell microplate was loaded into the analyzer for measurement. Measurements were normalized to cell number. Briefly, cells were fixed in 4% neutral buffered formalin, and then nuclei were stained (NucBlue, Thermo Fisher) and imaged at 10X using the BioTek Lionheart imager before being counted using Gen5 software.
For cells being illuminated, after the 90‐min incubation with BPD, PDP was performed at 690 nm, an irradiance of 10 mW/cm2, and a radiant exposure of 1 J/cm2 via full plate illumination. The cells were then incubated in 0% CO2 at 37°C for 2 h post illumination. The same protocol was followed for analyzing the energetics with the Seahorse analyzer, collecting the data, and fixing the cells.
Total intracellular ATP content
Total ATP content in cells at steady state was determined using the ATPlite Luminescence Assay (Revvity, Perkin Elmer) according to the manufacturer's protocol. Briefly, MDA‐MB‐231 and VBL‐MDA‐MB‐231 cells were cultured overnight in a 96‐well microplate at a cell density of 20,000 cells per well. Cells were lyzed with assay lysis buffer (50 μL per well) at room temperature for 5 min before incubating with the assay substrate solution (50 uL per well) for an additional 15 min. Luminescence from cells was measured using the Tecan Spark Multimode Microplate Reader. Intracellular ATP concentration is determined from luminescence based on a known ATP concentration standard curve and normalized to a count of 20,000 cells.
Cell surface expression and transport function of P‐gp
Flow cytometry studies were performed to check the expression and function of P‐gp as described by Calcagno et al. 43 Briefly, 500,000 cells were plated in 35‐mm dishes (Thermo Fisher) and were allowed to incubate overnight in 5% CO2 at 37°C. The following day, BPD was incubated with cells (0–2 μM) for 90 min in 5% CO2 at 37°C. Cells were then washed twice with PBS containing calcium and magnesium, and fresh media was added to each well. PDP was performed at 690 nm with an irradiance of 10 mW/cm2 and a radiant exposure of 1 J/cm2. The cells were then incubated in 5% CO2 at 37°C for 2 h postillumination. Cells were washed once with PBS and were then trypsinized and collected in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 5% v/v fetal bovine serum (Gibco). Cells were counted (Cellometer Auto 2000) and 300,000 cells were aliquoted into 5 mL round bottom polystyrene tubes.
To investigate P‐gp expression, cells were incubated with the MRK16 monoclonal antibody or IgG2a for 1 h at 37°C. Cells were washed with cold IMDM by centrifuging at 1500 rpm for 5 min at 4°C. The cells were incubated with anti‐mouse FITC‐conjugated secondary antibody for 30 min at 37°C, were washed again in IMDM, then resuspended in PBS containing 1% BSA. Flow cytometry was performed using a FACS Canto II (BD) to determine surface expression of P‐gp.
To investigate P‐gp function, cells were incubated with P‐gp substrates: rhodamine 123 (1.3 μM) for 45 min or calcein‐AM (0.5 μM) for 10 min at 37°C. 44 After washing with cold IMDM, cells were resuspended in PBS containing 1% BSA. Flow cytometry was performed using a FACS Canto II (BD) to analyze substrate retention. This flow cytometry assay measures how much fluorescent substrate remains in the cell at steady state. A high fluorescent signal indicates high substrate retention, and a low fluorescence signal indicates low substrate retention due to P‐gp‐mediated efflux. 43 The selected substrates have been well characterized for this assay. 45 , 46 The flow cytometry data were analyzed using FlowJo software (Tree Star, Inc. Ashland, OR).
Cell membrane permeability
DAPI is used to label dead cells for exclusion in flow cytometry assays. 47 It is well established that photodamage promotes cell membrane permeability, likely due to necrosis. 48 , 49 , 50 DAPI is inefficiently permeable to cells with an intact plasma membrane, and since the photodamage promotes cell membrane permeability, DAPI was used with flow cytometry to investigate the effect of PDP on membrane permeabilization to determine the true effect of PDP on residual live cells. 47 , 51 The same protocol as described above was followed with a few changes. Cells were plated and treated with BPD and PDP and then were incubated with MRK16 and P‐gp substrates as mentioned above, but instead of 5 mL polystyrene tubes, 300,000 cells were aliquoted into a 96‐well plate to perform the assay. After washing, 200 μL PBS containing 1% BSA was added per well. A volume of 1 μL of 0.2 mg/mL DAPI (Sigma Aldrich) in DI water was added to each well (final concentration 0.001 μg/μL), and the flow cytometry was performed using a SonyID7000 spectral analyzer to analyze the nuclei staining with DAPI. The flow cytometry data were analyzed using FlowJo software (Tree Star, Inc. Ashland, OR).
RESULTS
Chemoresistant VBL‐MDA‐MB‐231 cells have a higher basal metabolic demand than parental chemosensitive MDA‐MB‐231 cells
The aim of this study was to understand the mechanism of PDP as an approach to overcome P‐gp mediated MDR through studying cellular energetics since P‐gp is ATP‐dependent. We began by characterizing the level of cell surface expression of P‐gp in a TNBC chemosensitive and chemoresistant (vinblastine‐resistant) cell pair to understand the energetic needs of each cell line. We reasoned that this would help establish an understanding of the existing ATP demand in parental and P‐gp‐expressing cells. To assess the expression of P‐gp in MDA‐MB‐231 and VBL‐MDA‐MB‐231 cells, cells were stained with human MRK16 P‐gp‐specific monoclonal antibody, and the IgG2a isotype was used as a control. Our data show that VBL‐MDA‐MB‐231 cells exhibit 9.45‐times higher P‐gp expression (Figure 1A) compared to MDA‐MB‐231 cells (Figure 1B) based on the FITC fluorescence intensity with MRK16, compared to IgG2a measured by flow cytometry (Figure 1C). We next used rhodamine 123, a substrate for P‐gp, as a molecular probe to study the MDR phenotype and P‐gp function in VBL‐MDA‐MB‐231 cells. Figure 1D,E shows that rhodamine 123 accumulation is 21.2 times lower in resistant VBL‐MDA‐MB‐231 cells compared to MDA‐MB‐231 cells due to active efflux of this substrate by P‐gp in VBL‐MB‐MDA‐231 cells.
FIGURE 1.
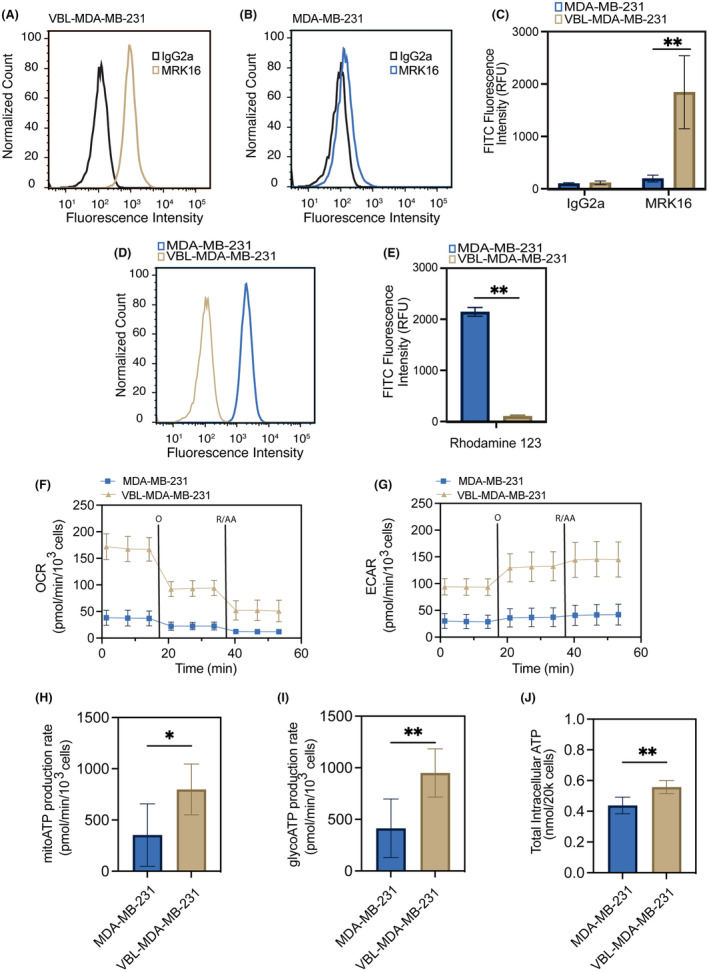
Chemoresistant cancer cells show increased cellular respiration and ATP production. Flow cytometry was used to analyze MDA‐MB‐231 (chemosensitive) and VBL‐MDA‐MB‐231 (chemoresistant) cells for cell surface P‐gp expression by P‐gp‐specific MRK16 antibody (fluorescent signal from FITC‐labeled secondary antibody) compared to the IgG2a negative isotype control (A–C) and P‐gp function using rhodamine 123 (D, E). (F) Oxygen consumption rate (OCR) and (G) extracellular acidification rate (ECAR) in response to sequential injection of oligomycin (O) and rotenone with antimycin A (R/AA). (H, I) Mitochondrial and glycolytic ATP production rates in MDA‐MB‐231 and VBL‐MDA‐MB‐231 cells were determined by Seahorse XF96 extracellular flux analyzer. VBL‐MDA‐MB‐231 cells exhibit increased ATP production rates compared to those of MDA‐MB‐231 cells. (J) Analysis of total intracellular ATP content by ATPLite luminescence assay reveals higher intracellular ATP content in VBL‐MDA‐MB‐231 cancer cells. Data presented as mean ± SD values of three independent experiments (*p ≤ 0.05, **p ≤ 0.01, two‐tailed t‐test).
Recognizing that overexpression of P‐gp requires high ATP production in cells, 52 we next characterized the baseline metabolic demand of VBL‐MDA‐MB‐231 and MDA‐MB‐231 cells. We used the Agilent Seahorse Real Time ATP assay to measure oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), allowing quantification of mitochondrial respiration and glycolysis. Our data show that VBL‐MDA‐MB‐231 cells have higher levels of OCR and ECAR compared to MDA‐MB‐231 cells (Figure 1F,G). Further analysis revealed that VBL‐MDA‐MB‐231 cells have a twofold higher mitochondrial (mito) ATP and glycolytic (glyco) ATP production rate, compared to MDA‐MB‐231 cells (Figure 1H,I). Finally, our results show that the total intracellular ATP content in VBL‐MDA‐MB‐231 cells was 1.27 times higher than in MDA‐MB‐231 cells (Figure 1J). Consistent with published reports, our results verify that VBL‐MDA‐MB‐231 cells with P‐gp overexpression are more effective in drug efflux, driven by increased intracellular ATP production.
Identifying the PDP dose and evaluating mitochondrial membrane depolarization in VBL‐MDA‐MB‐231 cells
A cytotoxicity assay using MTT reagent was used to assess the PDP efficacy. At time zero, there is no detectable photodamage to cells, however, 2‐h post‐illumination, photodamage‐induced cytotoxicity is expected to increase. At a fixed PDP light dose of 1 J/cm2, the IC50 of BPD for VBL‐MDA‐MB‐231 cells is 3.706 μM, which is almost three times higher than the IC50 of BPD for MDA‐MB‐231 (1.197 μM). At 1 J/cm2 and 2 μM BPD, PDP is cytotoxic to MDA‐MB‐231 cells, leading to approximately 75% photodamage‐induced cytotoxicity (Figure 2A), but results in approximately 30% photodamage‐induced cytotoxicity for VBL‐MDA‐MB‐231 cells (Figure 2B). This level of cell killing is within the appropriate range to evaluate sublethal effects of PDP on the remaining population, consistent with previous findings to evaluate sublethal effects. 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 We then found that BPD retention is more pronounced in MDA‐MB‐231 (Figure 2C) and significantly lower in VBL‐MDA‐MB‐231 by approximately twofold, due to P‐gp‐mediated efflux (Figure 2D). As expected, the addition of the P‐gp inhibitor, tariquidar, restores BPD retention in VBL‐MDA‐MB‐231 cells but not in MDA‐MB‐231 cells. Due to negative membrane potential, TMRE is known to accumulate in the mitochondria, so the TMRE assay was used to investigate PDP‐induced mitochondrial membrane potential depolarization. 53 Figure 2E,F shows that the PDP threshold of inducing mitochondrial membrane potential depolarization in MDA‐MB‐231 cells (1 J/cm2 × 1 μM) is at least twofold lower than that in resistant VBL‐MDA‐MB‐231 (1 J/cm2 × 2 μM) cells. This further verifies that VBL‐MDA‐MB‐231 requires a higher concentration of BPD because of its confirmed P‐gp substrate status.
FIGURE 2.
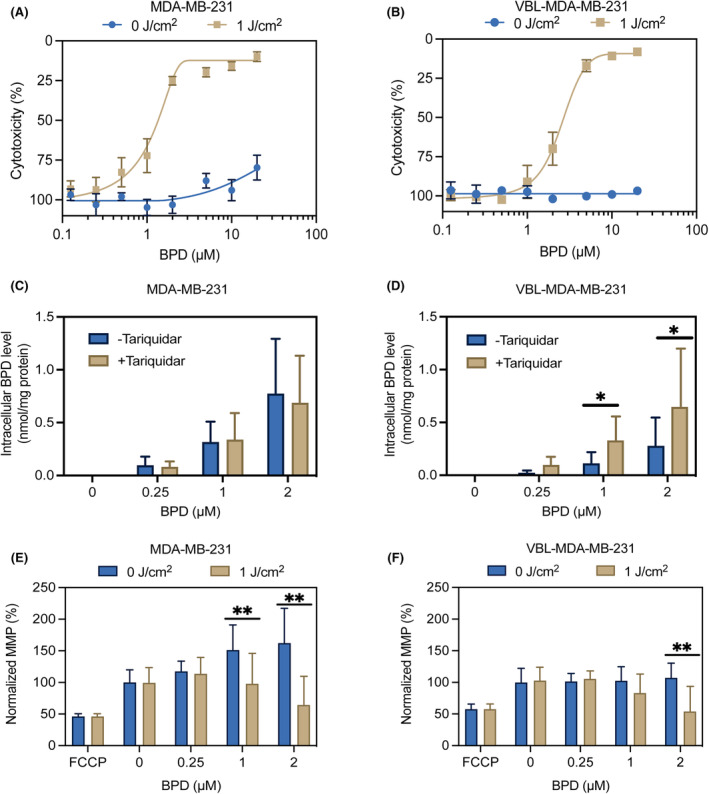
Photodynamic priming induces mitochondrial membrane potential depolarization. Cells were treated with BPD for 90 min prior to the PDP treatment. Cells were irradiated from the bottom up at 1 J/cm2 and then allowed to incubate for 2 h. Cytotoxicity, intracellular BPD concentration, and TMRE accumulations were measured 2‐h post‐PDP. Cytotoxicity was measured for (A) MDA‐MB‐231 and (B) VBL‐MDA‐MB‐231 cells using an MTT assay. BPD uptake was measured for both cell lines (C, D), with and without the P‐gp inhibitor, tariquidar, showing that BPD is a substrate for P‐gp. Mitochondrial membrane potential depolarization was measured in both cell lines (E, F) using a TMRE assay 2‐h post‐illumination (690 nm, 10 mW/cm2, 1 J/cm2). Data presented as mean ± SD values (n ≥ 3), (*p ≤ 0.05, **p ≤ 0.01, two‐tailed t‐test).
PDP decreases mitochondrial and glycolytic ATP levels in MDA‐MB‐231 and VBL‐MDA‐MB‐231 cell lines
The Agilent Seahorse Real Time ATP assay was used to evaluate changes in mitochondrial and glycolytic ATP production at 2 h after PDP. In the chemosensitive MDA‐MB‐231 cells, PDP at 1 J/cm2 and 0.25 μM BPD resulted in complete inhibition of mitochondrial ATP production (Figure 3A), but it did not significantly alter the glycolytic ATP production rate (Figure 3C). At a higher PDP dose of 1 J/cm2 and 1 μM BPD, a near complete inhibition of mitochondrial and glycolytic ATP production rates was observed in MDA‐MB‐231 cells. In resistant VBL‐MDA‐MB‐231 cells, the required PDP dose was two times higher (1 J/cm2 and 2 μM BPD) to induce complete inhibition of mitochondrial and glycolytic ATP production rates (Figure 3B,D), compared to MDA‐MB‐231 (1 J/cm2 and 1 μM BPD). Photosensitizer alone at 0.25–2 μM BPD did not significantly change the mitochondrial and glycolytic ATP production rates in the MDA‐MB‐231 and VBL‐MDA‐MB‐231 lines. Interestingly, light alone increased the mitochondrial and glycolytic ATP production rates in MDA‐MB‐231 cells by 1.56‐times and 2.05‐times, respectively. However, this effect with light alone was not observed in P‐gp‐expressing VBL‐MDA‐MB‐231 cells.
FIGURE 3.
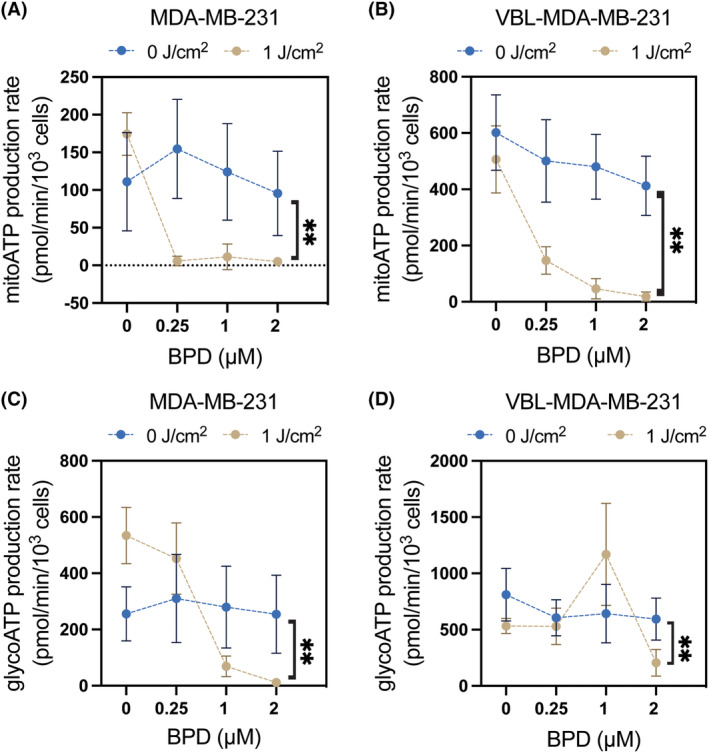
Photodynamic priming reduces mitochondrial and glycolytic ATP production rates. Cells were treated with BPD for 90 min prior to the PDP treatment. Cells were irradiated from the bottom up at 1 J/cm2 and then allowed to incubate for 2 h. Mitochondrial and glycolytic ATP production rates were measured using the Agilent Seahorse Real Time ATP Assay 2 h post‐PDP. Mitochondrial (A, B) and glycolytic (C, D) ATP production rates in MDA‐MB‐231 and VBL‐MDA‐MB‐231 cells were determined by Seahorse XF96 extracellular flux analyzer. Data presented as mean ± SD values (n ≥ 3, *p ≤ 0.05, **p ≤ 0.01, two‐tailed t‐test).
PDP did not significantly alter the P‐gp expression in cancer cells
We have previously shown that PDT could downregulate ABCG2 expression in pancreatic cancer cells. 28 Further investigation showed that BPD is a substrate for P‐gp and ABCG2 and that upon light activation, the proteins can be aggregated and rendered nonfunctional due to crosslinking, thus showing a decrease in their surface expression. 26 Here, we investigated if sub‐cytotoxic PDP would affect P‐gp expression in resistant VBL‐MDA‐MB‐231 cells to determine if direct P‐gp damage from crosslinking was responsible for any changes in efflux function. Using flow cytometry and the P‐gp‐specific monoclonal MRK16 antibody, Figure 4A,B shows that PDP at 1 J/cm2 and 2 μM BPD did not significantly alter P‐gp expression in MDA‐MB‐231 or VBL‐MDA‐MB‐231 cells. Quantification analysis of the fluorescent signal further revealed that light alone and PDP at 1 J/cm2 in the presence of 0–2 μM BPD did not significantly change the level of surface expression of P‐gp in both MDA‐MB‐231 and VBL‐MDA‐MB‐231 cells. These results suggest that any changes in P‐gp substrate retention are a result of modulation of the cellular energetics in the chemoresistant cell line compared to the chemosensitive cell line.
FIGURE 4.
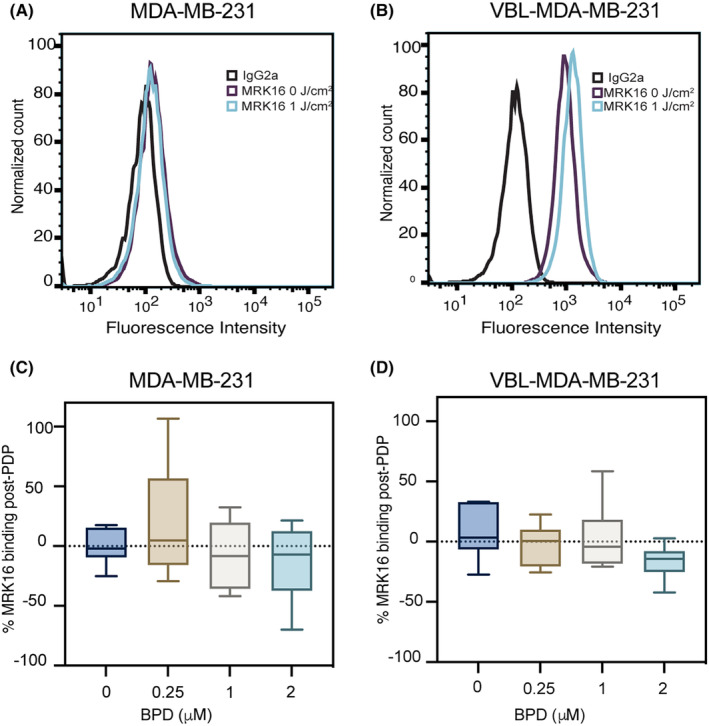
Photodynamic priming does not cause significant changes in the cell surface expression level of P‐gp. Flow cytometry was used to measure P‐gp expression using human P‐gp‐specific MRK16 antibody and IgG2a as a negative isotype control 2‐h post‐illumination (690 nm, 10 mW/cm2, 1 J/cm2) (A, B). MDA‐MB‐231 and VBL‐MDA‐MB‐231 cells exhibit no significant changes in fluorescence of MRK16 binding due to PDP (C, D). MRK16 binding was measured using a BD FACS Canto II flow cytometer and FITC fluorescence was analyzed using FlowJo software. Data presented as histogram, 2 μM BPD, 0 J/cm2 in red and 1 J/cm2 in orange for MDA‐MB‐231, 0 J/cm2 in light blue and 1 J/cm2 in dark blue for VBL‐MDA‐MB‐231, and values are given as mean ± SD (n > 3), ordinary one‐way ANOVA.
PDP increases P‐gp‐substrate retention in chemoresistant cancer cells but not in chemosensitive cancer cells
So far, we have shown that PDP at 1 J/cm2 and 2 μM BPD can decrease ATP production (Figure 3) but does not alter P‐gp expression (Figure 4) in resistant VBL‐MDA‐MB‐231 cells. Based on these results, we next tested if PDP could improve substrate accumulation by P‐gp in VBL‐MDA‐MB‐231 cells. Using flow cytometry and fluorescent P‐gp substrates (rhodamine 123 and calcein‐AM), we were able to assess the drug efflux function of P‐gp after sub‐cytotoxic PDT. We found that PDP at 1 J/cm2 and 2 μM BPD improves accumulation of rhodamine 123 and calcein (fluorescent product generated after removal of ester group of nonfluorescent calcein‐AM by cytoplasmic esterases) in VBL‐MDA‐MB‐231 cells by 150% (Figure 5A,B) and 228% (Figure 5F,G), respectively. Light alone (1 J/cm2) or a lower dose of PDP (1 J/cm2 with 0.25 μM or 1 μM) did not improve the accumulation of rhodamine 123 or calcein in VBL‐MDA‐MB‐231. DAPI staining showed that PDP resulted in 12% cell death in chemoresistant cells (Figure 5D) compared to its dark control (Figure 5C) at the highest concentration of BPD. Histograms of the overlapped, DAPI‐stained cell populations before (Figure 5H) and after PDP (Figure 5I) also confirm that this had no effect on rhodamine 123 or calcein retention in the chemoresistant cell line (Figure 5E,J).
FIGURE 5.
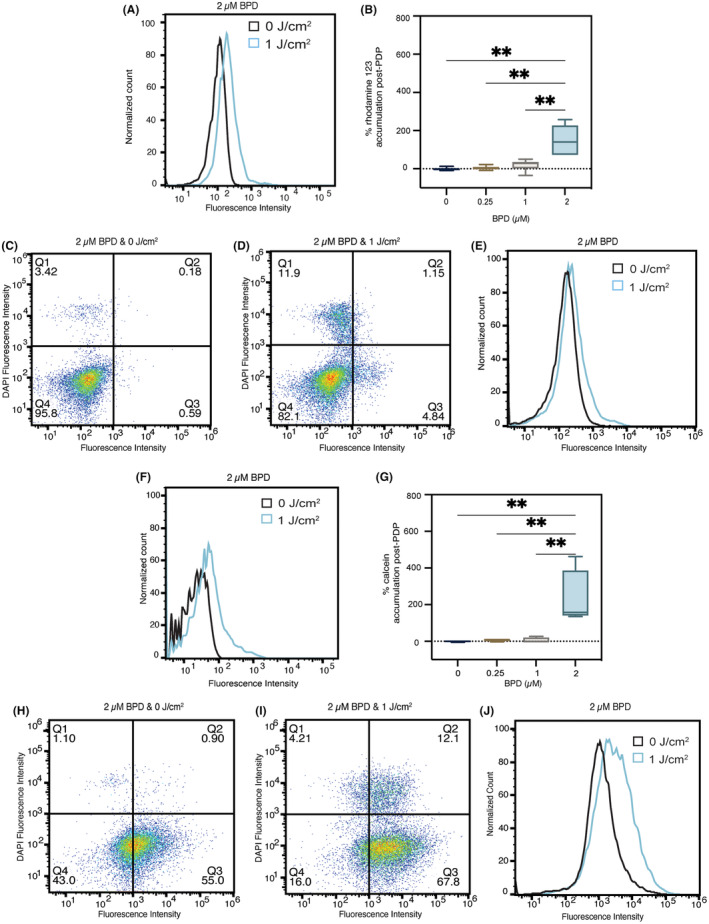
Photodynamic priming increases P‐gp substrate accumulation in chemoresistant cells. Flow cytometry was used to measure intracellular accumulation of rhodamine 123 and calcein generated from calcein‐AM. Accumulation of rhodamine 123 (A, B) and calcein (F, G) both increased in VBL‐MDA‐MB‐231 cells 2‐h post‐illumination. DAPI staining shows membrane permeability due to PDP treatment at 2 μM BPD and 1 J/cm2 (D (rhodamine 123), I calcein) compared to 2 μM BPD and 0 J/cm2 (C (rhodamine 123), H (calcein)). Overlapping histograms show substrate retention in cell populations with nuclear staining with DAPI (E (rhodamine 123), J (calcein)). Substrate accumulation was measured using a Sony ID7000 spectral analyzer and fluorescence was analyzed using FlowJo software. Data presented as histograms, with 2 μM BPD, 0 J/cm2 in black and 1 J/cm2 in blue and mean ± SD (n > 3), *p ≤ 0.05, **p ≤ 0.01, ordinary one‐way ANOVA.
In the chemosensitive MDA‐MB‐231 cell line, PDP at 1 J/cm2 in the presence of 0.25–2 μM is cytotoxic (Figure 6A,F) and did not significantly alter the accumulation of rhodamine 123 (p > 0.05) (Figure 6B) or calcein (Figure 6F). A PDP dose‐dependent decrease in calcein accumulation was observed in MDA‐MB‐231 cells, presumably due to membrane permeabilization and phototoxicity (Figure 6G). This was confirmed using DAPI staining, which requires membrane permeabilization to be internalized by cells. 51 The left shift in substrate fluorescence indicates a cell population with membrane permeabilization and increased DAPI uptake (>103 in DAPI fluorescence intensity) after PDP (Figure 6D,I) compared to its dark control (Figure 6C,H) at 2 μM BPD. Histograms of the overlapped, DAPI‐stained cell populations before and after PDP (Figure 6E,J), confirm that left shifts in fluorescence indicate cell populations that had increased levels of DAPI retention but showed no changes in substrate retention in the live cell population.
FIGURE 6.
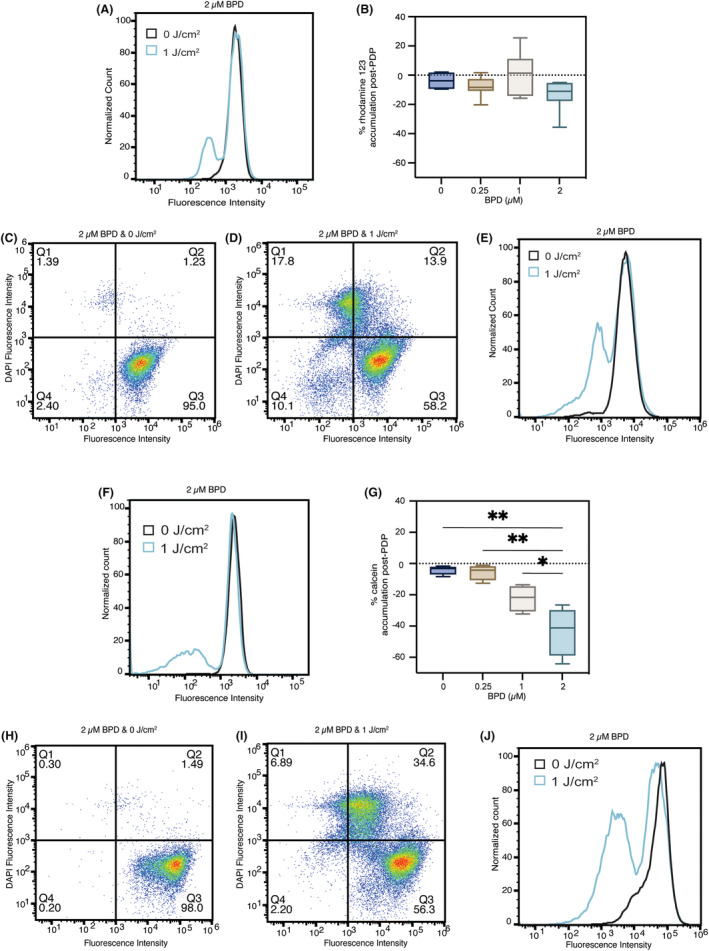
Photodynamic priming increases membrane permeability and decreases P‐gp substrate accumulation in chemosensitive cells. Flow cytometry was used to measure intracellular accumulation of rhodamine 123 and calcein. Accumulation of rhodamine 123 (A, B) and calcein (F, G) both decreased in MDA‐MB‐231 cells 2‐h post‐illumination. DAPI staining shows membrane permeability due to PDP treatment at 2 μM BPD and 1 J/cm2 (D (rhodamine 123), I (calcein)) compared to 2 μM BPD and 0 J/cm2 (C (rhodamine 123), H (calcein)). Overlapping histograms show substrate retention in cell populations with DAPI staining (E (rhodamine 123), J (calcein)). Substrate accumulation was measured using a Sony ID7000 spectral analyzer and fluorescence was analyzed using FlowJo software. Data presented as histograms, with 2 μM BPD, 0 J/cm2 in black and 1 J/cm2 in blue and mean ± SD (n > 3), *p ≤ 0.05, **p ≤ 0.01, ordinary one‐way ANOVA.
DISCUSSION
The phenomenon of MDR remains one of the most difficult challenges to successful cancer treatment in the clinic. Overexpression of P‐gp is seen in many human cancer types and is related to multidrug resistance and recurrence in cancers. 1 , 8 This overexpression can be intrinsic, but is often noticed post treatment with chemotherapeutic agents, making it difficult to kill cancer cells with typical chemotherapy. 54 Most inhibitors of P‐gp are too toxic to normal cells at the required dose, interact poorly with other chemotherapeutic drugs, or are non‐selective for ABC drug transporters present on the cancer cells alone. 8 Photochemical inhibition via PDP is an attractive strategy in the context of MDR because it is a more localized approach with minimal side effects. 22 Previous studies have used PDP as an investigative tool in different treatment contexts. PDP has been shown to be beneficial in increasing vessel permeability to improve drug delivery, and decreasing the incidence of distant metastases in an orthotopic mouse model. 27 More relevant to drug resistance in different cancers, PDP has also been investigated as a tool to decrease the function of the ABC drug transporters ABCB1 and ABCG2 by inducing protein crosslinking, causing the transporters to aggregate. 26 To our knowledge, PDP has never previously been explored in the context of cellular metabolism to overcome P‐gp‐mediated MDR.
TNBC makes up approximately 15% of all breast cancer cases. It is extremely aggressive, and patients tend to have poor survival rates due to the limited treatment options available. 55 PDT has been widely investigated for use to treat breast cancer. One study investigated PDT as a combination therapy with the CB2 agonist, JWH‐133, to inhibit tumor growth and found that the combination of PDT with CB2 agonist induced the most significant amount of cell death in MDA‐MB‐231 cells in vitro, and significantly decreased tumor volume and prolonged survival in vivo using the same cell line. 56 Sorrin et al. 25 found that the anti‐migratory activity of E‐type prostanoid receptor 4 (EP4), a major target implicated in breast cancer metastasis, can also be decreased through the combination of PDP using BPD. This evidence shows that while photochemical approaches are not enough on their own, photodynamic therapy and priming can be easily incorporated into the clinical workflow intraoperatively as an additional compliment to the treatment regimen. 57 In fact, many clinical trials highlight the feasibility of PDT in the clinic to treat breast cancer, featured in the review by Kim & Chang. 58 One phase I/IIa trial using BPD during a dose escalation study found that as the light dose increased from 20 J/cm2 to 50 J/cm2, histology found increasing markers of necrosis in patients in the PDT‐treated groups. Other sensitizers were also shown to be effective at treating breast cancer such as Temoporfin and Photofrin for chest wall invasion. In the trial with Photofrin, 67% of patients with >500 chest wall metastasis showed a complete or partial response to continuous low irradiance PDT. Although PDT has not yet been FDA approved to treat breast cancer, these studies exemplify its use as a stand‐alone treatment. Our work, however, further bolsters the evidence that PDP for TNBC has immense potential for clinical translation because of the different mechanisms through which it compliments combination treatments. PDP had never previously been investigated through the lens of cellular energetics and our study is the first in vitro proof of concept showing a BPD‐PDP synergy mechanism that is metabolism‐related using TNBC cell lines.
Previous assumptions that cancer cells use glycolysis as the main mode of ATP synthesis due to impaired mitochondrial function and overproduction of lactic acid have been challenged in recent years. 59 Studies have found that glycolysis in cancer cells inhibits OXPHOS, but mitochondrial function and OXPHOS‐mediated ATP generation can be restored if glycolysis is inhibited. 60 The process through which ATP is generated can change, since energy demand is a stimulus for energy production. 59 P‐gp overexpression increases the cell's demand for ATP, making chemoresistant cells an interesting model for developing treatments that target cellular metabolism. There is conflicting data in the literature on whether glycolytic or mitochondrial ATP plays a larger role in fueling ABC drug transporters. Various studies using glycolytic inhibition have proven that glycolysis is the main mode of ATP synthesis to fuel ABC drug transporters. 61 , 62 However, a more recent study has shown that mitochondrial respiration contributes to multiple functions in cancer cells, and that mitochondrial ATP is responsible for fueling P‐gp‐mediated drug efflux. 19 The use of photodynamic therapy is relevant in this context because photosensitizers can specifically affect the function of mitochondria, allowing us to unlock another level of utility in leveraging PDP in the clinic. The primary response of cells to PDP is cell death due to ROS generation. However, understanding that the light dose decreases exponentially as tissue depth increases, here we present one of the mechanisms of action by which a secondary, metabolism‐targeted response to PDP affects the residual live cells post‐treatment.
Photosensitizers are known to localize in the endoplasmic reticulum (ER), Golgi apparatus, plasma membrane, lysosomes, or mitochondria. 63 Photosensitizer selection could impact the mechanism of action for overcoming MDR using photochemical approaches. Since P‐gp is on the cell surface, if we used a photosensitizer that localized in the ER, we might induce damage in P‐gp synthesis to prevent mature, functional P‐gp translocation from the Golgi to the plasma membrane to prevent drug efflux. 64 Inward‐facing P‐gp is expressed on lysosomes and can be a major contributor to MDR by sequestering P‐gp substrates within the lysosomes to prevent their interaction with the intended molecular target. 65 , 66 , 67 Lysosome‐targeted photosensitizers could disrupt this sequestration to allow the drugs to localize properly. P‐gp is a transmembrane protein so disruption of the plasma membrane via plasma membrane‐targeted photosensitizers would render P‐gp non‐functional. Our previous work showed that BPD is a substrate for P‐gp and ABCG2, making it easy to target them with photochemistry to induce aggregation of the proteins and reductions in ATPase activity to inhibit their function when incubated with 2 μM BPD for 3 min at a low light dose (5 J/cm2). 26 We chose to use BPD as the photosensitizer because it is known to localize in the mitochondria and depolarize the mitochondrial membrane potential when illuminated, and our aim was to target ATP production to decrease P‐gp function. 29 Here, when we increase the incubation time with BPD to 90 min, we allow for the BPD to accumulate in the mitochondria, so that when illuminated, we can depolarize mitochondrial membrane potential by up to 60% in the parental chemosensitive cell line and 49.5% in the chemoresistant cell line. Since BPD is a substrate for P‐gp, we expected that using a higher concentration would be required to have an effect in the chemoresistant cell line, which is what our results show. Interestingly, we do not see the same result in the mitochondrial and glycolytic ATP production.
In the parental cell line, we observe significant increases in both mitochondrial and glycolytic ATP production rates at 0 μM BPD and 1 J/cm2, suggesting photo‐biomodulation effects. This is consistent with other studies showing that low fluences of red‐light photo‐stimulation can directly increase ATP production due to an increase in cytochrome c oxidase (COX) activity. 68 , 69 Interestingly, we do not observe this increase in ATP production in the chemoresistant cell line. COX, complex IV in the electron transport chain, has been reported to be a main photoreceptor in mitochondria using red light, which supports the observation of increased ATP. However, further experiments must be conducted to confirm this theory, as COX measurements may be inadequate as an indicator of increased ATP synthesis alone. 69 MitoATP production significantly decreases once we illuminate cells that have been treated with BPD in both cell lines, suggesting that PDP modulation of mitochondrial ATP production is not specific to P‐gp‐overexpressing cells. GlycoATP production also increased in the chemosensitive cell line at 0.25 μM BPD and 1 J/cm2, but then significantly decreases as we increase BPD concentration. This could suggest that 0.25 μM BPD at this light dose does not generate enough singlet oxygen to have an effect on glycolysis. We also did not observe any decrease in glycoATP production in the chemoresistant cell line until 2 μM BPD concentration, which suggests that the impact of BPD‐PDP is stronger on mitochondrial respiration versus glycolysis, although both modes are still implicated in ATP production in P‐gp over‐expressing cells.
Using MRK16 binding, we show that there are no significant changes in surface expression of P‐gp. MRK16 is an anti‐P‐gp antibody that binds to extracellular loops (ECLs) 1 and 4 of P‐gp and requires epitopes with amino acid residues on both the N‐ and C‐terminals of P‐gp for binding recognition. 70 , 71 We observe only slight decreases in MRK16 binding as we increase BPD concentration, suggesting that there is no damage to these specific loops of the extracellular domain of P‐gp due to PDP at this light dose and at these BPD concentrations. We previously showed that we can induce direct transporter damage by inducing crosslinking as a result of PDT with BPD, causing the transporter proteins to aggregate. 26 In the present study, we are using the same BPD concentration (2 μM), but we are using a much lower light dose, while our previous work used four times the fluence rate and five times the irradiance compared to this work. This emphasizes that PDP is a significantly gentler approach, while providing immense improvement in drug delivery.
We observed a significant increase in accumulation of rhodamine123 and calcein (due to decreased efflux of calcein‐AM) under the same PDP conditions (2 μM BPD, 1 J/cm2) in the chemoresistant cell line, suggesting that a decrease in ATP production results in reduction of function of P‐gp. We showed that at this BPD concentration and light dose, we are decreasing both mito‐ and glycoATP production, suggesting that ATP generated from both processes is necessary to fuel ABC drug transporter function. We also previously showed that when we increase the BPD concentration to 20 μM, and 1/20th of the fluence rate we used here, we can significantly reduce ATPase activity of ABCB1 and ABCG2, as a result of direct damage to these protein. 26 This shows that we can adjust the type of disruption to transporter function depending on the light doses and concentration of BPD. This is of great benefit because as we increase treatment depth, we see an exponential decrease in light dose, so although there would not be significant cytotoxicity in chemoresistant cells deeper within the tumor, we would still be able to increase the intracellular concentration of chemotherapeutics, thus achieving more cytotoxicity in these chemoresistant cells. We also showed that at this light dose, we observe approximately 75% cytotoxicity in the chemosensitive cells, demonstrating the twofold benefit of PDT and PDP in this context, where the non‐P‐gp‐expressing cell population is affected by the PDP treatment alone. This result speaks to the advantage of using a photochemical approach because it addresses the major treatment limitation of tumor heterogeneity. In this case, the different cell populations are all being affected by the light treatment, where the sensitive population is being destroyed directly, while the resistant population is being sensitized or “primed” for further treatment. A study from Mao et al. 18 highlights this importance where they use a mixed MDR tumor model by combining 3 T3 cells with P‐gp expressing KB‐8‐5‐11 to observe the PDT effect on drug accumulation in vivo. These tumors were implanted bilaterally in the flanks of mice, and one side received PDT using anti‐P‐gp‐conjugated IR700, while the other side was not illuminated as a dark control. This was followed by a single dose of Doxil 30 min later, on both sides. The results showed a 5.2‐fold increase in intracellular Doxil accumulation after PDT compared to the non‐PDT‐treated control. They also note that the Doxil preferentially killed the 3 T3 stromal cells whereas the P‐gp‐targeted PDT preferentially targets P‐gp expressing cells to increase the Doxil accumulation and thereby kill that cell population. Although we did not use a co‐culture model, we show individually that PDP can selectively increase substrate accumulation in P‐gp expressing cells by targeting cellular metabolism and while killing non‐Pgp‐expressing cells at the same light dose. This study from Mao et al. 18 also claims that a decrease in intracellular ATP is responsible for the synergistic effect of the combination of PDT and Doxil treatment, however they do not discuss the mechanism of subcellular organelle targeting as our study does using BPD as the chosen photosensitizer.
Based on this work and our previous work, we can now understand the effects of photodynamic priming on chemoresistant cells as a function of PDP dose. At the lowest PDP dose (0.125–1 J/cm2 × μM), we observe a decrease in P‐gp ATPase activity, which inhibits transporter function. As we increase from 1 to 10 J/cm2 × μM, we induce protein crosslinking, resulting in protein aggregation. Finally, at 2 J/cm2 × μM, we observe changes in ATP production due to mitochondrial modulation. Since tumors are heterogeneous, light dose and photosensitizer concentration distributions will not be the same throughout, but this study and our previous work show that we can achieve significant levels of improvement in ABC drug transporter inhibition using BPD‐PDP.
Although we show that PDP is a potential avenue for overcoming P‐gp‐mediated MDR, the main limitation of this approach is that the treatment effect is confined to areas that are being illuminated. This means that we will not see MDR control for distant or metastatic tumors. Many cancers, however, are diagnosed at an early stage, and if we treat the primary tumor with BPD‐PDP, we can improve drug delivery and hopefully reduce the metastatic potential, removing any need for distant tumor control as seen in multiple studies. 27 , 72 , 73 This was a main finding of the study by Huang et al. 27 where metastatic burden was observed in mice 60‐ and 120‐days post‐PDP treatment with a BPD nanoparticle to treat locally advanced pancreatic cancer (PDAC). The results showed complete inhibition of liver metastasis and significantly reduced distant organ metastases (<50 cancer cells) by day 60, and by day 120 distant metastases continued to reduce significantly. 27 In another study from Wang et al., 73 indocyanine green, (ICG) nanoparticles with anti‐PDL1 were being investigated in a metastatic breast cancer model (4 T1) to measure metastatic burden after PDP treatment of the primary tumor. This study found that metastatic lesions in the lungs decreased from approximately 10.8 ± 4.5 to 3.2 ± 3.4 lesions per lung. 73 These examples show that although the treatment is limited to the illuminated areas, the ability to improve drug delivery and control over distant metastases using PDP remains to be not only effective but also versatile in all the ways to gain tumor control in the context of MDR and overall treatment outcomes. Typical post‐surgical approaches include follow‐up rounds of chemotherapy, and including PDP in the surgical workflow is a promising method to ensure significant chemotherapy uptake in cancer cells that were not removed surgically. 74
CONCLUSION
We found that BPD‐PDP can reduce intracellular ATP levels required for the efflux function of P‐gp and improve intracellular substrate accumulation, which can aid in the development of new tools to overcome drug resistance in cancer. This is the first evidence of a novel way to induce stimulus‐responsive P‐gp inhibition using BPD‐PDP to improve substrate accumulation by up to twofold. Given the result of PDP affecting mitochondrial ATP more than glycolytic ATP, further studies of sub‐lethal PDP in vivo could enable more complete understanding of the Warburg effect and the contribution of mitochondrial ATP versus glycolytic ATP to fueling P‐gp function.
AUTHOR CONTRIBUTIONS
SA and HCH conceived the project and supervised the study, with help from AS. SA, HCH, AS, BL, and IR designed the experiments. IR and BL performed experiments and analyzed the data. IR, AS, SA, and HCH drafted the manuscript. SA, HCH and AS provided a critical revision of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
ACKNOWLEDGMENTS
We thank George Leiman and William Rhodes for editorial assistance. This work was supported by the NCI‐UMD Partnership for Integrative Cancer Research. Idrisa Rahman and Barry Liang were supported by the NCI‐UMD Partnership for Integrative Cancer Research and Andaleeb Sajid and Suresh V. Ambudkar were supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. This work utilized the NIH CCR/LGI Flow Cytometry Core, with training provided by Karen Wolcott. This work was also supported by the National Institutes of Health grants (R01CA260340, R21EB028508) and the National Science Foundation grant (2030253). We used Biorender.com to create the graphic for Graphical Abstract.
Rahman I, Liang B, Sajid A, Ambudkar SV, Huang H‐C. Photodynamic priming modulates cellular ATP levels to overcome P‐glycoprotein‐mediated drug efflux in chemoresistant triple‐negative breast cancer. Photochem Photobiol. 2025;101:188‐205. doi: 10.1111/php.13970
Contributor Information
Suresh V. Ambudkar, Email: ambudkar@mail.nih.gov.
Huang‐Chiao Huang, Email: hchuang@umd.edu.
DATA AVAILABILITY STATEMENT
All the data supporting the findings of this study are available within the article from the corresponding author.
REFERENCES
- 1. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug‐resistant cancer. Nat Rev Cancer. 2018;18(7):452‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dean M, Hamon Y, Chimini G. The human ATP‐binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42(7):1007‐1017. [PubMed] [Google Scholar]
- 3. Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55(1):3‐29. [DOI] [PubMed] [Google Scholar]
- 4. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP‐dependent transporters. Nat Rev Cancer. 2002;2(1):48‐58. [DOI] [PubMed] [Google Scholar]
- 5. Tamaki A, Ierano C, Szakacs G, Robey RW, Bates SE. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011;50(1):209‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9(1):105‐127. [DOI] [PubMed] [Google Scholar]
- 7. Robey RW, Massey PR, Amiri‐Kordestani L, Bates SE. ABC transporters: unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med Chem. 2010;10(8):625‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sajid A, Rahman H, Ambudkar SV. Advances in the structure, mechanism and targeting of chemoresistance‐linked ABC transporters. Nat Rev Cancer. 2023;23:762‐779. [DOI] [PubMed] [Google Scholar]
- 9. Theodoulou FL, Kerr ID. ABC transporter research: going strong 40 years on. Biochem Soc Trans. 2015;43(5):1033‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dai CL, Tiwari AK, Wu CP, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP‐binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68(19):7905‐7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mi YJ, Liang YJ, Huang HB, et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP‐binding cassette transporters. Cancer Res. 2010;70(20):7981‐7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tiwari AK, Sodani K, Wang SR, et al. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol. 2009;78(2):153‐161. [DOI] [PubMed] [Google Scholar]
- 13. Leonard GD, Polgar O, Bates SE. ABC transporters and inhibitors: new targets, new agents. Curr Opin Investig Drugs. 2002;3(11):1652‐1659. [PubMed] [Google Scholar]
- 14. Qadir M, O'Loughlin KL, Fricke SM, et al. Cyclosporin a is a broad‐spectrum multidrug resistance modulator. Clin Cancer Res. 2005;11(6):2320‐2326. [DOI] [PubMed] [Google Scholar]
- 15. Szalóki G, Krasznai ZT, Tóth Á, et al. The strong in vivo anti‐tumor effect of the UIC2 monoclonal antibody is the combined result of Pgp inhibition and antibody dependent cell‐mediated cytotoxicity. PLoS One. 2014;9(9):e107875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fowers KD, Kopeček J. Targeting of multidrug‐resistant human ovarian carcinoma cells with anti‐P‐glycoprotein antibody conjugates. Macromol Biosci. 2012;12(4):502‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao C, Qu P, Miley MJ, Zhao Y, Li Z, Ming X. P‐glycoprotein targeted photodynamic therapy of chemoresistant tumors using recombinant fab fragment conjugates. Biomater Sci. 2018;6(11):3063‐3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mao C, Li F, Zhao Y, Debinski W, Ming X. P‐glycoprotein‐targeted photodynamic therapy boosts cancer nanomedicine by priming tumor microenvironment. Theranostics. 2018;8(22):6274‐6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giddings EL, Champagne DP, Wu MH, et al. Mitochondrial ATP fuels ABC transporter‐mediated drug efflux in cancer chemoresistance. Nat Commun. 2021;12(1):2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cavalli LR, Varella‐Garcia M, Liang BC. Diminished tumorigenic phenotype after depletion of mitochondrial DNA1. Cell Growth Differ. 1997;8(11):1189‐1198. [PubMed] [Google Scholar]
- 22. de Silva P, Saad MA, Thomsen HC, Bano S, Ashraf S, Hasan T. Photodynamic therapy, priming and optical imaging: potential co‐conspirators in treatment design and optimization ‐ a Thomas Dougherty award for excellence in PDT paper. J Porphyr Phthalocyanines. 2020;24(11n12):1320‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhandari C, Moffat A, Fakhry J, et al. A single photodynamic priming protocol augments delivery of α‐PD‐L1 mAbs and induces immunogenic cell death in head and neck tumors. Photochem Photobiol. 2023;11:13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Silva P, Bano S, Pogue BW, Wang KK, Maytin EV, Hasan T. Photodynamic priming with triple‐receptor targeted nanoconjugates that trigger T cell‐mediated immune responses in a 3D in vitro heterocellular model of pancreatic cancer. Nanophotonics. 2021;10(12):3199‐3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sorrin AJ, Liu C, Cicalo J, et al. Photodynamic priming improves the anti‐migratory activity of prostaglandin E receptor 4 antagonist in cancer cells in vitro. Cancers (Basel). 2021;13(21):5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang BJ, Lusvarghi S, Ambudkar SV, Huang HC. Mechanistic insights into photodynamic regulation of adenosine 5′‐triphosphate‐binding cassette drug transporters. ACS Pharmacol Transl Sci. 2021;4(5):1578‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang HC, Rizvi I, Liu J, et al. Photodynamic priming mitigates chemotherapeutic selection pressures and improves drug delivery. Cancer Res. 2018;78(2):558‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang HC, Mallidi S, Liu J, et al. Photodynamic therapy synergizes with Irinotecan to overcome compensatory mechanisms and improve treatment outcomes in pancreatic cancer. Cancer Res. 2016;76(5):1066‐1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kessel D, Reiners JJ Jr. Enhanced efficacy of photodynamic therapy via a sequential targeting protocol. Photochem Photobiol. 2014;90(4):889‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cailleau R, Olivé M, Reeves WJ Jr. Breast tumor cell lines from pleural effusions. J Natl Cancer Inst. 1974;53(3):661‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huff LM, Lee JS, Robey RW, Fojo T. Characterization of gene rearrangements leading to activation of MDR‐1. J Biol Chem. 2006;281(48):36501‐36509. [DOI] [PubMed] [Google Scholar]
- 32. Baglo Y, Liang BJ, Robey RW, Ambudkar SV, Gottesman MM, Huang HC. Porphyrin‐lipid assemblies and nanovesicles overcome ABC transporter‐mediated photodynamic therapy resistance in cancer cells. Cancer Lett. 2019;457:110‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen Y, Li M, Sun F, et al. Low‐dose photodynamic therapy‐induced increase in the metastatic potential of pancreatic tumor cells and its blockade by simvastatin. J Photochem Photobiol B. 2020;207:111889. [DOI] [PubMed] [Google Scholar]
- 34. Saneesh Babu PS, Manu PM, Dhanya TJ, et al. Bis(3,5‐diiodo‐2,4,6‐trihydroxyphenyl)squaraine photodynamic therapy disrupts redox homeostasis and induce mitochondria‐mediated apoptosis in human breast cancer cells. Sci Rep. 2017;7:42126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kawczyk‐Krupka A, Sieroń‐Stołtny K, Latos W, et al. ALA‐induced photodynamic effect on vitality, apoptosis, and secretion of vascular endothelial growth factor (VEGF) by colon cancer cells in normoxic environment in vitro. Photodiagnosis Photodyn Ther. 2016;13:308‐315. [DOI] [PubMed] [Google Scholar]
- 36. Kawczyk‐Krupka A, Czuba ZP, Kwiatek B, Kwiatek S, Krupka M, Sieroń K. The effect of ALA‐PDT under normoxia and cobalt chloride (CoCl(2))‐induced hypoxia on adhesion molecules (ICAM‐1, VCAM‐1) secretion by colorectal cancer cells. Photodiagnosis Photodyn Ther. 2017;19:103‐115. [DOI] [PubMed] [Google Scholar]
- 37. Kawczyk‐Krupka A, Czuba Z, Latos W, et al. Influence of ALA‐mediated photodynamic therapy on secretion of interleukins 6, 8 and 10 by colon cancer cells in vitro. Photodiagnosis Photodyn Ther. 2018;22:137‐139. [DOI] [PubMed] [Google Scholar]
- 38. Udartseva OO, Zhidkova OV, Ezdakova MI, et al. Low‐dose photodynamic therapy promotes angiogenic potential and increases immunogenicity of human mesenchymal stromal cells. J Photochem Photobiol B. 2019;199:111596. [DOI] [PubMed] [Google Scholar]
- 39. Bulin AL, Broekgaarden M, Simeone D, Hasan T. Low dose photodynamic therapy harmonizes with radiation therapy to induce beneficial effects on pancreatic heterocellular spheroids. Oncotarget. 2019;10(27):2625‐2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Overchuk M, Harmatys KM, Sindhwani S, et al. Subtherapeutic photodynamic treatment facilitates tumor Nanomedicine delivery and overcomes Desmoplasia. Nano Lett. 2021;21(1):344‐352. [DOI] [PubMed] [Google Scholar]
- 41. Anbil S, Pigula M, Huang HC, et al. Vitamin D receptor activation and photodynamic priming enables durable low‐dose chemotherapy. Mol Cancer Ther. 2020;19(6):1308‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang F, Chopp M, Katakowski M, et al. Photodynamic therapy with Photofrin reduces invasiveness of malignant human glioma cells. Lasers Med Sci. 2002;17:280‐288. [DOI] [PubMed] [Google Scholar]
- 43. Calcagno AMK, Wu C‐P, Shukla S, Ambudkar SV. ABC drug transporters as molecular targets for the prevention of multidrug resistance and drug‐drug interactions. Curr Drug Deliv. 2007;4(4):324‐333. [DOI] [PubMed] [Google Scholar]
- 44. Sajid A, Lusvarghi S, Murakami M, et al. Reversing the direction of drug transport mediated by the human multidrug transporter P‐glycoprotein. Proc Natl Acad Sci U S A. 2020;117(47):29609‐29617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pe'triz J, Garcı'a‐Lo'pez J. Flow cytometric analysis of P‐glycoprotein function using rhodamine 123. Leukemia. 1997;11(7):1124‐1130. [DOI] [PubMed] [Google Scholar]
- 46. Legrand O, Simonin G, Perrot JY, Zittoun R, Marie JP. Both Pgp and MRP1 activities using calcein‐AM contribute to drug resistance in AML. Adv Exp Med Biol. 1999;3:161‐175. [DOI] [PubMed] [Google Scholar]
- 47. Wallberg F, Tenev T, Meier P. Analysis of apoptosis and necroptosis by fluorescence‐activated cell sorting. Cold Spring Harb Protoc. 2016;2016(4):87387. [DOI] [PubMed] [Google Scholar]
- 48. Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part two‐cellular signaling, cell metabolism and modes of cell death. Photodiagnosis Photodyn Ther. 2005;2(1):1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thompson SA, Aggarwal A, Singh S, Adam AP, Tome JPC, Drain CM. Compromising the plasma membrane as a secondary target in photodynamic therapy‐induced necrosis. Bioorg Med Chem. 2018;26(18):5224‐5228. [DOI] [PubMed] [Google Scholar]
- 50. Ros U, Pedrera L, Garcia‐Saez AJ. Partners in Crime: the interplay of proteins and membranes in regulated necrosis. Int J Mol Sci. 2020;21(7):2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chazotte B. Labeling nuclear DNA using DAPI. Cold Spring Harb Protoc. 2011;2011(1):5556. [DOI] [PubMed] [Google Scholar]
- 52. Seelig A. P‐glycoprotein: one mechanism, many tasks and the consequences for pharmacotherapy of cancers. Front Oncol. 2020;10:576559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. O'Reilly CM, Fogarty KE, Drummond RM, Tuft RA, Walsh JV. Quantitative analysis of spontaneous mitochondrial depolarizations. Biophys J. 2003;85(5):7‐3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bukowski K, Kciuk M, Kontek R. Mechanisms of multidrug resistance in cancer chemotherapy. Int J Mol Sci. 2020;21(9):3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Foulkes WD, Smith IE, Reis‐Filho JS. Triple negative breast cancer. N Engl J Med. 2010;363(20):10‐1948. [DOI] [PubMed] [Google Scholar]
- 56. Zhang J, Zhang S, Liu Y, et al. Combined CB2 receptor agonist and photodynamic therapy synergistically inhibit tumor growth in triple negative breast cancer. Photodiagnosis Photodyn Ther. 2018;24:185‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lamberti MJ, Vittar NB, Rivarola VA. Breast cancer as photodynamic therapy target: enhanced therapeutic efficiency by overview of tumor complexity. World J Clin Oncol. 2014;5(5):901‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim TE, Chang JE. Recent studies in photodynamic therapy for cancer treatment: from basic research to clinical trials. Pharmaceutics. 2023;15(9):2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zheng J. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (review). Oncol Lett. 2012;4(6):1151‐1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fantin VR, St‐Pierre J, Leder P. Attenuation of LDH‐A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425‐434. [DOI] [PubMed] [Google Scholar]
- 61. Nakano A, Tsuji D, Miki H, et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS One. 2011;6(11):e27222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bean JF, Qiu YY, Yu S, Clark S, Chu F, Madonna MB. Glycolysis inhibition and its effect in doxorubicin resistance in neuroblastoma. J Pediatr Surg. 2014;49(6):981‐984. [DOI] [PubMed] [Google Scholar]
- 63. Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one‐photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn Ther. 2004;1(4):279‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hano M, Tomášová L, Šereš M, Pavlíková L, Breier A, Sulová Z. Interplay between P‐glycoprotein expression and resistance to endoplasmic reticulum stressors. Molecules. 2018;23(2):227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yamagishi T, Sahni S, Sharp DM, Arvind A, Jansson PJ, Richardson DR. P‐glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem. 2013;288(44):31761‐31771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. al‐Akra L, Bae DH, Sahni S, et al. Tumor stressors induce two mechanisms of intracellular P‐glycoprotein‐mediated resistance that are overcome by lysosomal‐targeted thiosemicarbazones. J Biol Chem. 2018;293(10):3562‐3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu‐Kreyche P, Shen H, Marino AM, Iyer RA, Humphreys WG, Lai Y. Lysosomal P‐gp‐MDR1 confers drug resistance of Brentuximab Vedotin and its cytotoxic payload monomethyl Auristatin E in tumor cells. Front Pharmacol. 2019;10:749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ferraresi C, Hamblin MR, Parizotto NA. Low‐level laser (light) therapy (LLLT) on muscle tissue: performance, fatigue and repair benefited by the power of light. Photonics Lasers Med. 2012;1(4):267‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ferraresi C, Kaippert B, Avci P, et al. Low‐level laser (light) therapy increases mitochondrial membrane potential and ATP synthesis in C2C12 myotubes with a peak response at 3‐6 h. Photochem Photobiol. 2015;91(2):411‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vahedi S, Lusvarghi S, Pluchino K, et al. Mapping discontinuous epitopes for MRK‐16, UIC2 and 4E3 antibodies to extracellular loops 1 and 4 of human P‐glycoprotein. Sci Rep. 2018;8(1):12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nosol K, Romane K, Irobalieva RN, et al. Cryo‐EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc Natl Acad Sci U S A. 2020;117(42):26245‐26253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Banerjee SM, Acedo P, el Sheikh S, et al. Combination of verteporfin‐photodynamic therapy with 5‐aza‐2′‐deoxycytidine enhances the anti‐tumour immune response in triple negative breast cancer. Front Immunol. 2023;14:1188087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang D, Wang T, Yu H, et al. Engineering nanoparticles to locally activate T cells in the tumor microenvironment. Sci Immunol. 2019;4(37):6584. [DOI] [PubMed] [Google Scholar]
- 74. Komolibus K, Fisher C, Swartling J, Svanberg S, Svanberg K, Andersson‐Engels S. Perspectives on interstitial photodynamic therapy for malignant tumors. J Biomed Opt. 2021;26(7):70604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data supporting the findings of this study are available within the article from the corresponding author.