

ABSTRACT
Enhancing mesenchymal stromal cell (MSC) therapeutic efficacy through licensing with proinflammatory cytokines is now well established. We have previously shown that macrophage migration inhibitory factor (MIF)‐licensed MSCs exerted significantly enhanced therapeutic efficacy in reducing inflammation in house dust mite (HDM)‐driven allergic asthma. Soluble mediators released into the MSC secretome boast cytoprotective properties equal to those associated with the cell itself. In asthma, epithelial barrier damage caused by the inhalation of allergens like HDM drives goblet cell hyperplasia. Vascular endothelial growth factor (VEGF) plays a pivotal role in the repair and maintenance of airway epithelial integrity. Human bone marrow‐derived MSCs expressed the MIF receptors CD74, CXCR2, and CXCR4. Endogenous MIF from high MIF expressing CATT7 bone marrow‐derived macrophages increased MSC production of VEGF through the MIF CXCR4 chemokine receptor, where preincubation with CXCR4 inhibitor mitigated this effect. CATT7‐MIF licensed MSC conditioned media containing increased levels of VEGF significantly enhanced bronchial epithelial wound healing via migration and proliferation in vitro. Blocking VEGFR2 or the use of mitomycin C abrogated this effect. Furthermore, CATT7‐MIF MSC CM significantly decreased goblet cell hyperplasia after the HDM challenge in vivo. This was confirmed to be VEGF‐dependent, as the use of anti‐human VEGF neutralising antibody abrogated this effect. Overall, this study highlights that MIF‐licenced MSCs show enhanced production of VEGF, which has the capacity to repair the lung epithelium.
Keywords: airway epithelial, house dust mite, macrophage migration inhibitory factor, mesenchymal stromal cells, vascular endothelial growth factor
Licensing of MSCs with MIF requires CXCR4 receptor binding. Enhanced VEGF production by MIF‐licensed MSCs promotes wound healing in vitro and can be blocked using the VEGFR2 inhibitor SU‐5416. MIF‐licensed MSC conditioned medium significantly reduced goblet cell hyperplasia in house dust mite‐driven allergic airway inflammation in vivo.

1. Introduction
Mesenchymal stromal cells (MSCs) are renowned for their cytoprotective abilities elicited through secreted factors including miRNA [1], mitochondrial DNA [2, 3, 4], lipids (prostaglandin E2), extracellular vesicles (EV) [5, 6, 7], metabolites (kynurenine) and cytokines (TNF and IL‐6) [8, 9, 10]. Furthermore, newly emerging data outlines the importance of MSC‐derived apoptotic bodies in their therapeutic efficacy, making the secretome their main mechanism of action [11, 12, 13, 14, 15]. MSCs are known to elicit their anti‐apoptotic and pro‐regenerative effects through the production of vascular endothelial growth factor (VEGF) [16, 17, 18] and have been shown to enhance B cell survival in a VEGF‐dependent manner [19].
In preclinical models of lung disease, conditioned media from MSCs (MSC CM) has proven to be as potent as the cellular counterpart [5, 20, 21, 22, 23]. After lipopolysaccharide (LPS)‐induced injury, bone marrow‐derived MSC CM mitigated neutrophil influx and alternatively activated wound healing associated M2 alveolar macrophages, dampening lung injury in an IGF‐1 dependent manner [24]. Similarly, murine MSC CM rescued lung fibroblasts from a cigarette‐smoked‐induced lung injury, illustrating the positive role of MSC‐secreted factors in facilitating epithelial regeneration [22, 23, 25]. More recent studies also follow this narrative, demonstrating the potent effects of MSC CM in preclinical lung disease [26, 27, 28, 29].
Extensive literature illustrates the benefits of licensing MSCs prior to administration [30, 31, 32, 33, 34, 35, 36, 37], which can be carried out by modifying the environment in which these cells grow through exogenous stimulation, genetic manipulation, or even the addition of chemical agents. The asthmatic lung can act as a suitable environment for the activation of MSCs in vivo, as it contains a multitude of pro‐inflammatory cytokines including macrophage migration inhibitory factor (MIF) [34]. MIF has not only been found at elevated levels in the bronchoalveolar lavage fluid of asthma patients [38] but its level of expression has been linked to disease severity [39]. Low expression of MIF is associated with a low number of repeats of the tetranucleotide repeat polymorphism ‘CATT’ located within the promoter region of the MIF gene [40]. Using novel humanised MIF mice expressing the 7‐repeat allele termed CATT7, we have shown that high expression of human MIF drives airway inflammation following the house dust mite (HDM) challenge [41]. Our group has also demonstrated the ability of CATT7‐MIF licensing to enhance MSCs immunomodulatory effects in vivo [42].
Epithelial cells lining the airways play a key role in defence mechanisms against external pathogens and thus, disease [43]. In asthmatics, repetitive mechanical exacerbations due to inhaled agents or non‐specific stimuli can result in physical or biological injury of the airways and/or abnormal cycles of wound healing [44, 45, 46], where epithelial cell apoptosis and damage can drive further airway remodelling [47, 48]. MSCs have illustrated efficacy in resolving damage inflicted by these repeated exacerbations by repairing endothelial barrier integrity [49] and increasing wound healing [50].
This study sets out to investigate the capacity for MIF‐licensing to enhance the in vitro and in vivo cytoprotective functions of the MSC secretome and to identify the mechanisms involved using a clinically relevant HDM‐induced allergic airway model.
2. Materials and Methods
2.1. Ethical Approval and HPRA Compliance
All procedures involving the use of animals were carried out by licensed personnel. Ethical approval for all work was granted by the ethics committee of Maynooth University (BRESC‐2018‐13). Project authorisation was received from the HPRA (AE19124/P022), whereby animal experiments were carried out in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) criteria.
2.2. Cell Culture
Human bone marrow‐derived mesenchymal stromal cells (hBM‐MSCs) (RoosterBio Frederick, MD, USA) were expanded for two passages according to the manufacturer's instructions. Following this, MSCs were cultured and maintained in DMEM low glucose (Sigma‐Aldrich, Arklow, Wicklow, Ireland) supplemented with 10% (v/v) fetal bovine serum (BioSera, Cholet, France) and 1% (v/v) penicillin/streptomycin (Sigma‐Aldrich, Arklow, Wicklow, Ireland). Human alveolar epithelial cells (A549) and human normal bronchial epithelial cells (BEAS‐2B) were cultured in complete low glucose DMEM (Sigma‐Aldrich, Arklow, Wicklow, Ireland). All cells were incubated at 37°C/5% CO2/20% O2.
2.3. Generation of L929 Conditioned Media (M‐CSF)
L929 cells were thawed, seeded in complete RPMI‐1640 medium GlutaMAX (Gibco, Paisley, UK), and incubated at 37°C/5% CO2/20% O2 for 7 days. The supernatant was collected, centrifuged, and filtered (0.2 µm), and conditioned media containing M‐CSF was aliquoted and stored at −80°C. L929 conditioned media will be referred to as M‐CSF throughout the text.
2.4. Surface Staining of MIF Receptors
MSCs were seeded at 1 × 105 cells per well in six‐well plates. MSCs were stimulated with rhMIF (1, 10, or 100 ng/mL), or endogenous MIF (CATT7 CM) for 24 h. Cells were stained with CD74 (PE, BD Pharmingen, Berkshire, UK), CXCR2 (CD182) (PerCP‐eFluor 710, eBioscience, San Diego, CA, USA), or CXCR4 (CD184) (APC, eBioscience, San Diego, CA, USA) for 45 min. Cells were then washed in flow cytometry staining buffer and acquired using the Attune Nxt flow cytometer.
2.5. Animal Strains
A C57BL/6 mouse strain expressing the human high‐expression CATT7 MIF allele (MIFCATT7 [(C57BL/6NTac‐Miftm3884.1(MIF)Tac‐Tg(CAG‐Flpe)2Arte] mice) was created using vector‐based recombinant replacement of murine MIF by Taconic Biosciences (Rensselaer, NY, USA). The entire mouse MIF promoter has been deleted and replaced by inserting the human MIF promoter region. Validation of human but not murine MIF mRNA expression was verified by qPCR, and −794 CATT‐length dependent stimulated MIF production was confirmed in vivo [51]. Littermate wildtype (WT) and MIF−/− (MIF knockout) mice (R. Bucala, Yale School of Medicine, Yale University, New Haven, CT, USA) were used as controls. All mice were housed according to the HPRA SAP (Ireland) guidelines.
2.6. Model of HDM‐Induced Allergic Airway Inflammation
CATT7 transgenic humanised MIF mice were intranasally (I.N.) challenged with 25 µg of Dermatophagoides pteronyssinus (endotoxin content of 9937.5 EU/vial) (Greer Laboratories Inc, Lenoir, NC, USA) on days 0, 2, 4, 7, 9, 11, 14, 16, and 18.
2.7. Histological Analysis
On day 21 of the HDM model, lungs were harvested. Tissue was fixed in 10% (v/v) neutral buffered formalin (Sigma‐Aldrich) for 24 h, processed, and embedded in paraffin wax (Shandon Pathcentre, Runcorn, UK). For periodic acid Schiff (PAS) (Abcam, Cambridge, UK) staining, tissue sections (5 µm) were stained, air dried, and a coverslip mounted with DPX mounting media (Sigma‐Aldrich, Wicklow, Ireland). 20× images were taken using an Olympus BX51 light microscope. Images were scored by counting the number of PAS‐positive mucin‐producing goblet cells, relative to the diameter of the airway in a blinded manner.
2.8. Generation of CATT7 MIF Conditioned Media
Mice were humanely euthanised using the cervical dislocation technique on day 18, 4 h after the last HDM challenge. Bone marrow was isolated from the femur and tibia and centrifuged at 300g for 5 min and red blood cells were lysed (eBioscience, San Diego, CA, USA). Cells were seeded in two T175 flasks per mouse. Cells were grown for 72 h in complete RPMI‐1640 medium GlutaMAX (Gibco) supplemented with 20% L929 conditioned media. Supernatants were collected, centrifuged, and filter sterilised to remove cell debris. Aliquots were stored at −20°C. Aliquots were not freeze‐thawed. To account for the variability of human MIF levels between CATT7 mice and to verify that WT mice did not produce human MIF, supernatants were measured by human MIF ELISA (R&D) [41].
2.9. Generation of MSC Conditioned Media
hBM‐MSCs were cultured as described. Documented concentrations of rhMIF (1, 100, or 400 ng/mL) [52, 53] or conditioned media generated by bone marrow‐derived macrophages (BMDMs) from CATT7, WT, or MIF−/− mice were added with fresh cDMEM at a 1:1 ratio for 24 h. Cells were washed with warm PBS and replaced with serum‐free media. After 48 h, supernatants were collected and centrifuged to remove cell debris. Aliquots were stored at −20°C.
2.10. Generation of MIF Inhibited SCD‐19 Conditioned Media
SCD‐19 (3‐(2‐methylphenyl)‐1H‐isochromen‐1‐one) (Specs.net, the Netherlands) was reconstituted in 70% ethanol and diluted in PBS to a working concentration of 100 µM. SCD‐19 was added to BMDM‐derived CATT7 and WT supernatant for 1 h in a shaking incubator at 37°C before the supernatants were added at a 1:1 ratio into flasks containing human BM‐MSCs.
2.11. Wound Healing Assay
The underside of a six‐well plate (Sarstedt, Nümbrecht, Germany) was scratched with three horizontal lines using a scalpel and a ruler to allow for accurate analysis. A549 or BEAS‐2B cells were seeded out at a density of 1 × 105/mL/well. When cells are 60–80% confluent, a single perpendicular vertical scratch was made with a sterile p200 tip. Wells were washed with warm PBS to remove cell debris. cDMEM and MIF‐MSC conditioned media were added in a 1:1 ratio. On day 0, baseline measurements (100% open) were taken using Optika imaging software and a Nikon imaging microscope. Plates were incubated at 37°C/5% CO2/20% O2 for 48 h, or until one scratch had sufficiently closed. Cells were fixed with 10% neutral buffered formalin for 8 min, air dried, and stained with crystal violet (Sigma) for 4 min. ImageJ software was used to measure the percentage of wound closure of each image (Figure S1), relative to the 100% baseline measurements taken on day 0 (Table S1).
2.12. Use of VEGFR2 Inhibitor SU‐5416
To investigate if VEGF was facilitating MSCs’ ability to enhance wound closure in A549 and BEAS‐2B epithelial cells, a VEGFR2 inhibitor SU‐5416 (Tocris) was used. 10 µM of SU‐5416 or a DMSO vehicle control was added to A549 or BEAS‐2B cells for 4 h before the scratch was created and conditioned media was added.
2.13. Use of Cell Cycle Inhibitor Mitomycin C
BEAS‐2B cells were exposed to 10 µg/mL Mitomycin C (MMC), for 2 h, before being washed off with PBS. Cells were then scratched and a conditioned medium was added.
2.14. Preparation of CATT7 MSC CM for In Vivo Administration
MSC CM and CATT7 CM were generated as previously described. To concentrate the levels of human VEGF present, Amicon Ultra‐0.5 Centrifugal Filter Units (Sigma‐Aldrich) with a molecular weight cut‐off of 50 kDa were used as per the manufacturer's instructions. An anti‐human VEGF neutralising antibody (Bevacizumab Biosimilar) or human IgG1 isotype control was added to the concentrated conditioned media. 30 µL of conditioned media was administered intranasally on day 14 of the HDM model.
2.15. VEGF Elisa
MIF‐licensed MSC supernatants were collected and centrifuged at 300g for 5 min to remove debris, before being stored at −20°C. ELISAs were carried out according to the manufacturer's instructions (R&D Systems).
2.16. Gene Expression Analysis
At the 48 h timepoint, the cells were also harvested for gene expression studies of genes that indicate proliferation: pcna and mib1/ki67 (detailed in Table S2). Total RNA was extracted using TRIzol (Ambion Life Sciences) according to the manufacturer's instructions. cDNA synthesis was performed using the manufacturer's instructions (Quantobio cDNA synthesis kit). Real‐time‐polymerase chain reaction (RT‐PCR) was carried out using PerfeCta SYBR Green FastMix (Quantbio). Expression was quantified in relation to the housekeeper gene HPRT using the ΔCT method. The fold change in the relative gene expression was determined by calculating the 2−ΔΔCT values.
2.17. Statistical Analysis
Mice were randomised. Observers assessing end‐points were blinded to group assignments. Data for individual animals and independent experiments are presented as individual symbols. All data are presented as mean ± SEM. Results of two or more groups were compared by one‐way or two‐way analysis of variance (ANOVA) followed by the post hoc Tukey's multiple comparison test. GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA) was used for all statistical analyses.
3. Results
3.1. Human BM‐MSCs Constitutively Express Canonical and Non‐Canonical MIF Receptors
To explore the interaction between MIF and MSCs, we sought to determine whether MSCs express the classical (CD74) and non‐classical MIF receptors (CXCR2 and CXCR4) by flow cytometric analysis (Figure 1A). MSCs constitutively expressed MIF receptors CD74, CXCR2, and CXCR4, with CXCR4 most highly expressed (Figure 1). Interestingly, there was large inter‐donor variability in CXCR4 expression and this aligns with other studies [54].
FIGURE 1.
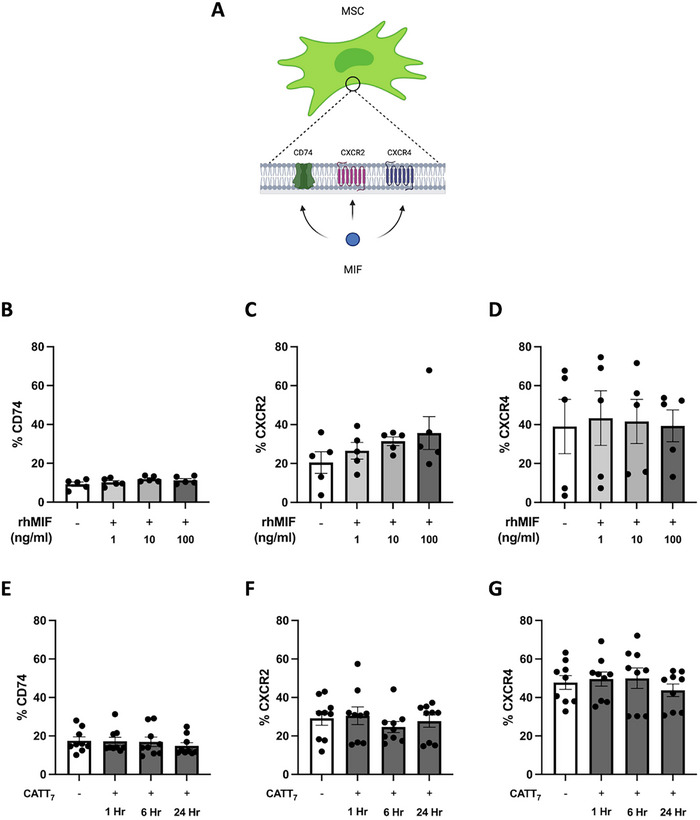
Human BM‐MSCs constitutively express canonical and non‐canonical MIF receptors. A, Human BM‐MSCs expressing MIF receptors CD74, CXCR2, and CXCR4 on their cell surface. MSCs were treated with different concentrations of rhMIF (1, 10, and 100 ng/mL) for 6 h, and percentage of (B) CD74, (C) CXCR2, (D) CXCR4 expression were measured by flow cytometry. Additionally, MSCs were treated with endogenous hMIF generated from BMDMs from CATT7 HDM challenged mice (CATT7 CM) where the percentage of (E) CD74, (F) CXCR2, (G) CXCR4 expression was assessed after endogenous hMIF stimulation at 6, 12, and 24 h timepoints. Data are presented as mean ± SEM and represent three independent experiments; no statistically significant differences were found by one‐way ANOVA.
Next, we investigated if MIF binding influences receptor expression. Recombinant human MIF (rhMIF) did not affect the MSC expression levels of MIF receptors CD74 (Figure 1B), CXCR2 (Figure 1C), or CXCR4 (Figure 1D). We also utilised endogenous MIF from our humanised CATT7 MIF mice as an alternative method of licensing MSCs (Figure 2A), a process which we have previously shown significantly enhances MSC therapeutic efficacy [42]. As expected, CATT7 mice that express the high MIF expression allele produce significantly higher levels of human MIF compared with WT controls (Figure 2B). Similar to the observations with rhMIF (Figure 1B–D), exposure to high levels of endogenous MIF from CATT7 CM across several timepoints (1, 6, and 24 h) had no effect on the percentage of CD74 (Figure 1E), CXCR2 (Figure 1F), and CXCR4 (Figure 1G) expression on the cell surface of MSCs. Thus, these data indicate that MSCs have the ability to interact with human MIF, through both the classical CD74 receptor, but also non‐classical chemokine CXCR2 and CXCR4 receptors.
FIGURE 2.
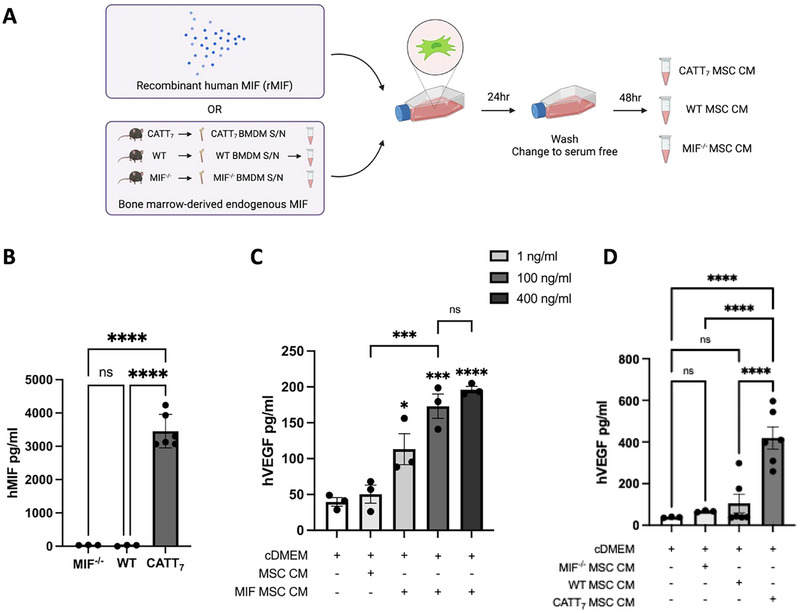
Recombinant or endogenous MIF stimulates MSC secretion of VEGF. A, Schematic depicting the generation of MIF MSC conditioned media using recombinant human MIF or endogenous human MIF generated from BMDMs of CATT7 transgenic mice that were challenged with HDM for 3 weeks. HDM‐challenged WT and MIF knockout (MIF−/−) mice that do not express human MIF were used as controls. B, Levels of hMIF detected in the BMDMs of MIF−/−, WT, and CATT7 mice challenged with 25 µg of HDM for 3 weeks. (n = 3–6 mice). (C) Human VEGF protein levels in MSC conditioned media supernatants, measured by ELISA, after licensing with different concentrations of recombinant human MIF (1, 100, and 400 ng/mL). (D) Human VEGF protein levels in MIF MSC conditioned media supernatants, using bone marrow‐derived supernatants from high MIF expressing CATT7 mice, MIF−/‐ mice, or WT mice (n = 3–6). Data are presented as mean ± SEM and represent three independent experiments. Statistical significance was determined by one‐way ANOVA; ns = non‐significant; *p < 0.05, ***p < 0.001, ****p < 0.0001.
3.2. Recombinant and Endogenous MIF Stimulates MSC Secretion of VEGF
Activation of MSCs boosts their cytoprotective effects through the production of soluble mediators [55, 56]. MSCs were licensed with different concentrations of rhMIF (1, 100, or 400 ng/mL) or with BMDM supernatants from HDM‐challenged human MIF‐expressing CATT7 mice, MIF−/− and WT mice (Figure 2A). As MIF is known to be stored in intracellular pools, being secreted only after stimulation [57, 58, 59, 60], high MIF‐expressing CATT7 mice, MIF−/− mice, and WT mice were exposed to a model of house dust mite‐induced acute allergic airway inflammation prior to generating BMDM supernatants. The supernatants from BMDMs isolated from WT, MIF−/−, and CATT7 mice were measured by human MIF ELISA, where only CATT7 BMDM supernatant contained high levels of human MIF, thus serving as a source of endogenous MIF (Figure 2B).
VEGF, an important soluble factor secreted by MSCs [16, 19, 61], is a trophic factor known to play a role in wound healing [62]. After licensing MSCs with rhMIF, human VEGF (hVEGF) levels in rhMIF‐MSC CM supernatants were measured by ELISA (Figure 2C). MSCs stimulated with all concentrations of rhMIF exhibited significantly elevated levels of hVEGF in a dose‐dependent manner. MSCs licensed with 100 and 400 ng/mL rhMIF secreted the highest hVEGF protein levels, with no statistical difference between the two concentrations (Figure 2C). Thus, 100 ng/mL of rhMIF was used to license MSCs for the remainder of the study.
Alternatively, after licensing MSCs with CATT7‐derived human MIF, WT‐derived murine MIF, or no MIF (MIF−/−), VEGF protein levels in the endogenous MIF‐MSC CM were measured by ELISA (Figure 2D). MIF−/− and murine MIF‐expressing WT mice did not significantly increase VEGF production by MSCs compared with cDMEM control (Figure 2D). Conversely, MSCs licensed with CATT7 supernatants containing human MIF displayed significantly elevated VEGF protein levels in the associated conditioned media, compared with those licensed with MIF−/− or WT supernatants. This shows that human, but not murine MIF, drives enhanced VEGF production by human MSCs (Figure 2D).
3.3. MIF Signaling Through CXCR4, but Not CXCR2 or CD74, Leads to Enhanced VEGF Production in MSCs
MIF licensing significantly increased MSC production of VEGF (Figure 2). As previously discussed, MIF signals through canonical (CD74) and non‐canonical receptors (CXCR2 and CXCR4), are all constitutively expressed on the surface of MSCs (Figure 1). Thus, to further validate the involvement of MIF in the licensing of MSC cytoprotective ability, MSCs were exposed to an anti‐CD74 neutralising antibody (10 mg/mL) or immunoglobulin G1 (IgG1) isotype control (10 mg/mL) for 30 min before being incubated with CATT7 CM for 24 h. Similarly, a CXCR2 chemokine receptor inhibitor (Reparixin; 40 µM) or a CXCR4 chemokine receptor inhibitor (AMD3100) (50 µg/mL) were used to determine if MIF's non‐canonical receptors were responsible for the MIF‐MSC interaction, resulting in enhanced VEGF production into the MSC secretome.
CATT7‐MSC CM had significantly increased levels of VEGF production compared with MSCs that were not licensed (Figure 3A). Interestingly, MSCs that were pre‐treated with the CXCR4 chemokine receptor inhibitor AMD3100 but not vehicle control prior to CATT7 CM licensing had significantly less VEGF production compared with MSCs that were licensing with CATT7 CM alone (Figure 3A), demonstrating that MIF licensing of MSCs was dependent on the CXCR4 receptor. No significant difference was noted between reparixin pre‐treated CATT7‐MSC CM and vehicle control CATT7‐MSC CM groups, indicating that MIF‐MSC signalling is independent of CXCR2 (Figure 3B). Lastly, MSCs pretreated with the CD74 neutralizing antibody prior to CATT7‐CM licensing did not have significantly different levels of VEGF compared with MSCs licensed with CATT7‐CM (Figure 3C). Similarly, the addition of an IgG1 isotype control prior to MSCs being licensed with CATT7‐CM had no significant effect on MSC production of VEGF, concluding that MIF also does not signal through the CD74 receptor on the surface of MSCs (Figure 3C). Thus, these data conclude that MIF signaling through CXCR4, but not CXCR2 or CD74 leads to enhanced VEGF production in MSCs.
FIGURE 3.
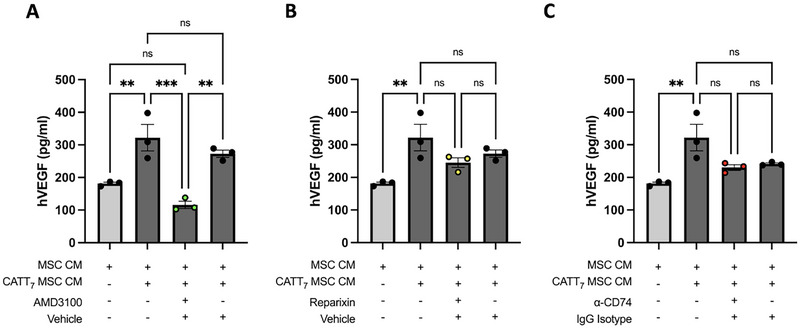
MIF signaling through CXCR4, but not CXCR2 or CD74 leads to enhanced VEGF production in MSCs. MIF MSC conditioned media was generated by licensing MSCs with endogenous human MIF secreted by BMDMs of HDM‐challenged CATT7 transgenic mice. To investigate the MIF receptor involved in enhanced VEGF production by MSCs, inhibitors of (A) CXCR4 (AMD3100), (B) CXCR2 (reparixin), or (C) a CD74 neutralizing antibody were used. Prior to endogenous MIF licensing, MSCs were exposed to inhibitors, vehicle control (DMSO), or IgG Isotype control for 30 min. After 24 h, media was removed and cells were washed with PBS before being replaced with serum‐free media for 48 h. VEGF production was measured by human VEGF ELISA. Data are presented as mean ± SEM and represent three independent experiments. Statistical significance was determined by one‐way ANOVA; ns = non‐significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3.4. Endogenous CATT7‐MIF MSC Conditioned Media Drives Bronchial Epithelial Wound Closure in a VEGF‐Dependent Manner
In the asthmatic lung, repeated exacerbations can inflict injury on the membrane epithelium of the lung. Furthermore, the role of VEGF in wound healing is established in a variety of different conditions [62, 63, 64], such as type 1 diabetes [65] and pulmonary fibrosis [66]. We have shown that the increased levels of VEGF observed in CM from rhMIF‐licensed MSCs (Figure 2C) and endogenous CATT7‐MSC CM (Figure 2D) drive airway epithelial cell wound closure in human alveolar basal epithelial cells; A549s (Figure S2B,C). Using the more physiologically relevant normal human bronchial epithelial cell line (BEAS‐2B), we show that CATT7‐MSC CM significantly increased the percentage of wound closure compared with WT‐MSC CM and cDMEM groups (Figure 4A) (Table S1). To validate the role of VEGF in bronchial epithelial wound closure, a potent and specific VEGFR2 inhibitor, SU‐5416 was used to block the VEGF receptor on the surface of epithelial cells prior to the addition of CATT7‐MSC CM (Figure 4A). Blocking VEGFR2 on BEAS‐2B cells attenuated the significant enhancement of wound closure mediated by CATT7‐MSC CM, but not WT‐MSC CM (Figure 4A,B), illustrating the importance of human CATT7 MIF licensing in MSC‐derived VEGF production and BEAS‐2B wound closure, and that murine MIF does not drive wound closure in a VEGF‐dependent manner. Similarly, the application of SU‐5416 to A549s demonstrated the important role of MIF licensing on MSC's cytoprotective abilities (Figure S2B,C). BEAS‐2B cells pretreated with the vehicle control prior to the addition of CATT7‐MIF MSC CM maintained a significant increase in percentage wound closure mediated by CATT7‐MIF MSC CM. The use of a VEGFR2 inhibitor had no off‐target, non‐specific effects on the general growth of these cells, as cDMEM wells treated with SU‐5416 had no significant difference in percentage wound closure compared with cDMEM alone (Figure 4A). The increase in wound closure associated with CM from CATT7‐MIF licensed MSCs is illustrated in Figure 4B. Furthermore, when the VEGFR2 was blocked using SU‐5416 but not vehicle control, the inhibition of wound closure was clear (Figure 4B).
FIGURE 4.
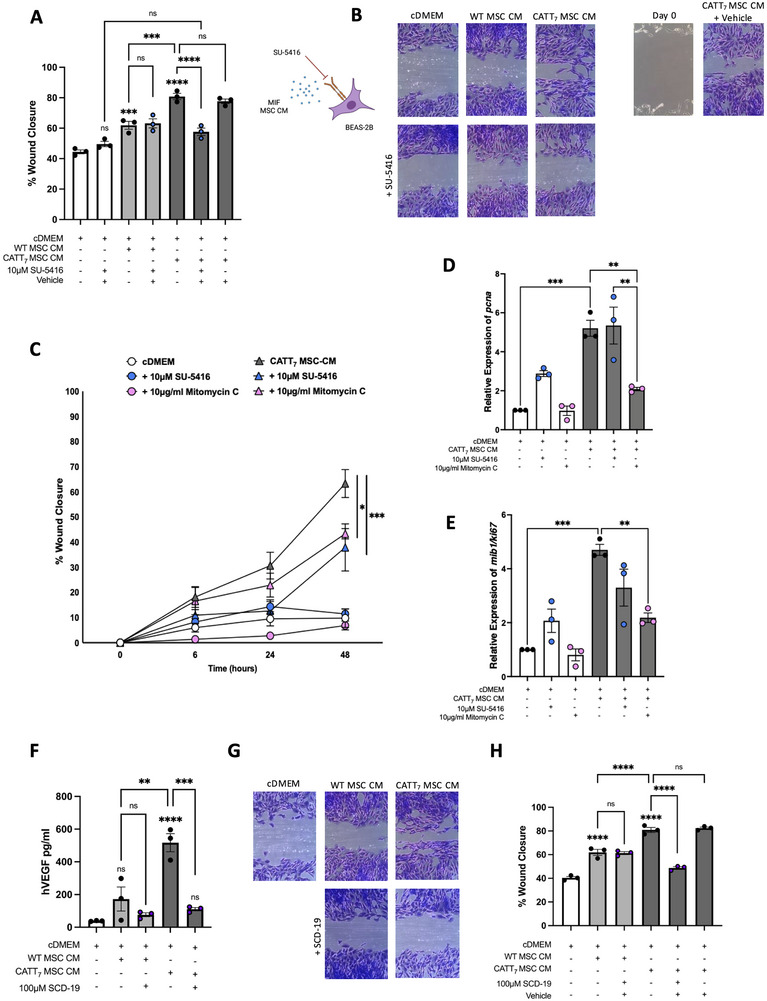
Endogenous CATT7‐MIF MSC conditioned media drives bronchial epithelial wound closure in a VEGF‐dependent manner. Using a human bronchial epithelial cell line (BEAS‐2B), a scratch assay was carried out to investigate the impact of MIF MSC CM in bronchial cell wound closure. A, B, A VEGFR2 inhibitor SU‐5416 (10 µM) or vehicle control (DMSO) was added to BEAS‐2B for 4 h prior to the addition of WT or CATT7 MIF MSC CM. C, The cell cycle inhibitor mitomycin C (10 µg/mL) was added to BEAS‐2B for 2 h before being washed off with PBS prior to the addition of cDMEM or CATT7 MIF MSC CM. Wound healing was quantified at 6, 24, and 48 h and cells were harvested for gene expression analysis of (D) pcna and (E) ki67 at 48 h. F–H, To block MIF activity, a MIF inhibitor SCD‐19 (100 µM) or vehicle control (DMSO) was added to WT and CATT7 BMDM supernatants for 1 h before being added to BEAS‐2B cells. After 48 h, or until the first scratch had closed, media was removed and cells were fixed with 10% neutral buffered formalin and stained with crystal violet before being imaged using Optika imaging software on a Nikon imaging microscope. Percentage wound closure relative to day 0 baseline was calculated using Image J software. Data are presented as mean ± SEM and represent three independent experiments. Statistical significance was determined by one‐way ANOVA in (A, D–F, H) and two‐way ANOVA in (C); ns = non‐significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To prove that conditioned media from another cell type could not facilitate enhanced wound closure in alveolar epithelial cells, or that a positive VEGF feedback loop in A549 conditioned media was encouraging self‐renewal of these cells, conditioned media from A549 cells was used as a negative control. A549 conditioned media did not significantly increase the percentage of wound closure in A549 epithelial cells (Figure S2B,C).
We have elucidated that human, but not murine, MIF drives VEGF production from MSCs (Figure 4F) and thus facilitates a significant increase in wound closure (Figure 4A,B). We performed experiments to determine the effect of SU‐5416 on the proliferation of BEAS‐2B cells. In wound closure experiments, the addition of SU‐5416 had no significant effect on BEAS‐2B cells cultured in cDMEM (Figure 4C); however, SU‐5416 significantly reduced wound closure mediated by CATT7 MSC‐CM (Figure 4C). We also used mitomycin C, a cell cycle antagonist to inhibit cell proliferation. We show that the addition of mitomycin C significantly reduced wound closure mediated by CATT7 MSC‐CM (Figure 4C). We also examined the expression of two genes associated with proliferation; pcna and ki67. CATT7 MSC‐CM significantly increased the expression of pcna and ki67 in BEAS‐2B cells (Figure 4D,E). While SU‐5416 had no significant effect on BEAS‐2B cells exposed to CATT7 MSC‐CM, the addition of mitomycin C significantly decreased the expression of pcna and ki67 (Figure 4D,E). Together, these findings support a role for both migration and proliferation in the enhancement of wound closure mediated by CATT7 MSC‐CM.
To fully elucidate this MIF‐associated effect on MSC CM increase in wound closure, an MIF inhibitor SCD‐19 was used to block MIF's biological activity prior to MSC licensing. When endogenous CATT7 MIF supernatants were incubated with 100 µM of SCD‐19 for 1 h prior to MSC licensing, MIF inhibition significantly decreased MSC‐mediated VEGF production compared with cDMEM controls (Figure 4F). Importantly, SCD‐19 did not affect WT‐derived MIF supernatants, as no significant difference in VEGF production was noted.
SCD‐19, but not vehicle control, effectively decreased the capacity for CATT7‐MSC CM to enhance wound closure in bronchial epithelial cells (Figure 4G,H). These data conclude that high levels of human MIF from CATT7 BMDM‐derived supernatants can license MSCs to produce increased levels of VEGF (Figures 2D and 4F), with increased efficacy than those licensed with recombinant MIF (Figure 2C). Following this narrative, conditioned media generated from endogenous human MIF‐licensed MSCs can significantly increase wound closure in BEAS‐2B cells in a VEGF‐dependent manner (Figure 4A,B), illustrating MIF's specific role through utilising a potent MIF antagonist SCD‐19 (Figure 4G,H).
3.5. VEGF Produced by MIF‐Licensed MSCs Reduces HDM‐Induced Goblet Cell Hyperplasia
VEGF, specifically through VEGFR2 (KDR) signalling, has previously been shown to protect against goblet cell metaplasia during processes of goblet cell renewal [67]. Thus, we set out to investigate the therapeutic capacity of VEGF present in CATT7‐MIF MSC CM in an HDM model of allergic airway inflammation. To elucidate the specific role of MSC‐derived VEGF in vivo, an anti‐human VEGF neutralising antibody or IgG isotype control was used (Figure 5A).
FIGURE 5.
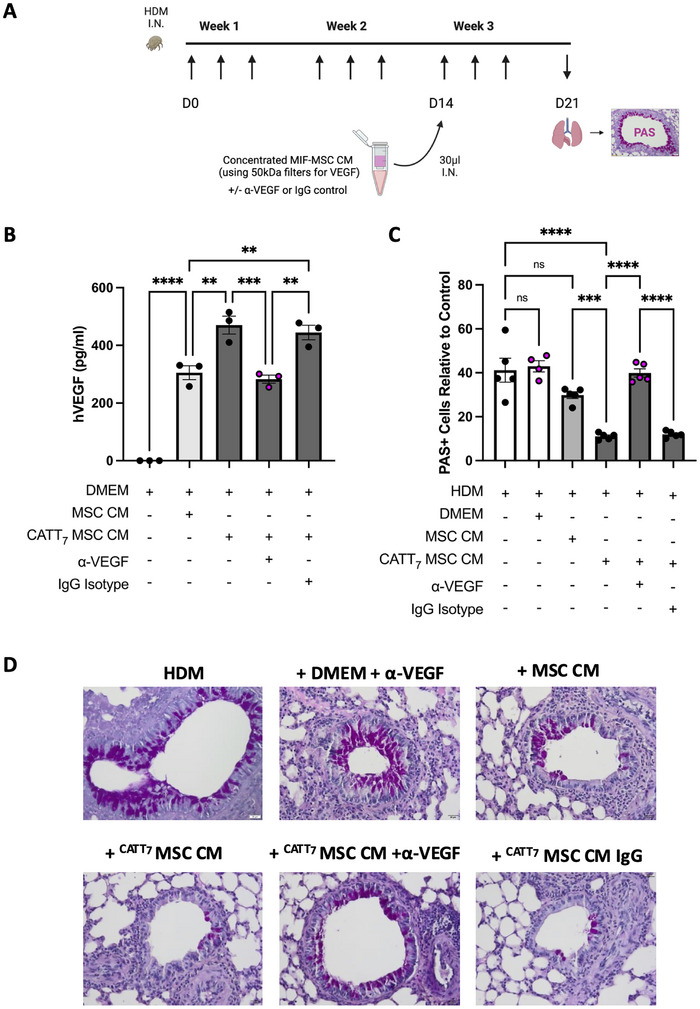
VEGF produced by MIF‐licensed MSCs reduces HDM‐induced goblet cell hyperplasia. A, CATT7 mice were challenged with 25 µg of HDM intranasally I.N. three times a week for 3 weeks. MIF MSC CM containing VEGF was concentrated using 50 kDa Amicon filters. A, VEGF‐neutralizing antibody (8 ng/mL) or IgG isotype control was added before intranasal administration on day 14. B, VEGF levels were measured by VEGF ELISA. C, D, Lungs were harvested on day 21 for histological analysis by staining goblet cells with periodic acid Schiff (PAS). Slides were imaged and the number of PAS‐positive cells (magenta) relative to the control were quantified using ImageJ software. N = 4–5 mice per group. Data are presented as mean ± SEM. Statistical significance was determined by one‐way ANOVA; ns = non‐significant, **p < 0.01, ***p < 0.001, ****p < 0.0001.
After MSC CM and CATT7‐MIF MSC CM were generated as previously described (Figure 2A), supernatants were concentrated using ultracentrifugal filters with a molecular cut‐off of 50 kDa (VEGF molecular weight: 45 kDa) to allow for intranasal administration on day 14 of the HDM model (Figure 5A). To neutralise MSC‐derived VEGF present in conditioned media, a bevacizumab biosimilar anti‐VEGF monoclonal antibody or IgG isotype control was added to the supernatant at 8 ng/mL prior to I.N. administration on day 14.
To ensure the efficacy of the VEGF neutralising antibody, VEGF protein in the MSC CM was measured using a human VEGF ELISA. As expected, CATT7 MSC CM contained significantly elevated levels of VEGF compared with that present in conditioned media from unlicensed MSCs (Figure 5B). CATT7 MSC CM + α‐VEGF had a significantly decreased concentration of VEGF present, compared with both CATT7 MSC CM, and CATT7 MSC CM + IgG isotype control (Figure 5B).
To investigate the therapeutic role of CATT7 MSC CM in a model of HDM‐induced allergic asthma, lungs were harvested on day 21 and processed for histological analysis, tissue sections were stained with PAS to visualise and measure levels of airway goblet cell hyperplasia. CATT7 MSC CM, but not MSC CM significantly reduced HDM‐induced goblet cell hyperplasia (Figure 5C,D), reiterating the importance of human MIF in licensing MSC's cytoprotective efficacy. However, administration of CATT7 MSC CM treated with α‐VEGF neutralising antibody, but not an IgG control antibody could no longer significantly decrease the number of PAS‐positive cells in HDM‐challenged mice, demonstrating the role of VEGF in epithelial protection against goblet cell hyperplasia. This study demonstrates that MIF‐licensing significantly increases VEGF production by MSCs which is responsible for the enhanced cytoprotective effects of CATT7‐licensed MSC CM in airway epithelial cells in vitro and in a clinically relevant pre‐clinical model of HDM‐induced allergic airway inflammation.
4. Discussion
This study set out to investigate the cytoprotective mechanisms associated with the MIF‐licensed MSC secretome in the context of epithelial injury in vitro and in vivo. Human BM‐MSCs express the canonical (CD74) and non‐canonical (CXCR2 and CXCR4) MIF receptors allowing MSCs to respond to MIF present in the microenvironment. Previously, we have demonstrated that MIF can promote the expansion and immunosuppressive function of human BM‐MSCs in vitro and significantly increase the retention of human BM‐MSCs in vivo in an HDM model of allergic asthma [42]. MIF signals through different receptors depending on the function. We have previously shown that CD74 is required for MIF‐licensed MSC immunomodulation and to significantly increase the retention of human BM‐MSCs in vivo in an HDM model of allergic asthma [42]. Differentially, CXCR4 has been shown to mediate MSC chemotaxis to MIF [68]. Here we show that MIF‐licensing significantly enhances MSC secretion of VEGF in a CXCR4‐dependent manner. In addition to rhMIF, endogenous human MIF produced by CATT7 BMDMs also had the capacity to license MSCs leading to significantly increased MSC‐derived VEGF secretion. Importantly, the use of WT BMDM CM to license MSCs did not have the same effect. This novel finding indicates that human, but not murine, MIF boosts the production of MSC‐derived VEGF. This supports the idea that the communication between exogenously administered MSCs and macrophages plays an important role in dictating MSC therapeutic effects.
MIF has previously been shown to upregulate VEGF production in the conditioned media of synovial fluid mononuclear cells [69] and endometrial stromal cells [70] promoting new blood vessel formation. Functionally, CATT7‐MIF licensed MSC CM exerted superior wound healing capacity in normal bronchial epithelial cells (BEAS‐2Bs) compared with naive MSC CM or WT‐licensed MSC CM. MSC CM has been previously shown to promote wound healing in airway epithelial cells via growth factors including HGF and KGF [23, 71].
Our group and others have highlighted the importance of licensing MSCs to enhance their therapeutic efficacy [42, 72, 73], however, less is understood about the influence of licensing approaches on MSC cytoprotective functions. Licensing approaches using IFNγ and hypoxia [74], or IFNγ and TNFα [75] have previously been used to enhance the cytoprotective/wound healing properties of MSC CM. VEGF production from MSCs has been shown to be increased by licensing MSCs with hypoxia [76, 77, 78, 79, 80], fibroblast growth factor‐2 [81], TGF‐α [82], IL‐1β [83], and LPS [84]. However, we are the first to demonstrate enhanced VEGF production from MSCs licensed with human MIF, a clinically relevant proinflammatory cytokine in a range of inflammatory diseases including asthma.
Blockade of MIF using the small molecule inhibitor, demonstrated the specificity of MIF in licensing MSCs to produce significantly increased levels of VEGF. This provides a novel mechanistic insight into how MIF can license MSCs and enhance their cytoprotective function, specifically their ability to provide protection against a dysregulated airway epithelial barrier and thus, also prevent the development of airway goblet cell hyperplasia after allergen challenge in vivo. The limitation of our study is that although we show a role for MSC‐CM‐derived VEGF, we have not identified the mechanism of how this MSC‐CM‐derived VEGF contributes to the repair of HDM‐induced epithelial damage in mice. Moreover, our findings of effects mediated by human VEGF on mouse epithelial cells in vivo add further complexity. However, human VEGF has been shown to act on mouse epithelial cells both in vitro and in vivo [85, 86].
VEGF has historically played a central role in epithelial repair and the maintenance of epithelial barrier integrity, where VEGF‐deficient mice had increased levels of bronchial and alveolar apoptosis [87]. VEGF enhanced wound healing, survival, and proliferation of airway epithelial cells [88, 89]. Furthermore, VEGF overexpression reduced bleomycin‐induced cell death in a model of idiopathic pulmonary fibrosis [66]. Interestingly, VEGF‐A and its receptor VEGFR2 have been shown to have a protective role in the defence against mucous cell metaplasia, commonly documented in asthma and cystic fibrosis [67]. In asthma, if VEGF‐A levels are decreased, the transcription factor Sox9 is upregulated, driving the club to goblet cell differentiation, exacerbating disease [67]. These studies, along with the data presented throughout this manuscript depict the protective role of VEGF in the repair and regulation of the airway epithelial barrier, decreasing goblet cell hyperplasia after the HDM challenge in vivo.
In the lung, there has been evidence of MSC‐derived VEGF having a protective role in acute lung injury [90, 91]; however, the effect of MSC‐secreted VEGF in allergic asthma is unknown. Thus, this manuscript is the first to demonstrate the impact of MSC‐VEGF on epithelial cells in vitro, and in vivo in a clinically relevant model of HDM‐induced acute allergic asthma.
Author Contributions
Hazel Dunbar performed research, analysed the data, designed the study, and wrote the manuscript. Ian J. Hawthorne performed research, analysed the data, and designed the study. Courteney Tunstead performed research and analysed the data. Molly Dunlop and Evelina Volkova performed research. Daniel J. Weiss and Claudia C. dos Santos contributed to the study design and data analysis. Seamas C. Donnelly and Michelle E. Armstrong provided reagents and contributed to the study design and data analysis. Karen English designed and supervised the study and wrote the manuscript. All authors approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202451205.
Supporting information
Supporting Information
Acknowledgements
This research was supported by an Irish Research Council Laureate Award to KE (IRCLA/2017/288). This publication has emanated from research supported in part by a COVID HEA research grant from Maynooth University, a research grant from Science Foundation Ireland (SFI) under grant 12/RI/2346, an infrastructure award supporting the Attune Nxt.
Open access funding provided by IReL.
Funding: Irish Research Council IRCLA/2017/288, Science Foundation Ireland 12/RI/2346, Higher Education Authority HEA COVID grant to Maynooth University
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Li C. L., Xu Z. B., Fan X. L., et al., “MicroRNA‐21 Mediates the Protective Effects of Mesenchymal Stem Cells Derived From iPSCs to Human Bronchial Epithelial Cell Injury Under Hypoxia,” Cell Transplantation 27, no. 3 (2018): 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahmad T., Mukherjee S., Pattnaik B., et al., “Miro1 regulates Intercellular Mitochondrial Transport & Enhances Mesenchymal Stem Cell Rescue Efficacy,” Embo Journal 33, no. 9 (2014): 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morrison T. J., Jackson M. V., Cunningham E. K., et al., “Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer,” American Journal of Respiratory and Critical Care Medicine 196, no. 10 (2017): 1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yao Y., Fan X. L., Jiang D., et al., “Connexin 43‐Mediated Mitochondrial Transfer of iPSC‐MSCs Alleviates Asthma Inflammation,” Stem Cell Reports 11, no. 5 (2018): 1120–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cruz F. F., Borg Z. D., Goodwin M., et al., “Systemic Administration of Human Bone Marrow‐Derived Mesenchymal Stromal Cell Extracellular Vesicles Ameliorates Aspergillus Hyphal Extract‐Induced Allergic Airway Inflammation in Immunocompetent Mice,” Stem Cells Translational Medicine 4, no. 11 (2015): 1302–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park K. S., Bandeira E., Shelke G. V., Lässer C., and Lötvall J., “Enhancement of Therapeutic Potential of Mesenchymal Stem Cell‐Derived Extracellular Vesicles,” Stem Cell Research & Therapy 10 (2019): 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dos Santos C. C., Lopes‐Pacheco M., English K., Rolandsson Enes S., Krasnodembskaya A., and Rocco P. R. M., “The MSC‐EV‐microRNAome: A Perspective on Therapeutic Mechanisms of Action in Sepsis and ARDS,” Cells 13, no. 2 (2024): 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. English K., “Mechanisms of Mesenchymal Stromal Cell Immunomodulation,” Immunology and Cell Biology 91, no. 1 (2013): 19–26. [DOI] [PubMed] [Google Scholar]
- 9. Szabłowska‐Gadomska I., Rudziński S., and Dymowska M., “Secretome of Mesenchymal Stromal Cells as a Possible Innovative Therapeutic Tool in Facial Nerve Injury Treatment,” BioMed Research International 2023 (2023): 8427200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xia J., Minamino S., Kuwabara K., and Arai S., “Stem Cell Secretome as a New Booster for Regenerative Medicine,” BioScience Trends 13, no. 4 (2019): 299–307. [DOI] [PubMed] [Google Scholar]
- 11. Liu J., Qiu X., Lv Y., et al., “Apoptotic Bodies Derived From Mesenchymal Stem Cells Promote Cutaneous Wound Healing via Regulating the Functions of Macrophages,” Stem Cell Research & Therapy 11, no. 1 (2020): 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Műzes G. and Sipos F., “Mesenchymal Stem Cell‐Derived Secretome: A Potential Therapeutic Option for Autoimmune and Immune‐Mediated Inflammatory Diseases,” Cells 11, no. 15 (2022): 2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pang S. H. M., D'Rozario J., Mendonca S., et al., “Mesenchymal Stromal Cell Apoptosis Is Required for Their Therapeutic Function,” Nature Communications 12, no. 1 (2021): 6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weiss D. J., English K., Krasnodembskaya A., Isaza‐Correa J. M., Hawthorne I. J., and Mahon B. P., “The Necrobiology of Mesenchymal Stromal Cells Affects Therapeutic Efficacy,” Frontiers in Immunology 10 (2019): 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. English K., “Apoptotic MSCs, COX2/PGE2 and Clinical Efficacy in Crohn Fistula,” Molecular Therapy 31, no. 12 (2023): 3364–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ge Q., Zhang H., Hou J., et al., “VEGF Secreted by Mesenchymal Stem Cells Mediates the Differentiation of Endothelial Progenitor Cells Into Endothelial Cells via Paracrine Mechanisms,” Molecular Medicine Reports 17, no. 1 (2018): 1667–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han Y., Yang J., Fang J., et al., “The Secretion Profile of Mesenchymal Stem Cells and Potential Applications in Treating human Diseases,” Signal Transduction and Targeted Therapy 7, no. 1 (2022): 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kozhukharova I., Minkevich N., Alekseenko L., Domnina A., and Lyublinskaya O., “Paracrine and Autocrine Effects of VEGF Are Enhanced in Human eMSC Spheroids,” International Journal of Molecular Sciences 23, no. 22 (2022): 14324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Healy M. E., Bergin R., Mahon B. P., and English K., “Mesenchymal Stromal Cells Protect Against Caspase 3‐mediated Apoptosis of CD19(+) Peripheral B Cells Through Contact‐dependent Upregulation of VEGF,” Stem Cells and Development 24, no. 20 (2015): 2391–2402. [DOI] [PubMed] [Google Scholar]
- 20. Cahill E. F., Tobin L. M., Carty F., Mahon B. P., and English K., “Jagged‐1 Is Required for the Expansion of CD4+ CD25+ FoxP3+ Regulatory T Cells and Tolerogenic Dendritic Cells by Murine Mesenchymal Stromal Cells,” Stem Cell Research & Therapy 6, no. 1 (2015): 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. English K., Ryan J. M., Tobin L., Murphy M. J., Barry F. P., and Mahon B. P., “Cell Contact, Prostaglandin E(2) and Transforming Growth Factor Beta 1 Play Non‐redundant Roles in human Mesenchymal Stem Cell Induction of CD4+CD25(High) Forkhead Box P3+ Regulatory T Cells,” Clinical and Experimental Immunology 156, no. 1 (2009): 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kennelly H., Mahon B. P., and English K., “Human Mesenchymal Stromal Cells Exert HGF Dependent Cytoprotective Effects in a Human Relevant Pre‐clinical Model of COPD,” Scientific Reports 6 (2016): 38207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cahill E. F., Kennelly H., Carty F., Mahon B. P., and English K., “Hepatocyte Growth Factor Is Required for Mesenchymal Stromal Cell Protection Against Bleomycin‐Induced Pulmonary Fibrosis,” Stem Cells Translational Medicine 5, no. 10 (2016): 1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ionescu L., Byrne R. N., van Haaften T., et al., “Stem Cell Conditioned Medium Improves Acute Lung Injury in Mice: In Vivo Evidence for Stem Cell Paracrine Action,” American Journal of Physiology Lung Cellular and Molecular Physiology 303, no. 11 (2012): L967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim S. Y., Lee J. H., Kim H. J., et al., “Mesenchymal Stem Cell‐conditioned media Recovers Lung Fibroblasts From Cigarette Smoke‐induced Damage,” American Journal of Physiology Lung Cellular and Molecular Physiology 302, no. 9 (2012): L891–908. [DOI] [PubMed] [Google Scholar]
- 26. Kruk D., Wisman M., Noordhoek J. A., et al., “Paracrine Regulation of Alveolar Epithelial Damage and Repair Responses by Human Lung‐Resident Mesenchymal Stromal Cells,” Cells 10, no. 11 (2021): 2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moreira A., Naqvi R., Hall K., et al., “Effects of Mesenchymal Stromal Cell‐conditioned media on Measures of Lung Structure and Function: A Systematic Review and Meta‐analysis of Preclinical Studies,” Stem Cell Research & Therapy 11, no. 1 (2020): 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su V. Y. F., Lin C. S., Hung S. C., and Yang K. Y., “Mesenchymal Stem Cell‐Conditioned Medium Induces Neutrophil Apoptosis Associated With Inhibition of the NF‐κB Pathway in Endotoxin‐Induced Acute Lung Injury,” International Journal of Molecular Sciences 20, no. 9 (2019): 2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Z., Hua Y., Ding Y., et al., “Conditioned Medium of Bone Marrow Mesenchymal Stem Cells Involved in Acute Lung Injury by Regulating Epithelial Sodium Channels via miR‐34c,” Frontiers in Bioengineering and Biotechnology 9 (2021): 640116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boland L., Burand A. J., Brown A. J., Boyt D., Lira V. A., and Ankrum J. A., “IFN‐γ and TNF‐α Pre‐licensing Protects Mesenchymal Stromal Cells From the Pro‐inflammatory Effects of Palmitate,” Molecular Therapy 26, no. 3 (2018): 860–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boyt D. T., Boland L. K., Burand A. J., Brown A. J., and Ankrum J. A., “Dose and Duration of Interferon γ Pre‐licensing Interact With Donor Characteristics to Influence the Expression and Function of Indoleamine‐2,3‐dioxygenase in Mesenchymal Stromal Cells,” Journal of the Royal Society, Interface 17, no. 167 (2020): 20190815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. AÉS C., MRR S., Alencar‐Silva T., Carvalho J. L., and Saldanha‐Araujo F., “Mesenchymal Stem Cells Immunomodulation: The Road to IFN‐γ Licensing and the Path Ahead,” Cytokine & Growth Factor Reviews 47 (2019): 32–42. [DOI] [PubMed] [Google Scholar]
- 33. Cheng H. Y., Anggelia M. R., Lin C. H., and Lin C. F., “Preconditioned Mesenchymal Stromal Cells to Improve Allotransplantation Outcome,” Cells 10, no. 9 (2021): 2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dunbar H., Weiss D. J., Rolandsson Enes S., Laffey J. G., and English K., “The Inflammatory Lung Microenvironment; a Key Mediator in MSC Licensing,” Cells 10, no. 11 (2021): 2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Pedro M. Á., Gómez‐Serrano M., Marinaro F., et al., “IFN‐Gamma and TNF‐Alpha as a Priming Strategy to Enhance the Immunomodulatory Capacity of Secretomes From Menstrual Blood‐Derived Stromal Cells,” International Journal of Molecular Sciences 22, no. 22 (2021): 12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hackel A., Vollmer S., Bruderek K., Lang S., and Brandau S., “Immunological Priming of Mesenchymal Stromal/Stem Cells and Their Extracellular Vesicles Augments Their Therapeutic Benefits in Experimental Graft‐versus‐host Disease via Engagement of PD‐1 Ligands,” Frontiers in Immunology 14 (2023): 1078551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Noronha N C., Mizukami A., Caliári‐Oliveira C., et al., “Priming Approaches to Improve the Efficacy of Mesenchymal Stromal Cell‐based Therapies,” Stem Cell Research & Therapy 10, no. 1 (2019): 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mizue Y., Ghani S., Leng L., et al., “Role for Macrophage Migration Inhibitory Factor in Asthma,” PNAS 102, no. 40 (2005): 14410–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Plant B. J., Gallagher C. G., Bucala R., et al., “Cystic Fibrosis, Disease Severity, and a Macrophage Migration Inhibitory Factor Polymorphism,” American Journal of Respiratory and Critical Care Medicine 172, no. 11 (2005): 1412–1415. [DOI] [PubMed] [Google Scholar]
- 40. Baugh J. A., Chitnis S., Donnelly S. C., et al., “A Functional Promoter Polymorphism in the Macrophage Migration Inhibitory Factor (MIF) Gene Associated With Disease Severity in Rheumatoid Arthritis,” Genes and Immunity 3, no. 3 (2002): 170–176. [DOI] [PubMed] [Google Scholar]
- 41. Dunbar H., Hawthorne I. J., Tunstead C., Armstrong M. E., Donnelly S. C., and English K., “Blockade of MIF Biological Activity Ameliorates House Dust Mite‐induced Allergic Airway Inflammation in Humanized MIF Mice,” Faseb Journal 37, no. 8 (2023): e23072. [DOI] [PubMed] [Google Scholar]
- 42. Hawthorne I. J., Dunbar H., Tunstead C., et al., “Human Macrophage Migration Inhibitory Factor Potentiates Mesenchymal Stromal Cell Efficacy in a Clinically Relevant Model of Allergic Asthma,” Molecular Therapy 31, no. 11 (2023): 3243–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Altorki N. K., Markowitz G. J., Gao D., et al., “The Lung Microenvironment: An Important Regulator of Tumour Growth and Metastasis,” Nature Reviews Cancer 19, no. 1 (2019): 9–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hsieh A., Assadinia N., and Hackett T. L., “Airway Remodeling Heterogeneity in Asthma and Its Relationship to Disease Outcomes,” Frontiers in Physiology 14 (2023): 1113100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tatler A. L., “Asthmatic Airway Remodeling: Long Overlooked but Too Important to Ignore,” Annals of Translational Medicine 11, no. 2 (2023): 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Varricchi G., Ferri S., Pepys J., Poto R., Spadaro G., Nappi E., et al., “Biologics and Airway Remodeling in Severe Asthma,” Allergy 77, no. 12 (2022): 3538–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ke X., Do D. C., Li C., et al., “Ras Homolog family Member A/Rho‐associated Protein Kinase 1 Signaling Modulates Lineage Commitment of Mesenchymal Stem Cells in Asthmatic Patients Through Lymphoid Enhancer‐binding Factor 1,” Journal of Allergy and Clinical Immunology 143, no. 4 (2019): 1560–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu X., Yu L., Guo B., Chen R., and Qiu C., “A Narrative Review of Research Advances in Mesenchymal Stem Cell Therapy for Asthma,” Annals of Translational Medicine 8, no. 21 (2020): 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pati S., Khakoo A. Y., Zhao J., et al., “Human Mesenchymal Stem Cells Inhibit Vascular Permeability by Modulating Vascular Endothelial Cadherin/β‐Catenin Signaling,” Stem Cells and Development 20, no. 1 (2011): 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou B. R., Xu Y., Guo S. L., et al., “The Effect of Conditioned media of Adipose‐derived Stem Cells on Wound Healing After Ablative Fractional Carbon Dioxide Laser Resurfacing,” BioMed Research International 2013 (2013): 519126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shin J. J., Fan W., Par‐Young J., et al., “MIF Is a Common Genetic Determinant of COVID‐19 Symptomatic Infection and Severity,” Qjm 116, no. 3 (2023): 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mawhinney L., Armstrong M. E., O' Reilly C., et al., “Macrophage Migration Inhibitory Factor (MIF) Enzymatic Activity and Lung Cancer,” Molecular Medicine 20, no. 1 (2015): 729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tynan A., Mawhinney L., Armstrong M. E., et al., “Macrophage Migration Inhibitory Factor Enhances Pseudomonas aeruginosa Biofilm Formation, Potentially Contributing to Cystic Fibrosis Pathogenesis,” Faseb Journal 31, no. 11 (2017): 5102–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trivedi A., Lin M., Miyazawa B., et al., “Inter‐ and Intra‐donor Variability in Bone Marrow–derived Mesenchymal Stromal Cells: Implications for Clinical Applications,” Cytotherapy 26, no. 9 (2024): 1062–1075. [DOI] [PubMed] [Google Scholar]
- 55. Lynch K., Treacy O., Chen X., et al., “TGF‐β1‐Licensed Murine MSCs Show Superior Therapeutic Efficacy in Modulating Corneal Allograft Immune Rejection in Vivo,” Molecular Therapy 28, no. 9 (2020): 2023–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tunstead C., Volkova E., Dunbar H., et al., “The ARDS Microenvironment Enhances MSC‐induced Repair via VEGF in Experimental Acute Lung Inflammation,” Molecular Therapy [Internet], https://www.sciencedirect.com/science/article/pii/S1525001624005252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bacher M., Metz C. N., Calandra T., et al., “An Essential Regulatory Role for Macrophage Migration Inhibitory Factor in T‐cell Activation,” PNAS 93, no. 15 (1996): 7849–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bernhagen J., Mitchell R. A., Calandra T., Voelter W., Cerami A., and Bucala R., “Purification, Bioactivity, and Secondary Structure Analysis of Mouse and human Macrophage Migration Inhibitory Factor (MIF),” Biochemistry 33, no. 47 (1994): 14144–14155. [DOI] [PubMed] [Google Scholar]
- 59. Calandra T., Bernhagen J., Mitchell R. A., and Bucala R., “The Macrophage Is an Important and Previously Unrecognized Source of Macrophage Migration Inhibitory Factor,” Journal of Experimental Medicine 179, no. 6 (1994): 1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Calandra T. and Roger T., “Macrophage Migration Inhibitory Factor: A Regulator of Innate Immunity,” Nature Reviews Immunology 3, no. 10 (2003): 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Arutyunyan I., Fatkhudinov T., Kananykhina E., et al., “Role of VEGF‐A in Angiogenesis Promoted by Umbilical Cord‐derived Mesenchymal Stromal/Stem Cells: In Vitro Study,” Stem Cell Research & Therapy 7 (2016): 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Johnson K. E. and Wilgus T. A., “Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair,” Adv Wound Care (New Rochelle) 3, no. 10 (2014): 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. DiPietro L. A., “Angiogenesis and Wound Repair: When Enough Is Enough,” J Leukoc Biol 100, no. 5 (2016): 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shams F., Moravvej H., Hosseinzadeh S., et al., “Overexpression of VEGF in Dermal Fibroblast Cells Accelerates the Angiogenesis and Wound Healing Function: In Vitro and in Vivo Studies,” Scientific Reports 12, no. 1 (2022): 18529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. White M. J. V., Briquez P. S., White D. A. V., and Hubbell J. A. V.‐A., “PDGF‐BB and HB‐EGF Engineered for Promiscuous Super Affinity to the Extracellular Matrix Improve Wound Healing in a Model of Type 1 Diabetes,” Npj Regen Med 6, no. 1 (2021): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Murray L. A., Habiel D. M., Hohmann M., et al., “Antifibrotic Role of Vascular Endothelial Growth Factor in Pulmonary Fibrosis,” JCI Insight 2, no. 16: e92192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jiang M., Fang Y., Li Y., et al., “VEGF Receptor 2 (KDR) Protects Airways From Mucus Metaplasia Through a Sox9‐dependent Pathway,” Developmental Cell 56, no. 11 (2021): 1646–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lourenco S., Teixeira V. H., Kalber T., Jose R. J., Floto R. A., and Janes S. M., “Macrophage Migration Inhibitory Factor‐CXCR4 Is the Dominant Chemotactic Axis in human Mesenchymal Stem Cell Recruitment to Tumors,” Journal of Immunology 194, no. 7 (2015): 3463–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim H. R., Park M. K., Cho M. L., Yoon C. H., Lee S. H., Park S. H., Leng L., Bucala R., Kang I., Choe J., and Kim H. Y., “Macrophage migration inhibitory factor upregulates angiogenic factors and correlates with clinical measures in rheumatoid arthritis” J Rheumatol. 34 no. 5: (2007): 927–936. [PubMed] [Google Scholar]
- 70. Veillat V., Carli C., Metz C. N., Al‐Abed Y., Naccache P. H., and Akoum A., “Macrophage Migration Inhibitory Factor Elicits an Angiogenic Phenotype in human Ectopic Endometrial Cells and Triggers the Production of Major Angiogenic Factors via CD44, CD74, and MAPK Signaling Pathways,” Journal of Clinical Endocrinology and Metabolism 95, no. 12 (2010): E403–412. [DOI] [PubMed] [Google Scholar]
- 71. Curley G. F., Hayes M., Ansari B., et al., “Mesenchymal Stem Cells Enhance Recovery and Repair Following Ventilator‐induced Lung Injury in the Rat,” Thorax 67, no. 6 (2012): 496–501. [DOI] [PubMed] [Google Scholar]
- 72. Carty F., Dunbar H., Hawthorne I. J., et al., “IFN‐γ and PPARδ Influence the Efficacy and Retention of Multipotent Adult Progenitor Cells in Graft vs Host Disease,” Stem Cells Translational Medicine 10, no. 11 (2021): 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Corbett J. M., Hawthorne I., Dunbar H., et al., “Cyclosporine A and IFNγ Licencing Enhances human Mesenchymal Stromal Cell Potency in a Humanised Mouse Model of Acute Graft versus Host Disease,” Stem Cell Research & Therapy 12, no. 1 (2021): 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang S., Umrath F., Cen W., Salgado A. J., Reinert S., and Alexander D., “Pre‐Conditioning With IFN‐γ and Hypoxia Enhances the Angiogenic Potential of iPSC‐Derived MSC Secretome,” Cells 11, no. 6 (2022): 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu C., Xu Y., Lu Y., et al., “Mesenchymal Stromal Cells Pretreated With Proinflammatory Cytokines Enhance Skin Wound Healing via IL‐6‐dependent M2 Polarization,” Stem Cell Research & Therapy 13, no. 1 (2022): 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fierro F. A., O'Neal A. J., Beegle J. R., et al., “Hypoxic Pre‐conditioning Increases the Infiltration of Endothelial Cells Into Scaffolds for Dermal Regeneration Pre‐seeded With Mesenchymal Stem Cells,” Frontiers in Cell and Developmental Biology 3 (2015): 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ishiuchi N., Nakashima A., Doi S., et al., “Hypoxia‐preconditioned Mesenchymal Stem Cells Prevent Renal Fibrosis and Inflammation in Ischemia‐reperfusion Rats,” Stem Cell Research & Therapy 11, no. 1 (2020): 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lagonda C. A., Tjahjadi F. B., Fauza D., and Kusnadi Y., “Hypoxia Increases Vegf Secretion in Multiple Sources of Mesenchymal Stem Cell,” Cytotherapy 20, no. 5 (2018): S44–5. [Google Scholar]
- 79. Bader A. M., Klose K., Bieback K., et al., “Hypoxic Preconditioning Increases Survival and Pro‐Angiogenic Capacity of Human Cord Blood Mesenchymal Stromal Cells in Vitro,” PLoS ONE 10, no. 9 (2015): e0138477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Razban V., Lotfi A. S., Soleimani M., Ahmadi H., Massumi M., Khajeh S., et al., “HIF‐1α Overexpression Induces Angiogenesis in Mesenchymal Stem Cells,” BioResearch Open Access 1, no. 4 (2012): 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gorin C., Rochefort G. Y., Bascetin R., et al., “Priming Dental Pulp Stem Cells With Fibroblast Growth Factor‐2 Increases Angiogenesis of Implanted Tissue‐Engineered Constructs Through Hepatocyte Growth Factor and Vascular Endothelial Growth Factor Secretion,” Stem Cells Translational Medicine 5, no. 3 (2016): 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang Y., Crisostomo P. R., Wang M., Markel T. A., Novotny N. M., and Meldrum D. R., “TGF‐alpha Increases human Mesenchymal Stem Cell‐secreted VEGF by MEK‐ and PI3‐K‐ but Not JNK‐ or ERK‐Dependent Mechanisms,” American Journal of Physiology Regulatory, Integrative and Comparative Physiology 295, no. 4 (2008): R1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koch D. W., Berglund A. K., Messenger K. M., Gilbertie J. M., Ellis I. M., and Schnabel L. V., “Interleukin‐1β in Tendon Injury Enhances Reparative Gene and Protein Expression in Mesenchymal Stem Cells,” Front Vet Sci 9 (2022): 963759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yao Y., Zhang F., Wang L., et al., “Lipopolysaccharide Preconditioning Enhances the Efficacy of Mesenchymal Stem Cells Transplantation in a Rat Model of Acute Myocardial Infarction,” Journal of Biomedical Science 16, no. 1 (2009): 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kokki E., Karttunen T., Olsson V., Kinnunen K., and Ylä‐Herttuala S., “Human Vascular Endothelial Growth Factor A165 Expression Induces the Mouse Model of Neovascular Age‐Related Macular Degeneration,” Genes (Basel) 9, no. 9 (2018): 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mujagic E., Gianni‐Barrera R., Trani M., Patel A., Gürke L., Heberer M., et al., “Induction of Aberrant Vascular Growth, but Not of Normal Angiogenesis, by Cell‐based Expression of Different Doses of human and Mouse VEGF Is Species‐dependent,” Hum Gene Ther Methods 24, no. 1 (2013): 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tang K., Rossiter H. B., Wagner P. D., and Breen E. C., “Lung‐targeted VEGF Inactivation Leads to an Emphysema Phenotype in Mice,” J Appl Physiol (1985) 97, no. 4 (2004): 1559–1566. discussion 1549. [DOI] [PubMed] [Google Scholar]
- 88. Mura M., Han B., Andrade C. F., et al., “The Early Responses of VEGF and Its Receptors During Acute Lung Injury: Implication of VEGF in Alveolar Epithelial Cell Survival,” Critical Care (London, England) 10, no. 5 (2006): R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ohwada A., Yoshioka Y., Iwabuchi K., Nagaoka I., Dambara T., and Fukuchi Y., “VEGF Regulates the Proliferation of Acid‐Exposed Alveolar Lining Epithelial Cells,” Thorax 58, no. 4 (2003): 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ahn S. Y., Park W. S., Kim Y. E., et al., “Vascular Endothelial Growth Factor Mediates the Therapeutic Efficacy of Mesenchymal Stem Cell‐derived Extracellular Vesicles Against Neonatal Hyperoxic Lung Injury,” Experimental & Molecular Medicine 50, no. 4 (2018): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen Q., Lin J., Deng Z., and Qian W., “Exosomes Derived From Human Umbilical Cord Mesenchymal Stem Cells Protect Against Papain‐induced Emphysema by Preventing Apoptosis Through Activating VEGF‐VEGFR2‐Mediated AKT and MEK/ERK Pathways in Rats,” Regenerative Therapy 21 (2022): 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.