

ABSTRACT
IL1‐β plays a central role in inflammation but its biological action needs to be tightly controlled. Such negative regulation can be exerted by the decoy receptor IL1R2. However, IL1R2 biology in immune cells remains poorly characterized, in particular in monocytes. Using conditional deficient mice, we show that Il1r2 deficiency in monocytes does not affect their steady‐state life cycle but dysregulates their trafficking to inflamed tissues in models of peritonitis and neuro‐inflammation. Mechanistically, we found that Il1r2 deficiency in monocytes increases CCL2 secretion in the inflamed peritoneum, thereby amplifying monocyte recruitment from blood. In autoimmune neuro‐inflammation, Il1r2 deficiency in monocytes exacerbates disease severity. Our findings suggest that the specific action of IL1R2 in monocytes contributes to a feedback mechanism for fine‐tuning the numbers of recruited monocytes during inflammation.
Keywords: IL1‐β, IL1R2, inflammation, leukocyte trafficking, monocyte
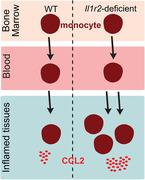
Il1r2 deficiency in monocytes dysregulates their trafficking to inflamed tissues through increased production of the monocyte chemoattractant CCL2.
1. Introduction
IL1‐β plays a central role in inflammation and immune responses [1]. However, dysregulation of the IL1‐β pathway can induce tissue damage and is associated to various human inflammatory pathologies [2]. Therefore, IL1‐β biological action needs to be tightly regulated. A major player in IL1‐β negative regulation is the decoy receptor IL1R2 [3, 4]; however, a full understanding of its biology in immune cells is lacking.
IL1R2 can be expressed on the cell surface or released in a soluble form and acts as a dominant negative and scavenger [5]. At steady‐state, IL1R2 is highly expressed by neutrophils but is also found on monocytes [6, 7, 8]. In vivo studies of IL1R2 biology in myeloid cells remain scarce. In models of rheumatoid arthritis, Il1r2 deficiency increased disease symptoms [8, 9], and it was further shown that neutrophil‐expressed IL1R2 dampened inflammatory cytokine secretion by fibroblasts [9]. By contrast, the role of IL1R2 in monocytes is poorly understood, in particular in physiological settings.
Monocytes originate from the bone marrow and circulate in the blood [10, 11]. Two main populations of monocytes have been described, with a precursor–product relationship [10, 11]. In the mouse, classical Ly6C+ monocytes convert into nonclassical Ly6ClowCD43+ monocytes, which mostly remain in the vasculature [12, 13]. Monocyte egress from the bone marrow is dependent on the chemokine receptor CCR2 and its ligand CCL2 [14, 15, 16]. Ly6C+ circulating monocytes can then migrate into peripheral tissues where they differentiate into macrophages or dendritic cells (DC), both in steady‐state and during inflammation [10, 17, 18]. Monocyte migration from blood into inflamed tissues has been found to be mostly dependent on CCL2 and/or CCL5, depending on inflammatory contexts [16, 19, 20, 21]. During inflammation, monocytes recruited to inflamed tissues not only act as precursors for macrophages and DC but also secrete large amounts of pro‐inflammatory or pro‐repair mediators depending on the context [10]. Dysregulation of monocyte trafficking can lead to tissue damage or pathology, including chronic inflammatory disorders and fibrosis [10]. Yet, how numbers of recruited monocytes are fine‐tuned during inflammation remains unclear. Here, we show that IL1R2 acts as a negative regulator of monocyte recruitment to inflamed tissues.
2. Results and Discussion
To address the role of IL1R2 in monocytes, we generated mice with a conditional deletion of Il1r2 in monocytes, using a strain with YFP expression and tamoxifen‐inducible Cre recombinase expression under the control of the Cx3cr1 promoter. To assess whether Il1r2 deficiency had an impact on monocyte homeostasis, we evaluated monocyte abundance in steady‐state conditions using flow cytometry. We also included neutrophils for comparison. We found no difference between Cx3cr1*Il1r2‐deficient and WT mice in Ly6C+ monocytes numbers in the bone marrow (Figure 1A,B) and blood (Figure 1C,D), showing that their generation and maintenance were unchanged. There was also no difference in CD43+ monocytes numbers, suggesting that Ly6C+ monocytes conversion into CD43+ monocytes was unaffected by Il1r2 deficiency. CCR2 expression was also similar between Il1r2‐deficient and WT monocytes (Figure 1E). Collectively, these results indicate that the homeostatic life cycle of Il1r2‐deficient monocytes is unaltered.
FIGURE 1.
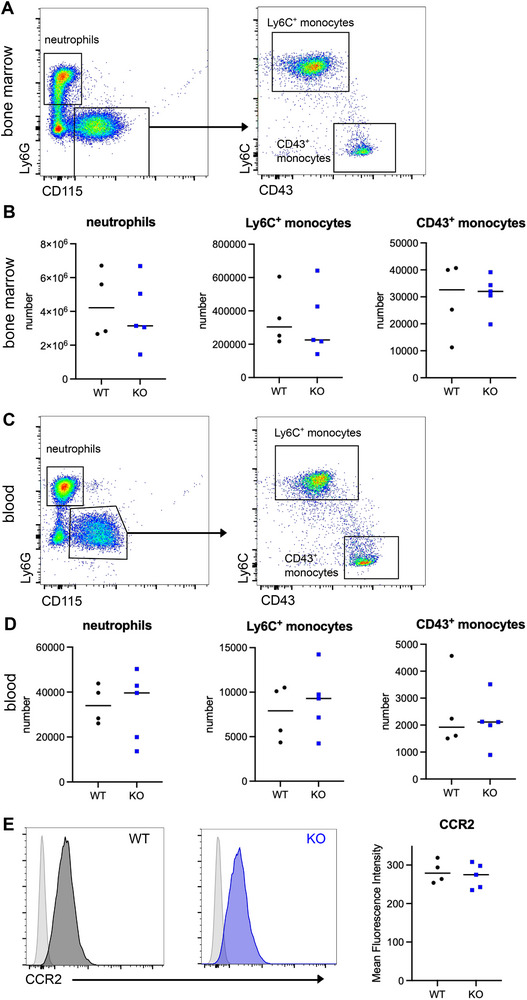
Lack of IL1R2 expression in monocytes does not impact their steady‐state life cycle. Bone marrow (A, B, E) and blood (C and D) from Cx3cr1*Ili1r2‐deficient mice or WT littermates were analyzed. (A and C) Gating strategy. Gated on live singlets TCR‐β−CD19−CD11b+ cells. Representative results are shown. (B) Number of monocytes and neutrophils in bone marrow from one leg. Median is shown (n = 4–5 mice in two independent experiments). (D) Number of monocytes and neutrophils in 50 µL of blood. Median is shown (n = 4–5 mice in two independent experiments). (E) CCR2 expression on Ly6C+ monocytes from bone marrow. Representative results are shown. Light gray histograms represent fluorescence‐minus‐one controls. Mean fluorescence intensity, median is shown (n = 4–5 mice in two independent experiments).
We then sought to address the potential role of IL1R2 in monocytes during inflammation. We first used a sterile peritonitis model, on the basis of the thioglycolate intraperitoneal injection. In this model, IL1‐β is secreted by recruited neutrophils within 4 h [22]. In addition, we have previously evidenced in this model the recruitment of Ly6C+ monocytes in 24–48 h and their progressive differentiation into monocyte‐derived macrophages (mo‐Mac) or monocyte‐derived DCs (mo‐DC) [23]. We verified which cell populations were targets of Il1r2 deletion in the inflamed peritoneum by taking advantage of YFP expression in Cre+ cells. We assessed YFP staining by flow cytometry in cells from the peritoneal lavage (Figure 2A and Figure S1). We detected YFP as expected in monocytes and monocyte‐derived cells, but not in neutrophils. We also detected YFP in around 50% of cDC2, suggesting that cDC2 may be partially deleted for Il1r2. Nevertheless, because cDC2 represent a minor cell population compared to monocytes (Figure 2B), the contribution to the observed phenotype of Il1r2 deficiency in cDC2 should be negligible. Neutrophils abundance in inflamed peritoneum was similar in Cx3cr1*Il1r2‐deficient and WT mice (Figure 2B), suggesting that their recruitment during the early phase of inflammation was unaffected. By contrast, monocytes, mo‐DC, and mo‐Mac were significantly more abundant in Cx3cr1*Il1r2‐deficient mice compared to WT littermates, whereas resident macrophages were unchanged (Figure 2B). These results indicate an increased recruitment of Il1r2‐deficient monocytes into the inflamed peritoneum. Consistent with this, CCL2 concentration in the inflamed peritoneum was higher in Cx3cr1*Il1r2‐deficient mice compared to WT littermates (Figure 2C), whereas IL‐1β was virtually undetectable at this time point in the peritoneum (Figure 2D). We then assessed cell numbers in blood during peritonitis. We found no difference in neutrophils, Ly6C+ monocytes or CD43+ monocytes numbers (Figure 2E), suggesting that monocyte egress from bone marrow was similar between Cx3cr1*Il1r2‐deficient or WT mice. Blood concentration of CCL2 and IL‐1β was unchanged between Cx3cr1*Il1r2‐deficient mice and WT littermates (Figure 2F,G). Collectively, these results support the idea that Il1r2 deficiency in infiltrating monocytes augments CCL2 production specifically in the inflamed peritoneum, thereby amplifying monocyte recruitment from blood.
FIGURE 2.
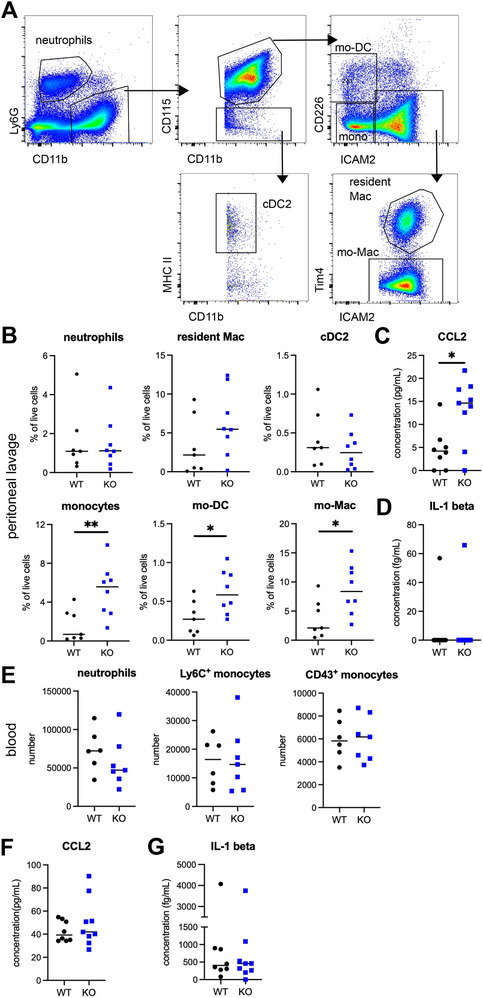
Il1r2‐deficient monocytes show increased recruitment into inflamed peritoneum. Peritonitis was induced in Cx3cr1*Il1r2‐deficient mice and WT littermates by injection of thioglycolate. (A) Gating strategy for peritoneal lavage analysis. Neutrophils were gated as Ly6G+CD11b+. CD11b+CD115+ cells were separated into monocytes, mo‐DC, mo‐Mac (ICAM2+Tim4−), and resident macrophages (Res Mac, ICAM2+TIM4+). cDC2 were gated as CD11b+CD115−MHCII+ cells. (B) Abundance of indicated cell types in peritoneal lavage (B) after 48 h. Median is shown (n = 6–8 mice in two independent experiments). Mann–Whitney test, *p < 0.05, **p < 0.01. (C and D) CCL2 and IL‐1β concentration in peritoneal lavage after 48 h. Median is shown (n = 8–9 mice in two independent experiments). (E) Abundance of indicated cell types in 50 µL of blood after 48 h. Median is shown (n = 6–8 mice in two independent experiments). (F and G) CCL2 and IL‐1β concentration in peritoneal lavage after 48 h. Median is shown (n = 8–9 mice in two independent experiments). mo‐DC, monocyte‐derived dendritic cell.
In our previous work [23], we observed that Il1r2 expression was increased during human monocyte differentiation specifically in cells committed to DC differentiation. In addition, reanalysis of transcriptomic data showed that Il1r2 was also more expressed in vivo in peritoneal mo‐DC compared to mo‐Mac, in both mouse (Immgen dataset) and human [24] (Figure S2A). These observations could indicate a role for IL1R2 in monocyte differentiation into mo‐DC. However, we found no difference between Cx3cr1*Il1r2‐deficient and WT mice in the relative proportion of mo‐DC versus mo‐Mac, neither in steady‐state nor inflamed peritoneum (Figure S2B). These results suggest that Il1r2 expression is not involved in monocyte fate commitment toward mo‐DC versus mo‐Mac. Instead, Il1r2 may be part of a gene program preferentially expressed in DC populations. Consistent with this hypothesis, Il1r2 was found in transcriptomic signatures of various DC populations in single‐cell RNA‐seq studies [25, 26, 27, 28].
Finally, we addressed the impact of Il1r2 deficiency in monocytes in a model of neuro‐inflammation, experimental autoimmune encephalomyelitis (EAE), in which monocyte recruitment [29, 30, 31] and IL1‐β production [32, 33] play major roles in the pathology. We first analyzed immune cell populations in lymph nodes draining the site of autoantigen immunization during the initiation phase, before disease onset. We verified which cell populations were targets of Il1r2 deletion in the inflamed lymph nodes by assessing YFP expression (Figure 3A and Figure S3A). Similar to the peritoneum, we detected YFP expression in monocytes and mo‐DC and at low levels in cDC2. Monocyte and mo‐DC numbers into lymph nodes 7 days after induction were increased in Cx3cr1*Il1r2‐deficient mice compared to WT mice (Figure 3A,B), whereas other cell types were unaffected (Figure S3B). The abundance of autoantigen‐specific CD4 T cells was also increased in the lymph nodes of Cx3cr1*Il1r2‐deficient mice (Figure 3C), indicating enhanced priming of pathogenic T cells in deficient mice. This is consistent with the fact that monocyte‐derived cells are the main antigen‐presenting cells involved in this model [34, 35]. These observations suggest that Il1r2 deficiency in monocytes increases monocyte recruitment into inflamed lymph nodes. Finally, Cx3cr1*Il1r2‐deficient mice were more sensitive to pathology than WT littermates, with higher incidence and more severe symptoms (Figure 3D,E). Overall, these results demonstrate that Il1r2 deficiency in monocytes dysregulates their recruitment into inflamed lymph nodes and exacerbates pathogenesis in a model of autoimmune inflammation.
FIGURE 3.
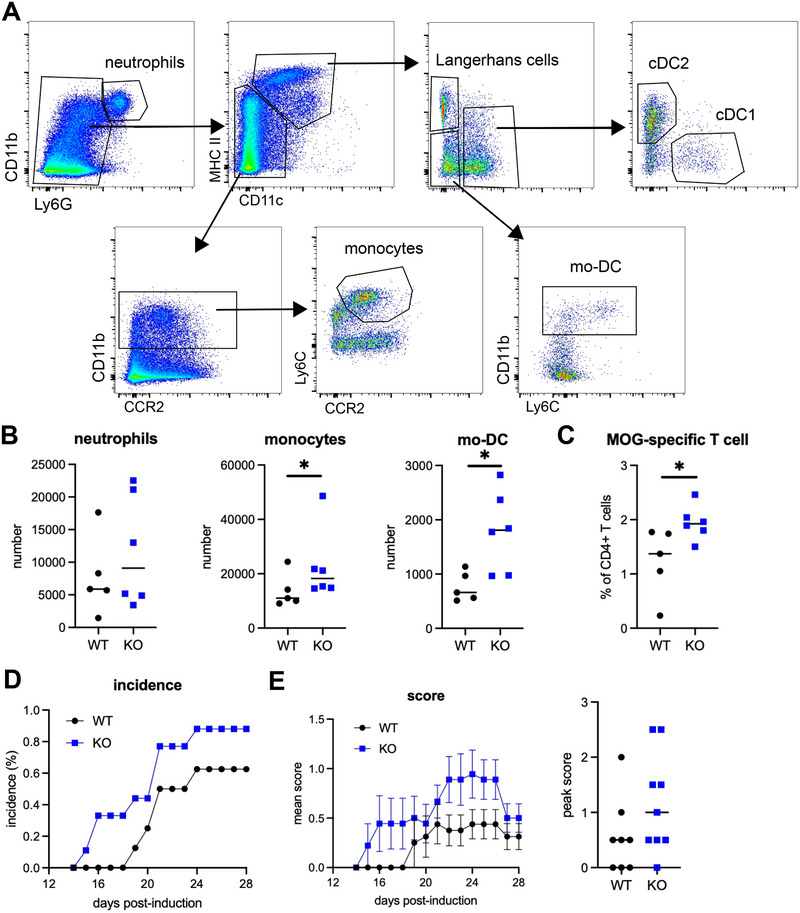
Lack of IL1R2 expression in monocytes worsens autoimmune neuro‐inflammation. Experimental autoimmune encephalomyelitis (EAE) was induced in Cx3cr1*Il1r2‐deficient mice or WT littermates by injection of MOG peptide. (A–C) Lymph nodes draining the site of MOG injection were analyzed 7 days after immunization. (A) Gating strategy for lymph node cells. (B) Numbers of indicated cell types in lymph nodes. Median is shown (n = 5–6 mice in two independent experiments). Mann–Whitney test, *p < 0.05. (C) Abundance of MOG‐specific CD4 T cells in lymph nodes. Median is shown (n = 5–6 mice in two independent experiments). Mann–Whitney test, *p < 0.05. (D) Percentage of mice having developed symptoms. (E) Mean clinical score over time. Bars represent SEM. Peak clinical score (n = 8–9 mice in two independent experiments). DC, dendritic cells; mo‐DC, monocyte‐derived DCs; MOG, myelin oligodendrocyte glycoprotein.
In conclusion, we show that Il1r2 deficiency in monocytes does not affect their steady‐state life cycle but dysregulates their trafficking to inflamed tissues, potentially aggravating monocyte‐driven inflammatory damage. On the basis of our results, we propose a model whereby monocytes, following their recruitment to inflamed tissues, participate through IL1R2 expression in the local dampening of IL1‐β activity, thereby limiting the production of chemo‐attractants and the infiltration of additional monocytes, and ultimately favoring the resolution of inflammation. Of note, IL1‐β signaling was shown to induce CCL2 production in multiple cell types in vivo [36]. Mechanistically, IL1‐β activity may be neutralized by decreasing IL1‐β local availability. Indeed, inflammatory mediators were shown to induce IL1R2 shedding from the cell surface of monocyte ex vivo [7].
Our findings reveal a new facet of the anti‐inflammatory actions of IL1R2 [3, 4, 5] and shed light onto its specific role in monocyte biology.
3. Data Limitations and Perspectives
Because the expression of the Cre recombinase is not entirely restricted to monocytes and monocyte‐derived cells, we cannot exclude that the observed phenotype may be influenced by partial Il1r2 deficiency in cDC2, especially in inflamed lymph nodes where cDC2 are present in the same order of magnitude as monocytes. In addition, because microglia are also targeted in this genetic construction [12] and express IL1R2 [37], we cannot exclude that Il1r2 deficiency in microglia might also contribute to the observed exacerbated neuro‐inflammation. Finally, another limitation of the study is the fact that we could not detect IL1‐β in the inflamed peritoneum at the time point analyzed. Further work would be needed to definitely demonstrate that Il1r2 deficiency in monocytes affects IL1‐β production, by analyzing shorter time points after the induction of inflammation.
4. Materials and Methods
4.1. Animals
Cx3cr1‐creERT2 (B6.129P2(Cg)‐Cx3cr1tm2.1(cre/ERT2)Litt /WganJ) mice were obtained from Jackson Laboratory (strain 021160) [38]. Il1r2‐flox mice (C57BL/6N‐A<tm1Brd>Il1r2< tm1c(EUCOMM)Wtsi>/WtsiPh) were obtained from the INFRAFRONTIER/EMMA consortium (strain EM:14887, www.infrafrontier.eu) [39, 40], and associated primary phenotypic information can be found at www.mousephenotype.org. Cx3cr1*Il1r2 strain was generated in‐house by crossing Cx3cr1‐creERT2 and Il1r2‐flox mice. All mice were on C57BL/6J background. Mice were maintained under specific pathogen‐free conditions at the animal facility of Institut Curie in accordance with institutional guidelines. Mice were housed in a 12 h light/12 h dark environment, with ad libitum access to water and food. Cx3cr1*Il1r2 female and male mice (Il1r2flox/flox Cre± and Cre−/− littermates) were used and sacrificed at age 8–14 weeks. All animal procedures were in accordance with the guidelines and regulations of the French Veterinary Department and have been approved by the local ethics committee (authorization APAFIS #25217‐2020042522586261 v1). To avoid confounding effects of circadian rhythms on monocyte biology, mice were always sacrificed for analysis at the same time of day (90 min after light onset).
4.2. Flow Cytometry
Cells were stained for 30–45 min on ice with indicated antibody cocktails supplemented with Fc block (BD Biosciences) in sterile FACS buffer: PBS (Eurobio Scientific, #CS1PBS01‐01) containing 0.5% BSA (Euromedex, #04‐100‐812‐C) and 2 mM EDTA (Invitrogen, #15575‐038). After washing with FACS buffer, cells were resuspended in FACS buffer containing DAPI (Fischer Scientific, 100 ng/mL). Cells were acquired on a ZE5 instrument (Bio‐Rad) using volumetric counting. Supervised analysis was performed using FlowJo software v10 (FlowJo LLC).
4.3. Bone Marrow and Blood Cells Analysis
For steady‐state analysis, all mice (Cre± and Cre−/− littermates) were treated with 5 mg of tamoxifen (Sigma, #T5648) resuspended in corn oil (Sigma, #C8267) by oral gavage, on Days 0 and 3 and sacrificed on Day 4. Bone marrows were flushed out from leg bones and filtered using 40 µm cell strainers. Blood of 50 µL was analyzed. Bone marrow and blood cell suspensions were treated with sterile red blood cells lysis buffer Hybri‐Max (Sigma, #R7757) for 3 min at room temperature.
Cells were stained with anti‐TCRβ BUV737 (BD Bioscience, clone H57‐597), anti‐CD19 BV480 (BD Bioscience clone 1D3), anti‐Ly6G APC‐Cy7 (Biolegend, clone 1A8), anti‐CCR2 BV711 (BD Bioscience, clone 475301), anti‐CD43 BB700 (BD Bioscience, clone S7), anti‐CD11b PeDazzle594 (BD Bioscience, clone M1/70), anti‐Ly6C PeCy7 (Biolegend, clone HK1.4), and anti‐CD115 APC (BD Bioscience, clone AFS98).
4.4. Peritoneal Cells Analysis
For experimental peritonitis, all mice (Cre± and Cre−/− littermates) were treated with 5 mg of tamoxifen resuspended in corn oil by oral gavage, on Days 0 and 3. On Day 5, mice received a third gavage of tamoxifen and were injected intraperitoneally with 1 mL of sterile 3.8% brewer's thioglycolate medium (Sigma, #B2551). Thioglycolate was sterilized by autoclaving at 15 lbs. pressure (121°C) for 15 min. Mice were analyzed on Day 7 (48 h after thioglycolate injection). Peritoneal lavage was recovered by intraperitoneal injection of 3 mL of sterile PBS.
Cells were stained with anti‐CD115 BUV 395 (BD Bioscience, clone AFS98), anti‐Ly6G APC‐Cy7 (Biolegend, clone 1A8), and anti‐ICAM2 biotin (Biolegend, clone 3C4) followed by staining with streptavidin‐BV605 (BD Bioscience), anti‐MHC II BV650 (Biolegend, clone M5/114.15.2), anti‐CD226 PE‐Dazzle594 (Biolegend, clone 10E5), anti‐CD11b PerCP‐Cy5.5 (BD Bioscience, clone M1/70), and anti‐TIM4 Alexa647 (Biolegend, clone RMT4‐54).
4.5. Experimental Autoimmune Encephalomyelitis
All mice (cre± and cre−/− littermates) were treated with 5 mg of tamoxifen resuspended in corn oil by oral gavage on Days 0, 3, 5, and 7. On Day 5, mice were immunized subcutaneously in the back with 100 µg myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide (sb‐PEPTIDE) emulsified in Incomplete Freud's Adjuvant (Invivogen) supplemented with 4 mg/mL desiccated Mycobacterium tuberculosis (H37RA, Sigma). Mice were injected intraperitoneally with 200 ng of pertussis toxin from Bordetella pertussis (Calbiochem) on Days 5 and 7 (0 and 48 h after immunization). Mice were examined daily for clinical signs. In agreement with the local ethics committee, mice were scored as follows: 0 healthy; 0.5 tail weakness; 1 limp tail; 1.5 tail paralysis and hindlimb weakness; 2 tail paralysis and limping of one hindlimb; 2.5 tail paralysis and limping of both hindlimbs; 3 paralysis of tail and both hindlimbs; 3.5 paralysis of tail and both hindlimbs, and weakness in forelimbs. Score 3 was predefined as the humane endpoint of the experiment.
4.6. Lymph Node Cells Analysis
EAE was induced as above. Inguinal lymph nodes were collected 7 days post MOG immunization. For tetramer staining, lymph nodes were dissociated by forcing through a 40 µm cell strainer. Cells were incubated for 3 h at 37°C in RPMI GlutaMax (Gibco, #61870‐010) containing 10% fetal calf serum (Biosera, #FB‐1001/500) in the presence of PE‐conjugated MOG tetramer (I‐A(b) GWYRSPFSRVVH, 2.7 mg/mL) or control tetramer (I‐A(b) PVSKMRMATPLLMQA, 2.7 mg/mL) (both obtained from the NIH tetramer core facility). After washing, cells were stained with anti‐CD8 BUV395 (BD Bioscience, clone 53‐6.7), anti‐TCRβ BUV737 (BD Bioscience, clone H57‐597), anti‐CD19 BV480 (BD Bioscience clone 1D3), anti‐CD4 PerCPCy5.5 (BD Bioscience, clone RM4‐5), and anti‐CD11b PeCy7 (BD Bioscience, clone M1/70). Cells were gated on live single CD19−CD11b−CD8−TCRβ+CD4+ cells.
For flow cytometry of myeloid cells, lymph nodes were cut into small pieces and incubated for 30 min at 37°C in sterile digestion mix: RPMI containing 0.4 mg/mL DNAse I (Sigma) and 0.5 mg/mL collagenase d (Roche). Cell suspensions were then filtered using 40 µm cell strainers. Cells were stained with anti‐CCR2 BV711 (BD Biosciences, clone 475301), anti‐CD19 BV480 (BD Biosciences, clone 1D3), anti‐CD3 BV480 (BD Biosciences, clone 500A2), anti‐NK1.1 BV480 (BD Bioscience, clone PK136), anti‐SiglecF BV480 (BD Bioscience, clone E50‐2440), anti‐CD11c BV785 (Biolegend, clone N418), anti‐Ly6G BV605 (Biolegend, clone 1A8), anti‐CD11b PerCP‐Cy5.5 (BD Bioscience, clone M1/70), anti‐MHC II BV650 (Biolegend, clone M5/114.15.2), anti‐Ly6C Alexa700 (Biolegend, clone HK1.4), anti‐XCR1 AF647 (BioLegend, clone ZET), anti‐CD26 PE (BioLegend, clone H194‐112), and anti‐EpCAM APCFire750 (BioLegend, clone G8.8). Cells were gated on live single NK1.1−SiglecF−CD19− TCRβ− cells.
4.7. Cytokine Measurement
Blood was collected in micro‐sample tubes with lithium heparin (Sarstedt, #41.1393.005) and left at room temperature for 3 h. After centrifugation (450 g, 10 min), serum was collected for analysis and kept at −20°C. CCL2 concentration was measured in serum and in peritoneal lavage using CBA (BD Biosciences). The limit of detection was 10 pg/mL. IL1‐β concentration was measured using Enhanced Sensitivity CBA (BD Biosciences). The limit of detection was 274 fg/mL.
4.8. Analysis of Public Datasets
Datasets were downloaded from GEO and Immgen (www.immgen.org).
4.9. Statistical Analysis
Statistical tests were performed using Prism v10 (GraphPad Software). Mann–Whitney test was used for all data. Absence of stars indicates “not significant.” N corresponds to the number of independent biological replicates analyzed.
Author Contributions
Adeline Cros: investigation, writing–review and editing. Elodie Segura: investigation, conceptualization, formal analysis, supervision, writing–original draft, writing–review and editing.
Conflicts of Interest
The authors declare no conflicts of interest.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202451468.
Supporting information
Supporting Information
Acknowledgments
This work was funded by INSERM, Institut Curie, Agence Nationale de la Recherche (ANR‐10‐LABX‐0043, ANR‐10‐IDEX‐0001‐02 PSL), and Fondation ARSEP (Grant #1277). The authors thank the Wellcome Sanger Institute and its funders (www.sanger.ac.uk/mouseportal) for providing the Il1r2 mutant mouse line, and EMMA partner Institute of Molecular Genetics of the Czech Academy of Sciences from which the mouse was received. The authors wish to thank the Flow Cytometry Core and the In vivo Experiments Platform of Institut Curie.
Funding: This work was funded by INSERM, Institut Curie (core funding), Agence Nationale de la Recherche (Grant ANR‐10‐LABX‐0043, ANR‐10‐IDEX‐0001‐02 PSL), Wellcome Sanger Institute, Institut National de la Santé et de la Recherche Médicale (core funding), and Fondation ARSEP (Grant #1277).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Mantovani A., Dinarello C. A., Molgora M., and Garlanda C., “Interleukin‐1 and Related Cytokines in the Regulation of Inflammation and Immunity,” Immunity 50 (2019): 778–795, http://www.cell.com/article/S1074761319301293/fulltext, 10.1016/J.IMMUNI.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Broderick L. and Hoffman H. M., “IL‐1 and Autoinflammatory Disease: Biology, Pathogenesis and Therapeutic Targeting,” Nature Reviews Rheumatology 18 (2022): 448–463, https://www.nature.com/articles/s41584‐022‐00797‐1, 10.1038/s41584-022-00797-1. 2022 18:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Supino D., Minute L., Mariancini A., Riva F., Magrini E., and Garlanda C., “Negative Regulation of the IL‐1 System by IL‐1R2 and IL‐1R8: Relevance in Pathophysiology and Disease,” Frontiers in Immunology 13 (2022): 804641, https://www.frontiersin.org, 10.3389/FIMMU.2022.804641/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peters V. A., Joesting J. J., and Freund G. G., “IL‐1 Receptor 2 (IL‐1R2) and Its Role in Immune Regulation,” Brain, Behavior, and Immunity 32 (2013): 1–8, 10.1016/J.BBI.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colotta F., Re F., Muzio M., et al., “Interleukin‐1 Type II Receptor: A Decoy Target for IL‐1 That is Regulated by IL‐4,” Science 261 (1993): 472–475, https://www.science.org/, 10.1126/SCIENCE.8332913. [DOI] [PubMed] [Google Scholar]
- 6. Martin P., Palmer G., Vigne S., et al., “Mouse Neutrophils Express the Decoy Type 2 Interleukin‐1 Receptor (IL‐1R2) Constitutively and in Acute Inflammatory Conditions,” Journal of Leukocyte Biology 94 (2013): 791–802, https://pubmed.ncbi.nlm.nih.gov/23817563/, 10.1189/JLB.0113035. [DOI] [PubMed] [Google Scholar]
- 7. Giai C., Gonzalez C. D., Sabbione F., et al., “ Staphylococcus aureus Induces Shedding of IL‐1RII in Monocytes and Neutrophils,” Journal of Innate Immunity 8 (2016): 284–298, 10.1159/000443663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shimizu K., Nakajima A., Sudo K., et al., “IL‐1 Receptor Type 2 Suppresses Collagen‐Induced Arthritis by Inhibiting IL‐1 Signal on Macrophages,” Journal of Immunology 194 (2015): 3156–3168, 10.4049/jimmunol.1402155. [DOI] [PubMed] [Google Scholar]
- 9. Martin P., Palmer G., Rodriguez E., et al., “Deficiency in IL‐1 Receptor Type 2 Aggravates K/BxN Serum Transfer‐Induced Arthritis in Mice But Has no Impact on Systemic Inflammatory Responses,” Journal of Immunology 198 (2017): 2916–2926, https://pubmed.ncbi.nlm.nih.gov/28235865/, 10.4049/JIMMUNOL.1600855. [DOI] [PubMed] [Google Scholar]
- 10. Guilliams M., Mildner A., and Yona S., “Developmental and Functional Heterogeneity of Monocytes,” Immunity 49 (2018): 595–613, 10.1016/j.immuni.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 11. Trzebanski S. and Jung S., “Plasticity of Monocyte Development and Monocyte Fates,” Immunology Letters 227 (2020): 66–78, https://www.sciencedirect.com/science/article/pii/S016524782030362X, 10.1016/j.imlet.2020.07.007. [DOI] [PubMed] [Google Scholar]
- 12. Yona S., Kim K.‐W., Wolf Y., et al., “Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages Under Homeostasis,” Immunity 38 (2013): 79–91, https://www.sciencedirect.com/science/article/pii/S1074761312005481, 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mildner A., Schönheit J., Giladi A., et al., “Genomic Characterization of Murine Monocytes Reveals C/EBP$β$ Transcription Factor Dependence of Ly6C− Cells,” Immunity 46 (2017): 849–862, 10.1016/J.IMMUNI.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 14. Tsou C.‐L., Peters W., Si Y., et al., “Critical Roles for CCR2 and MCP‐3 in Monocyte Mobilization From Bone Marrow and Recruitment to Inflammatory Sites,” Journal of Clinical Investigation 117 (2007): 902–909, 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Serbina N. V. and Pamer E. G., “Monocyte Emigration From Bone Marrow During Bacterial Infection Requires Signals Mediated by Chemokine Receptor CCR2,” Nature Immunology 7 (2006): 311–317, 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 16. Lu B., Rutledge B. J., Gu L., et al., “Abnormalities in Monocyte Recruitment and Cytokine Expression in Monocyte Chemoattractant Protein 1–Deficient Mice,” Journal of Experimental Medicine 187 (1998): 601–608, 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trzebanski S. and Jung S., “Plasticity of Monocyte Development and Monocyte Fates,” Immunology Letters 227 (2020): 66–78, 10.1016/j.imlet.2020.07.007. [DOI] [PubMed] [Google Scholar]
- 18. Rigamonti A., Villar J., and Segura E., “Monocyte Differentiation Within Tissues: A Renewed Outlook,” Trends in Immunology 44 (2023): 999–1013, 10.1016/J.IT.2023.10.005. [DOI] [PubMed] [Google Scholar]
- 19. Shi C. and Pamer E. G., “Monocyte Recruitment During Infection and Inflammation,” Nature Reviews Immunology 11 (2011): 762–774, 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ajuebor M. N., Flower R. J., Hannon R., et al., “Endogenous Monocyte Chemoattractant Protein‐1 Recruits Monocytes in the Zymosan Peritonitis Model,” Journal of Leukocyte Biology 63 (1998): 108–116, 10.1002/jlb.63.1.108. [DOI] [PubMed] [Google Scholar]
- 21. Paul D., Ge S., Lemire Y., et al., “Cell‐Selective Knockout and 3D Confocal Image Analysis Reveals Separate Roles for Astrocyte‐and Endothelial‐Derived CCL2 in Neuroinflammation,” Journal of Neuroinflammation 11 (2014): 10, https://pmc/articles/PMC3906899/, 10.1186/1742-2094-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fukui S., Fukui S., Van Bruggen S., et al., “NLRP3 inflammasome Activation in Neutrophils Directs Early Inflammatory Response in Murine Peritonitis,” Scientific Reports 12 (2022): 1–9, https://www.nature.com/articles/s41598‐022‐25176‐4, 10.1038/s41598-022-25176-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Villar J., Ea Ouaknin L., Cros A., and Segura E., “Monocytes Differentiate Along Two Alternative Pathways During Sterile Inflammation,” EMBO Reports 24 (2023): e56308, https://onlinelibrary.wiley.com/doi/full/, 10.15252/embr.202256308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Segura E., Touzot M., Bohineust A., et al., “Human Inflammatory Dendritic Cells Induce Th17 Cell Differentiation,” Immunity 38 (2013): 336–348, 10.1016/j.immuni.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 25. Rodrigues P. F., Trsan T., Cvijetic G., et al., “Progenitors of Distinct Lineages Shape the Diversity of Mature Type 2 Conventional Dendritic Cells,” Immunity 57 (2024): 1567–1585.e5, http://www.cell.com/article/S1074761324002607/fulltext, 10.1016/j.immuni.2024.05.007. [DOI] [PubMed] [Google Scholar]
- 26. Hughes T. K., Wadsworth M. H., Gierahn T. M., et al., “Second‐Strand Synthesis‐Based Massively Parallel scRNA‐Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies,” Immunity 53 (2020): 878–894, 10.1016/J.IMMUNI.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakamizo S., Dutertre C. A., Khalilnezhad A., et al., “Single‐Cell Analysis of Human Skin Identifies CD14+ Type 3 Dendritic Cells Co‐Producing IL1B and IL23A in Psoriasis,” Journal of Experimental Medicine 218 (2021): e20202345, 10.1084/JEM.20202345/212481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao Y., Li H., Li Z., et al., “Single‐Cell Analysis Reveals the Heterogeneity of Monocyte‐Derived and Peripheral Type‐2 Conventional Dendritic Cells,” Journal of Immunology 207 (2021): 837–848, 10.4049/JIMMUNOL.2100094. [DOI] [PubMed] [Google Scholar]
- 29. Ajami B., Bennett J. L., Krieger C., McNagny K. M., and Rossi F. M. V., “Infiltrating Monocytes Trigger EAE Progression, but Do Not Contribute to the Resident Microglia Pool,” Nature Neuroscience 14 (2011): 1142–1149, https://www.nature.com/articles/nn.2887, 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 30. Mildner A., Mack M., Schmidt H., et al., “CCR2+Ly‐6Chi Monocytes Are Crucial for the Effector Phase of Autoimmunity in the Central Nervous System,” Brain 132 (2009): 2487–2500, 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- 31. Ge S., Shrestha B., Paul D., et al., “The CCL2 Synthesis Inhibitor Bindarit Targets Cells of the Neurovascular Unit, and Suppresses Experimental Autoimmune Encephalomyelitis,” Journal of Neuroinflammation 9 (2012): 1–13, https://jneuroinflammation.biomedcentral.com/articles/, 10.1186/1742-2094-9-171/FIGURES/7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ronchi F., Basso C., Preite S., et al., “Experimental Priming of Encephalitogenic Th1/Th17 Cells Requires Pertussis Toxin‐Driven IL‐1β Production by Myeloid Cells,” Nature Communications 7 (2016): 1–11, https://www.nature.com/articles/ncomms11541, 10.1038/ncomms11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paré A., Mailhot B., Lévesque S. A., et al., “IL‐1$β$ Enables CNS Access to CCR2hi Monocytes and the Generation of Pathogenic Cells Through GM‐CSF Released by CNS Endothelial Cells,” PNAS 115 (2018): E1194–E1203, 10.1073/pnas.1714948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Villar J., Cros A., De Juan A., et al., “ETV3 and ETV6 Enable Monocyte Differentiation Into Dendritic Cells by Repressing Macrophage Fate Commitment,” Nature Immunology 24 (2022): 84–95, https://www.nature.com/articles/s41590‐022‐01374‐0, 10.1038/s41590-022-01374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Amorim A., De Feo D., Friebel E., et al., “IFNγ and GM‐CSF Control Complementary Differentiation Programs in the Monocyte‐to‐Phagocyte Transition During Neuroinflammation,” Nature Immunology 23 (2022): 217–228, 10.1038/s41590-021-01117-7. [DOI] [PubMed] [Google Scholar]
- 36. Liu X., Nemeth D. P., McKim D. B., et al., “Cell‐Type‐Specific Interleukin 1 Receptor 1 Signaling in the Brain Regulates Distinct Neuroimmune Activities,” Immunity 50 (2019): 317–333.e6, 10.1016/j.immuni.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pinteaux E., Parker L. C., Rothwell N. J., and Luheshi G. N., “Expression of Interleukin‐1 Receptors and Their Role in Interleukin‐1 Actions in Murine Microglial Cells,” Journal of Neurochemistry 83 (2002): 754–763, 10.1046/j.1471-4159.2002.01184.x. [DOI] [PubMed] [Google Scholar]
- 38. Parkhurst C. N., Yang G., Ninan I., et al., “Microglia Promote Learning‐Dependent Synapse Formation Through BDNF,” Cell 155 (2019): 1596–1609, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf, 10.1016/j.cell.2013.11.030.Microglia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Skarnes W. C., Rosen B., West A. P., et al., “A Conditional Knockout Resource for the Genome‐Wide Study of Mouse Gene Function,” Nature 474 (2011): 337–344, https://pubmed.ncbi.nlm.nih.gov/21677750/, 10.1038/NATURE10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meehan T. F., Chen C. K., Koscielny G., et al., “INFRAFRONTIER–Providing Mutant Mouse Resources as Research Tools for the International Scientific Community,” Nucleic Acids Research 43 (2015): D1171–D1175, https://pubmed.ncbi.nlm.nih.gov/25414328/, 10.1093/NAR/GKU1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.