

Abstract
Mavacamten, the first drug in the class of β‐cardiac myosin modulator, is used for the treatment of patients with hypertrophic cardiomyopathy. This orally administered drug demonstrates wide interpatient variability in pharmacokinetics parameters, due in part to variant CYP2C19 alleles. Individuals who are CYP2C19 poor metabolizers have increased exposure and are at increased risk of reduced cardiac hypercontractility. To ensure the safety of all patients, European Medicines Agency recommends CYP2C19 preemptive genotyping, and consecutively, to adapt maintenance and initial mavacamten doses, and to manage drug–drug interactions, according to CYP2C19 phenotype. In this article, we summarize evidence from the literature supporting the association between CYP2C19 phenotype and pharmacological features of mavacamten and provide, beyond biologic guidelines, therapeutic recommendations for the use of mavacamten based on CYP2C19 and CYP3A4/CYP3A5 genotype.
Symptomatic obstructive hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a complex disorder, that is caused by dysfunction of the cardiac sarcomere, resulting in excessive cardiac myosin–actin cross‐bridging. HCM has an estimated prevalence from 1 in 500 1 to 1 in 3000 2 in the US population. It is frequently inherited as an autosomal dominant trait with variable penetrance. In about 40% of all HCM patients, pathogenic variations occur in genes encoding for sarcomeric proteins such as myosin‐binding protein 3 and beta‐myosin heavy chain (MYH7). 3 Approximately two‐thirds of the patients with HCM have obstruction of the left ventricular outflow tract (LVOT), a major determinant of symptoms and outcomes. Patients with symptomatic obstructive hypertrophic cardiomyopathy (oHCM) present a spectrum of symptoms, ranging from dyspnea, palpitations to atrial fibrillation, ventricular tachycardia, stroke, heart failure, and sudden cardiac death. 4 Management has primarily involved oral beta‐blockers (such as propranolol, nadolol, and pindolol), and/or non‐dihydropyridine calcium channel blockers (verapamil/diltiazem), that slow heart rate and have modest negative inotropic action. In the second line, disopyramide, antiarrhythmic, may be added because of its additional negative inotropic effect. 5 In cases of inefficiency to pharmacological treatment, septal reduction therapy (septal myectomy or alcohol septal ablation) is recommended for patients with symptomatic oHCM. These procedures require substantial operator experience, which is limited to a few medical centers and associated with serious cardiovascular morbidity and mortality. 6
In 2022, mavacamten is the first molecule in the class of cardiac myosin modulators, approved by the US Food and Drug Administration (FDA) for the treatment of adults with symptomatic oHCM. The European Medicines Agency (EMA) granted approval for mavacamten in 2023 7 and the medication has since become available in France and Belgium under a pre‐authorization procedure. 8
Mavacamten pharmacodynamics
Mavacamten is an allosteric, reversible, and selective inhibitor of the ATPase activity of cardiac myosin. Its mechanism of action involves decreasing the number of myosin–actin interactions by inhibiting the ATPase of myosin, which ultimately reduces myocardial hyper‐contractility. 9 This medication decreases the LVOT obstruction gradient in patients with oHCM. Its treatment is also associated with the reduction in baseline systolic function, resulting in reduced cardiac contractility. Mavacamten therapy should be monitored using echocardiography measurements, assessing the Left ventricular outflow tract gradient (LVOTg) after the Valsalva maneuver for efficacy and the left ventricular ejection fraction (LVEF) for safety.
During the Phase II clinical trial (PIONEER‐HCM), the concentration‐effect relationship was highlighted showing that mavacamten concentration is linked with both LVOTg and LVEF measurements. 10 After 12 weeks, mavacamten substantially reduced LVOT obstruction at trough plasma concentrations between 350 and 695 ng/mL, with LVEF reduction in a concentration‐dependent manner. 10 Plasma concentrations above 695 ng/mL were associated with a reduction in LVEF below 50% (reversible at arrest). These results have conducted the mavacamten dose adjustments guided by echocardiographic (LVOTg and LVEF measurements) and therapeutic drug monitoring (TDM) in the phase III clinical trial (EXPLORER‐HCM), 11 with a common starting dose of 5 mg/day, until a maximum of 15 mg/day. This dosing‐adjustment schedule was based on maintaining mavacamten plasma concentration range between 350 and 700 ng/mL for 85% of patients. In this phase III clinical trial, mavacamten treatment should be discontinued when: (1) mavacamten plasma concentration was above 1,000 ng/mL, (2) LVEF fell below 50% or (3) QT interval prolonged (> 15% baseline). 10 , 11 In the pivotal EXPLORER‐HCM study, LVEF reduction to < 50% was not highly correlated with the mavacamten plasma concentrations: 57% events of LVEF < 50% occurred with mavacamten plasma concentrations < 700 ng/mL and 43% occurred with mavacamten concentrations > 700 ng/mL). 11 Nonetheless, in the event of ineffectiveness or toxicity attributable to the treatment, the mavacamten trough concentration may be monitored to estimate the patient's exposure to the drug.
Mavacamten pharmacokinetics
Mavacamten is highly metabolized in the liver, predominantly via the cytochromes (CYP) 2C19 (74%), 3A4 (18%), and 2C9 (8%), with only 3% of the drug eliminated unchanged. 12 The metabolites exhibit very low plasma levels (3–4% of that of mavacamten). This metabolization leads to high inter‐individual drug exposure variability. The CYP2C19 gene has 35 haplotypes described in PharmVar, 13 resulting in different levels of phenotype. In Caucasian 2% are poor metabolizer, 26% intermediate, 40% normal or extensive, 27% rapid, and 5% ultrarapid. 14 , 15 Elimination half‐life of mavacamten strongly depends on CYP2C19 phenotype: 23 days for poor metabolizer compared 7 to 10 days for CYP2C19 intermediate metabolizers, 9 days for normal metabolizers, 8 days for CYP2C19 rapid metabolizers and 6 days for CYP2C19 ultrarapid metabolizers. 7 Clearance is affected by CYP2C19 phenotype: AUC/dose was 4.6‐fold higher in poor metabolizers and 1.6‐fold lower in ultra‐rapid metabolizers compared with normal metabolizers. 7
Considering the influence of CYP2C19 phenotype on mavacamten pharmacokinetics, the EMA decided to guide the initial and maximal doses of mavacamten based on the CYP2C19 genotype (poor metabolizer vs. others or non‐poor) by testing for *2‐ or *3‐star alleles (section 5.2 of ref. [7]). Drug interactions management also depends on the CYP2C19 phenotype.
FDA recommendations differ from those of EMA: CYP2C19 genotyping is not required, 16 due to the lack of validated laboratories and to avoid delaying the drug's availability. CYP2C19 phenotype is therefore not taken into account in the maintenance and initial doses, or in the management of drug–drug interactions. 17 As the CYP2C19 phenotype is not known and mavacamten is partly metabolized via CYP3A4, the FDA contraindicates co‐prescribing strong CYP3A4 inhibitors, moderate to strong CYP3A4 inducers and/or strong‐to‐moderate CYP2C19 inhibitors or inducers, for all patients. According to the FDA, the echocardiographic monitoring conducted 4‐weekly will quickly detect the few susceptible patients, around 2% (poor CYP2C19 metabolizers) before serious adverse effects occur. The detection of two successive LVEF measurements below 50% will definitively contraindicate the medication. Using echocardiographic measurements and mavacamten exposure from all clinical trials, an exposure–response models and population pharmacokinetic models were constructed to simulate echo‐guided dose titration for mavacamten without requiring TDM. 18 The best regimen guided by echocardiography measures only was determined in this simulation study to have a similar safety and efficacy profile to that of a TDM/echo‐guided regimen (used in EXPLORER‐HCM) for mavacamten in patients with obstructive HCM, and is currently approved by the FDA. 18
The French‐speaking Network of Pharmacogenetic (RNPGx) is proposing guidelines to (i) precisely outline the dose adjustment of mavacamten and its pharmacogenetic relevance; (ii) identify the pharmacogenetic variant of interest with three levels of recommendations: essential, advisable, and possibly helpful. 19 This work is based on a scientific review of literature on mavacamten clinical trials, pharmacological, and genetic studies, and on the Clinical Pharmacogenetics Implementation Consortium (CPIC) data for CYP2C19, CYP3A4, and CYP3A5 haplotypes definitions, alleles frequencies and functional impact.
DOSE ADJUSTMENT OF MAVACAMTEN AND PHARMACOGENETICS
Principle
Mavacamten is available in non‐bioequivalent capsules in four different dosages: 2.5, 5, 10 and 15 mg. The EMA approved a dose titration scheme based on a 4‐weekly echocardiographic assessment of Valsalva LVOT gradient (target between 20 and 30 mmHg) for efficacy measurement and LVEF (target ≥ 55%) for safety.
The CYP2C19 metabolic status determines the mavacamten starting daily dose in Europe. The dose is continued if LVEF falls above 50% and the LVOTg above 20 mmHg (absence of over‐efficacy). Mavacamten must be stopped for 4 weeks if LVEF is below 50%. Then it can be reintroduced at a lower dose, if after 4 weeks LVEF returns to over 50%. Definitive discontinuation is recommended when two incidences of LVEF below 50% have occurred 4 weeks apart at the lower dose (2.5 mg/day).
During the maintenance phase, which requires echocardiographic assessment every 12 weeks, the dose may be increased if efficacy has not been achieved (LVOTg > 30 mmHg and LVEF ≥ 55%). The maximum dose will depend on CYP2C19 metabolizer status (see below).
Impact of CYP2C19 metabolizer phenotype
The aim of pre‐therapeutic genotyping is to ensure the safety of all patients, by avoiding mavacamten overexposure, which can lead to systolic dysfunction in CYP2C19 poor metabolizers. After the administration of a single 15 mg dose of mavacamten, overexposure was observed in poor metabolizers, with a 241% increase in the area under the curve (AUC from zero to infinity), compared with CYP2C19 normal metabolizers. 7 As mentioned earlier, the mean half‐life of mavacamten is also substantially prolonged in CYP2C19 poor metabolizers, compared with CYP2C19 normal metabolizers (23 days vs. 6–10 days). From phases I, II, and III clinical trial data, the exposure–response model and population pharmacokinetics model were used to construct the echocardiography‐guided regimen, recommended in the mavacamten prescribing information. These models show that the poor CYP2C19 metabolizer group presents more frequently an LVEF below 50% (2–4% of simulated patients in this group). 18 Thus, according to EMA recommendations, CYP2C19 genotyping is required to determine the starting and maximum dose of mavacamten in accordance with the CYP2C19 phenotype. 20
- In CYP2C19 poor metabolizers:
-
⚬Recommended starting dose: 2.5 mg/day
-
⚬Maximum dose: 5 mg/day
-
⚬
- In intermediate, normal, rapid, or ultra‐rapid CYP2C19 metabolizers:
-
⚬Recommended starting dose: 5 mg/day
-
⚬Maximum dose: 15 mg/day
-
⚬
- In patients of unknown CYP2C19 metabolizer phenotype:
-
⚬Recommended starting dose: 2.5 mg/day
-
⚬Maximum dose: 5 mg/day
-
⚬
The dose is then adjusted according to the presence of drug–drug interactions (Figure 1 ).
Figure 1.

Principles of mavacamten dose adjustment based on CYP2C19 metabolizer status and drug–drug interactions. CYP, cytochrome; LVEF, left ventricular ejection fraction; LVOTg, left ventricular outflow tract gradient.
Pharmacokinetic drug–drug interactions
Combined use of a strong CYP2C19 inhibitor and a strong CYP3A4 inhibitor which inhibit both of the main metabolic pathways of mavacamten is contraindicated with mavacamten to avoid overexposure that could cause heart failure due to systolic dysfunction. In physiologically‐based pharmacokinetic (PB/PK) models, the simulation with voriconazole 400–800 mg/day (strong CYP2C19 inhibitor and moderate/strong CYP3A4 inhibitor) increases mavacamten AUC0–24h of 3.02 to 5.62‐fold, according to CYP2C19 phenotype. 21 , 22 The simulation with fluconazole 200 mg/day (strong CYP3A4 inhibitor and moderate CYP2C19 inhibitor) increases the mavacamten AUC0–24h of 3.10 to 4.13‐fold, according to CYP2C19 phenotype. 17
In CYP2C19 poor metabolizer phenotype
In poor CYP2C19 metabolizers, mavacamten is metabolized predominantly by CYP3A4 and CYP3A5. 12 The introduction of CYP3A4 inhibitors/inducers, or modification of their dose, may affect mavacamten clearance and increase/decrease its plasma concentrations. Thus, strong CYP3A4 inhibitors are contraindicated in this phenotypic group. The introduction or increased dose of moderate CYP3A4 inhibitors requires a reduction in the mavacamten dose to 2.5 mg/day or discontinue treatment for 4 weeks, if the lower dose was used (see Table 1 ).
Table 1.
Mavacamten dose adjustment for CYP2C19 poor metabolizers with CYP2C19 and/or CYP3A4 inhibitors, according to the product information and with regard to pharmacokinetic parameters described
| Concomitant treatment Inhibitors | Ratio AUC (mavacamten + inhibitor)/AUC mavacamten | Mavacamten dose adjustment for CYP2C19 poor metabolizers |
|---|---|---|
|
Strong CYP2C19 inhibitor + Strong CYP3A4 inhibitor: Fluconazole‐Voriconazole |
 Contra‐indicated Contra‐indicated |
|
|
Strong CYP2C19 inhibitor: Fluvoxamine – Ticlopidine – |
[1.00] |
No adjustment of the starting dose of 2.5 mg
|
|
Strong CYP3A4 inhibitor: Ceritinib – Clarithromycin – Cobicistat – Erythromycin – Idelalisib – Itraconazole – Ketoconazole ‐ Ritonavir – Tucatinib |
[1.96–2.27] ketoconazole 400 mg/day with mavacamten 15 mg/day for 8 and 210 days (PB‐PK model) 16 [1.72–2.14] itraconazole 200 mg/day with mavacamten 15 mg/day for 8 and 210 days (PB‐PK model) 16 |
 Contra‐indicated Contra‐indicated |
|
Moderate or weak CYP2C19 inhibitor: Clopidogrel – Esomeprazole – Fluoxetine – Moclobemide – Omeprazole |
[1.00] |
No adjustment of the starting dose of 2.5 mg
|
|
Moderate or weak CYP3A4 inhibitor: Amiodarone – Diltiazem – Dronedarone – Ethinylestradiol – Gestodene – Grapefruit juice – Nifedipine – Nitrendipine – Quetiapine – Verapamil |
[1.49–1.62] diltiazem 60 mg 3×/day (PB‐PK model) 22 |
No adjustment of the starting dose of 2.5 mg Initiating or increasing the dose of a moderate or weak inhibitor during mavacamten treatment:
|
In PB/PK models, strong CYP3A4 inhibitors such as itraconazole 200 mg/day or ketoconazole 400 mg/day predicted a twofold increase in mavacamten AUC0–24h. 17 In addition, moderate CYP3A4 inhibitors (such as diltiazem 60 mg 3×/day) were predicted to increase AUC0–24h of 55%, in poor CYP2C19 metabolizers, while other metabolizers saw little change on the pharmacokinetic parameters. 23
The introduction of a weak CYP3A4 inhibitor (such as cimetidine 400 mg 2×/day) has little influence on the pharmacokinetic parameters of mavacamten, regardless of the CYP2C19 phenotype. 23 Conversely, in poor CYP2C19 metabolizers, the introduction of a strong CYP3A4 and CYP2C19 inducer, such as rifampicin 600 mg/day for 7 days reduces mavacamten AUC0–24h of 69%, requiring clinical and echocardiographic assessment before adjusting the mavacamten dose (Table 2 ).
Table 2.
Mavacamten dose adjustment for CYP2C19 poor metabolizers with CYP2C19 and/or CYP3A4 inducers, according to the product information and with regard to pharmacokinetic parameters described
| Concomitant treatment inducers | Ratio AUC (mavacamten + inducer)/AUC mavacamten | Mavacamten dose adjustment for CYP2C19 poor metabolizers |
|---|---|---|
|
Strong CYP2C19 inducer and strong CYP3A4 inducer: Carbamazepine – Efavirenz – Enzalutamide – St. John's wort – Phenobarbital – Phenytoin – Primidone – Rifampicin |
[0.29–0.33] Rifampin 600 mg/day (PB‐PK model) 22 [0.33–0.39] Rifampin 600 mg/day with mavacamten 5 and 15 mg/day (PB‐PK model) 16 [0.68–0.72] Carbamazepine (PB‐PK model) 22 0.68 Efavirenz 600 mg/day with mavacamten 15 mg/day for 60 days (PB‐PK model) 16 |
Initiating or increasing the dose of a strong inducer during mavacamten treatment:
|
|
Moderate or weak CYP3A4 inducer: Dexamethasone – Etravirine – Modafinil – Nevirapine – Oxcarbazepine – Perampanel – Rifabutin |
Discontinuation or decreasing dose of a strong inducer during mavacamten treatment:
|
|
|
Moderate or weak CYP2C19 inducer: Dexamethasone – Enzalutamide – Prednisone |
No adjustment of the starting dose of 2.5 mg
|
In CYP2C19 non‐poor metabolizer phenotype
In poor CYP2C19 metabolizers, mavacamten exposure increased 3.4‐fold compared with normal CYP2C19 metabolizers 7 ; therefore, co‐administration of strong CYP2C19 inhibitors in normal metabolizers is equivalent to 100% inhibition of the enzyme, and predicts a similar 3.4‐fold increase in AUC0–24h, across normal CYP2C19 metabolizers. In PB/PK models, strong CYP2C19 inhibition was predicted to increase AUCinf < 98% across normal or rapid CYP2C19 metabolizers, after modeling with ticlopidine 219.57 mg 2×/day. 23 Indeed, strong CYP2C19 inhibitors will result in “phenoconversion,” a change in phenotype predicted by the genotype, and will necessitate a significant reduction in mavacamten dose. The initial dose should be reduced to 2.5 mg/day when combined with a strong CYP2C19 inhibitor. Or upon introduction of a strong CYP2C19 inhibitor, the mavacamten dose must be significantly reduced from 15 mg to 5 mg, from 10 mg to 2.5 mg, and from 5 mg to 2.5 mg. For patients on a low dose of 2.5 mg/day, mavacamten may need to be discontinued for 4 weeks, with an echocardiographic assessment (LVEF) performed at 4 weeks (see Table 3 ). 7
Table 3.
Mavacamten dose adjustment for intermediate, normal, rapid, or ultra‐rapid CYP2C19 metabolizers with CYP2C19 and/or CYP3A4 inhibitors according to the product information and with regard to pharmacokinetic parameters described
| Concomitant treatment inhibitors | Ratio AUC (mavacamten + inhibitor)/AUC mavacamten | Mavacamten dose adjustment for CYP2C19 non‐poor metabolizers |
|---|---|---|
|
Strong CYP2C19 inhibitor + Strong CYP3A4 inhibitor Fluconazole‐ Voriconazole |
[3.10–4.13] Fluconazole 200 mg/day with mavacamten 15 mg/day for 60 days (PB‐PK model) 16 |
 Contra‐indicated Contra‐indicated |
|
Strong CYP2C19 inhibitor: Fluvoxamine – Ticlopidine |
[1.68–2.26] Ticlopidine 219.57 mg 2×/day (PB‐PK model) 22 [3–3.4] AUC ratio between poor metabolizer and Normal metabolizer (PK‐pop) for mavacamten 15 and 5 mg 16 |
Start mavacamten at a dose of 2.5 mg/day
|
|
Strong CYP3A4 inhibitor: Ceritinib – Clarithromycin – Cobicistat – Erythromycin – Idelalisib – Itraconazole – Ketoconazole Ritonavir – Tucatinib |
[1.18–1.31] Itraconazole 200 mg/day (PB‐PK model) 22 |
No dose adjustment. Starting dose 5 mg/day
|
|
Moderate or weak CYP2C19 inhibitor: Clopidogrel – Esomeprazole – Fluoxetine – Moclobemide – Omeprazole |
[1.41–1.63] Omeprazole 40 mg/day (phase I study and PB‐PK model) 22 , 24 |
No dose adjustment. Starting dose 5 mg/day
|
|
Moderate or weak CYP3A4 inhibitor: Amiodarone – Diltiazem – Dronedarone – Ethinylestradiol – Gestodene – Grapefruit juice – Nifedipine – Nitrendipine – Quetiapine – Verapamil |
[1.13–1.19] Diltiazem 60 mg 3×/day (phase I study and PB‐PK model) 22 , 24 |
No dose adjustment. Starting dose 5 mg/day
No dose adjustment. Monitor LVEF after 4 weeks, then resume patient follow‐up and titration schedule. Adjust mavacamten dose according to clinical assessment |
Moderate CYP2C19 inhibitors predicted a 1.5‐fold increase AUCinf, such as omeprazole 40–80 mg/day, leading to a one‐step dose reduction of mavacamten in non‐poor CYP2C19 metabolizers. 23 Conversely, strong CYP3A4 and CYP2C19 induction predicted a reduction in AUC0–24h by 87%, such as rifampicin 600 mg/day for 7 days, leading to clinical and echocardiographic assessment 4 weeks after inducer introduction, before adjusting the mavacamten dose (see Table 4 ).
Table 4.
Mavacamten dose adjustment for intermediate, normal, rapid, or ultra‐rapid CYP2C19 metabolizers with CYP2C19 and/or CYP3A4 inducers, according to the product information and with regard to pharmacokinetic parameters described
| Concomitant treatment inducers | Ratio AUC (mavacamten + inducer)/ AUC Mavacamten | Mavacamten dose adjustement for CYP2C19 non‐poor metabolizers |
|
Strong inducer of CYP2C19 and CYP3A4: Carbamazepine – Efavirenz – Enzalutamide – St. John's wort – Phenobarbital – Phenytoin – Primidone – Rifampicin |
[0.38–0.41] Rifampin 600 mg/day (PB‐PK model) 22 0.13 Rifampin 600 mg/day with mavacamten 5 and 15 mg/day (PB‐PK model) 16 [0.86–0.88] Carbamazepine (PB‐PK model) 22 [0.61–0.63] Efavirenz 600 mg/day with mavacamten 15 mg/day (PB‐PK model) 16 |
Initiating or increasing dose of a potent inducer during treatment with mavacamten: Monitor LVOT gradient and LVEF 4 weeks later. Adjust mavacamten dose according to clinical assessment, then resume patient follow‐up and titration schedule |
|
Moderate or weak CYP2C19 inducer: Dexamethasone – Enzalutamide – Prednisone |
Discontinuation or decreasing dose of strong CYP2C19 and CYP3A4 inducers, moderate or weak CYP2C19 inducers during mavacamten treatment: Reduce mavacamten by one dose level when treatment is at 5 mg or more. Maintain mavacamten dose at 2.5 mg. Monitor LVEF 4 weeks later, then resume patient follow‐up and titration schedule. Adjust mavacamten dose according to clinical assessment |
|
|
Moderate or weak CYP3A4 inducer: Cyclophosphamide – Dexamethasone – Etravirine – Modafinil – Nevirapine – Oxcarbazepine – Perampanel – Rifabutin |
No dose adjustment. Starting dose 5 mg/day
|
Mavacamten pharmacokinetic parameters are mainly derived from preclinical studies 24 or phase I drug–drug interaction studies in healthy volunteers, 25 which included few poor CYP2C19 metabolizers and no comorbidities (hepatic or cardiac insufficiency). Indeed, the increase in mavacamten AUC with moderate to strong CYP2C19 and CYP3A4 inhibitors led to the exclusion of these drug associations during clinical trials. Only two phase I studies assessed drug–drug interactions with mavacamten. In these phase I studies with 29 healthy participants (60% normal CYP2C19 metabolizers, 40% rapid CYP2C19 metabolizers), treated in part with omeprazole 20 mg/day (weak CYP2C19 inhibitor), mavacamten AUC was increased by ~ 50%. 25 In another study involving 25 healthy participants (54% normal CYP2C19 metabolizers, 31% intermediate CYP2C19 metabolizers), treated in part with verapamil 240 mg/day (moderate CYP3A4 inhibitor), mavacamten AUC was modestly increased by 15%. 25 Thus, mavacamten can be co‐administered with weak CYP2C19 and moderate CYP3A4 inhibitors in ultra‐rapid, rapid, normal, and intermediate CYP2C19 metabolizers, but these interactions have not been evaluated in poor CYP2C19 metabolizers phenotype.
Analysis of pharmacokinetic interactions must take into account the potency of inducers and inhibitors on the two cytochromes, CYP2C19 and CYP3A4, as well as the patient's CYP2C19 phenotype and mavacamten dose. The development of TDM could be an additional tool for adapting mavacamten dose in drug–drug interaction situations. In real‐world population, various concomitant medications will be prescribed, such as antifungals, antivirals, antibiotics, and selective serotonin reuptake inhibitors susceptible to inhibit or induce CYP2C19 and/or CYP3A4.
Among proton pump inhibitors, their ability to inhibit CYP2C19 varies between the dose used (moderate to weak inhibitor), and the drug compound, they are so not all equivalent. Indeed, rabeprazole and pantoprazole have the lowest potency of CYP2C19 inhibition, compared with omeprazole or esomeprazole. 26 Consequently, these will induce less pharmacokinetic changes, as already demonstrated in association with clopidogrel 21 , 22 , 27 , 28 or in association with voriconazole. 29 , 30 Similarly, macrolides, bacteriostatic antibiotics known for their potential of CYP3A4 inhibition, are not all equivalent: erythromycin and clarithromycin are stronger CYP3A4 inhibitors than azithromycin or josamycin. 31 To April 3, 2024, the worldwide pharmacovigilance database has reported LVEF decrease in patient's treated concomitantly with mavacamten and clarithromycin 32 whose CYP2C19 phenotype is unknown.
Pharmacokinetic and inflammation
The phenotype which is predicted by the patient's genotype could be influenced by drug–drug interactions, but also modified by inflammation. Indeed, inflammation can reduce the expression and activity of CYP, especially CYP2C19 and 3A4, 33 leading to a diminished clearance of drug substrate, as already described with voriconazole that is metabolized by both CYP2C19 and 3A4. 34 , 35 , 36 , 37 Although this factor has not yet been evaluated in patients treated with mavacamten, such phenomena of inflammation‐induced phenoconversion could lead to increased mavacamten plasma concentrations and higher risk of cardiac dysfunction.
Pharmacodynamic drug interactions
When mavacamten is associated with other negative inotrope drugs, additive effects that reduce cardiac contractility are to be expected. One case of mavacamten misuse has been reported in the Vigibase database, 32 involving an association of beta‐blockers and diltiazem, in which LVEF decrease was reported. Few cases of misuse have been reported up to April 3, 2024, with 15 reports of drug–drug interactions out of 1,025 cases: one case of palpitations in association with metoprolol, and one case of dyspnea in association with diltiazem.
The safety of concomitant use of mavacamten with disopyramide, or use of mavacamten with beta‐blockers in association with verapamil or diltiazem, has not been established, because it has not been evaluated in previous clinical trials. Close medical monitoring for signs of heart failure (dyspnea, fatigue, palpitations, worsening or onset of arrhythmia) and systolic dysfunction (reduced LVEF) is therefore recommended in combination therapy.
Initiation of negative inotropes, or increasing the dose of negative inotropes with mavacamten, requires close medical monitoring with LVEF assessments, until stable doses and clinical response are achieved.
PHARMACOGENETIC TARGETS FOR MAVACAMTEN
Method for determining recommendation levels per RNPGx guidelines
We are discussing below the different levels of recommendations for testing genetic variants (star allele) during mavacamten prescription: Essential, advisable, or possibly helpful. 19 The RNPGx guidelines determine these recommendation levels based on three main criteria: functionality (the impact of the star allele on enzymatic function for cytochromes), clinical evidence (the impact of star allele on drug response or toxicity), and the presence of expert consensus. Table S1 provides the specific level of evidence for each star allele.
Essential test
CYP2C19 *2 and *3 alleles
More than 10 clinical trials involving mavacamten genotyped the CYP2C19 gene and mainly found patients carrying *2, *3, and *17 alleles due to their prevalence in the general population. 17 They also identified other rare alleles such as *4, *6, *8, and *9. The data from these studies were used to generate pharmacokinetic modeling and exposure–response modeling. These models showed that poor CYP2C19 metabolizers have a higher proportion of episodes of LVEF < 50% if the dose is similar for all patients, regardless of their CYP2C19 phenotype.
Genotyping CYP2C19*2 and *3 alleles is considered essential by RNPGx because of the established functionality, 38 the clinical impact of these alleles in the simulated 18 models, the EMA recommendations in the context of mavacamten prescription 7 and their frequency in the general population. 17
They are specifically identified with these genetic variants below:
NM_000769.4: c.681G>A (rs4244285, CYP2C19*2 allele)
NM_000769.4: c.636G>A (rs4986893, CYP2C19*3 allele)
Both are non‐functional variants. 38 The first one generates a splice defect, associated with a premature stop codon. The second one is a nonsense single‐nucleotide polymorphism (SNP), associated with the synthesis of an inactive truncated protein, mainly observed in the Asian population, with a frequency of 6%.
These alleles are already important for other pharmacogenetic indications, such as clopidogrel treatment, 39 antidepressants, 40 and voriconazole. 41
Advisable test
CYP2C19 *17 allele
Patients carrying *17 allele, whether in a heterozygous or homozygous state are classified as rapid or ultrarapid metabolizers, respectively. The previous exposure–response models used to assess the safest and more efficiency echo‐guided dose titration schedule for mavacamten showed that 35% of ultrarapid metabolizers had decreased LVOTg compared with 50% for other metabolizers, after 12 weeks of treatment, with similar dose schedule. 18 This result suggests underexposure for CYP2C19*17 carrier and that an increase in the dose of mavacamten could be pertinent to reach efficacy faster. However, dedicated clinical trials are needed to evaluate this new dose schedule.
Regarding the established functionality, the clinical impact in the simulated models, the frequency in the population 38 but the lack of expert agreement, the CYP2C19*17 allele is considered advisable by RNPGx in this context.
It is identified with the genetic variant: NM_000769.4: c.‐806 C>T; rs12248560.
We note that the identification of this SNP could lead to misinterpretation with the very rare non‐functional CYP2C19*4 allele. This *4 haplotype is defined by the combination of the promoter variation described above and a second SNP suppressing the initiation codon NM_000769.4: c.1A>G, rs28399504.
Possibly helpful test
CYP2C19 (other non‐functional haplotypes)
CYP3A4*22 and *20 alleles
CYP3A5*3, *6 and *7 alleles
CYP2C: TG haplotype
Below, we discuss other genetic variants that have not been investigated in clinical trials with mavacamten. But due to their functionalities, 42 , 43 , 44 , 45 these variants may modify the expression or function of cytochromes involved in mavacamten metabolism, as CYP2C19, CYP3A4, CYP3A5, and CYP2C:TG haplotype. Therefore, they could contribute to the mavacamten pharmacokinetic variability. Due to the lack of clinical evidence and expert agreement, the level of recommendations is “possibly helpful” in accordance with the criteria from the RNPGx.
We have considered various situations:
Adverse drug reaction in non‐poor CYP2C19 metabolizer after genotyping CYP2C19 *2 and *3
We cross‐referenced the CPIC CYP2C19 non‐functional allelic frequencies and poor metabolizer data, to evaluate the occurrence of non‐functional CYP2C19 haplotypes unrelated with *2 *3 haplotypes. The results are compiled in Figure 2 . It summarizes that poor metabolizers are mainly explained by the *2 and *3 alleles (> 90%) except in sub‐Saharan African and African American populations in which these two loss‐of‐function alleles account for only 53% and 71% of poor metabolizers, respectively (see Figure 2 ). These two populations frequently carry the CYP2C19 *35 allele which is recorded as non‐functional but with limited evidence. 42 , 46 The SNP defining *35 is a deep intronic variant (NM_000769.4:c.332‐23A>G; rs12769205) leading to alternative splicing by disrupting the branch point, but functional analysis did not confirm the deleterious effect. 42 Other loss‐of‐function alleles are found at a lower frequency, (e.g., CYP2C19*5, CYP2C19*9 and CYP2C19*10) or associated with a limited level of evidence (i.e. CYP2C19*6 *7 *8 *22 *24 and the two copy number variation *36 and *37).
Figure 2.
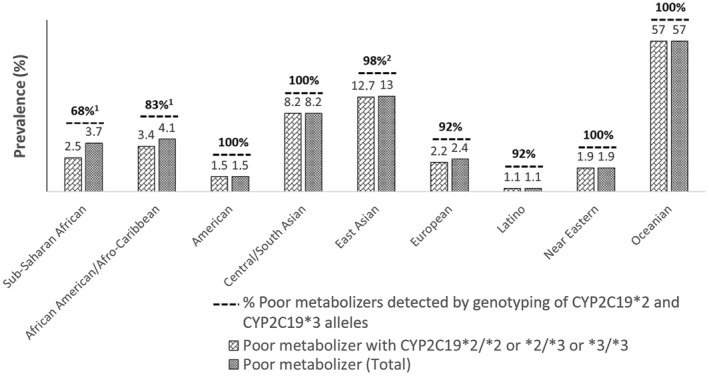
Prevalence of CYP2C19 poor metabolizers across different populations. Allelic frequency from the CPIC data. 15 Alleles > 1/1,000 in minor allele frequency excepted *2 and *3 are: African1 (*35, *9, *10), East Asian2 (*5).
Aside from the non‐functional alleles, CPIC defined a « likely poor metabolizer » category which concerns patients carrying « “decreased function” » haplotypes such as CYP2C19*9. These alleles require further investigation to evaluate their impact on mavacamten safety and efficacy.
CYP3A investigation in CYP2C19 poor metabolizers
In vitro studies indicate that mavacamten is secondarily metabolized by CYP3A4 and CYP3A5 in same proportion, 12 which are polymorphic. Genetic polymorphisms in CYP3A4 and CYP3A5 could affect mavacamten clearance (especially in poor CYP2C19 metabolizers) and contribute to inter‐individual variability. The influence of genetic polymorphisms in CYP3A4 and CYP3A5 has not been assessed in clinical studies; nevertheless, CYP3A genetic variability should be considered in this situation particularly in the same way as CYP3A4 strong inhibitors are contraindicated, in poor CYP2C19 metabolizers.
CYP3A4*22 and *20 alleles are defined as non‐functional and identified as relevant for the pharmacogenetics of antipsychotic medications. 47 The CYP3A4*22 allele, defined by the intronic variant NM_017460.5(CYP3A4): c.522‐191C>T, is a non‐functional allele associated with a cryptic splice site. Its frequency is ~ 10% and 0.3–0.4% in the Caucasian population in the heterozygous or homozygous state, respectively. The CYP3A4*20 allele is defined by the NM_017460.6: c.1461dup variant, associated with a duplication of the exon 13. Its allelic frequency is high in Hispanic population (3.8%). 43 In poor metabolizers of CYP2C19, CYP3A4 becomes the main enzyme metabolizing mavacamten and its enzymatic activity alteration may lead to overexposure. Further studies are needed to evaluate their impact on mavacamten efficacy and safety.
More than 80% of the Caucasian population are CYP3A5 poor metabolizers because they carry a null haplotype of CYP3A5. 48 Consequently, patients with CYP3A5 expressor phenotype (“Normal metabolizer”) and poor CYP2C19 metabolizer may experience lower efficacy due to a dose titration limited to 5 mg. Indeed, in patients who are poor CYP2C19 metabolizers, CYP3A4 and CYP3A5 become the primary enzymes responsible for metabolizing mavacamten. As a result, CYP3A5 expressors may have reduced mavacamten exposure compared with CYP3A5 non‐expressors, for the same dose. The non‐functional alleles are CYP3A5*3 defined by the variant NM_000777.5: c.219‐237A>G, rs776746 more frequent in Caucasians, 49 and CYP3A5*6 (NM_000777.5: c.624G>A; rs10264272) and CYP3A5*7 (NM_000777.5: c.1035dup; rs41303343) more frequent in African. 44
CYP2C: TG haplotype
A recent haplotype CYP2C18 NM_000772.3:c.*31T (rs2860840) and NM_000772.2:c.819+2182G (rs11188059), referred to as “CYP2C:TG, was associated with ultrarapid metabolism of various CYP2C19 substrates, such as omeprazole 50 ), sertraline, 51 and escitalopram. 52 However, other recent studies have called into question the impact of this haplotype, in 222 healthy patients, and after in vitro analysis with different CYP2C19 substrates 53 or with clopidogrel. 54 Considering all available data, there is insufficient evidence supporting genotyping CYP2C:TG before mavacamten therapy.
Analysis methods
Targeted genotyping strategy is suitable for CYP2C19 pharmacogenetic analysis of mavacamten treatment, including specifically the *2 and *3 alleles, and potentially *17. The available targeted analysis techniques for CYP2C19 are standard: Sanger sequencing, multiplex PCR with strip hybridization, PCR with Taqman® probe, and LAMP technique. 55 The more comprehensive high‐throughput sequencing analysis strategy can be considered as a secondary option, but its cost–benefit ratio has not been defined at the time of these recommendations.
For the essential test, allele frequencies vary across different ethnicities (Figure 2 ). However, in a targeted approach, CYP2C19*2 and *3 should be tested in all ethnic groups, as no other star alleles are currently considered essential for mavacamten management according to these recommendations. Notably, the CYP2C19*35 allele, which is common in African populations, has a limited level of evidence (a single publication 42 ), and its influence on mavacamten dosing requires further study before clinical use.
SUMMARY OF RECOMMENDATION LEVELS
After analyzing the available data, RNPGx recommends genotyping for clinical use both null alleles CYP2C19*2 and CYP2C19*3 before initiating mavacamten treatment, for every ethnicities. These alleles are associated with poor metabolizer status for the CYP2C19, in order to limit mavacamten overexposure and ensure patient safety (Figure 3 and Table 5 ). These two‐star alleles are the own with enough level of evidence: functional, clinical, and are recommended by the EMA.
Figure 3.

French‐speaking network of pharmacogenetics (RNPGx) recommendations for clinical use of mavacamten. Green Star = Essential Test; Yellow star = advisable test; Red star = possibly helpful test. (Picard et al. 19 )
Table 5.
Summary of recommendation levels for pharmacogenetic testing in mavacamten therapy from RNPGx
| Level | Pharmacogenetic test | Details |
|---|---|---|
| Essential | CYP2C19 *2 and *3 alleles | Stratification for starting and maximum dose prescription, as well as dose adjustment based on drug–drug interactions |
| Advisable | CYP2C19 *17 alleles | Dose adjustment (efficiency) |
| Possibly helpful |
Other non‐functional CYP2C19 alleles (details in Table S2 ) |
Adverse drug reaction associated with overexposure after genotyping CYP2C19 *2 and *3 alleles |
|
Possibly helpful (CYP2C19 poor metabolizers) |
CYP3A5 alleles*3, *6 and *7 | Dose adjustment (efficiency) |
| CYP3A4 alleles*22 and *20 | Adverse drug reaction after dose adjustment in CYP2C19 poor metabolizers |
The “advisable” and “possibly helpful” tests cannot currently be used to guide mavacamten treatment due to the lack of clinical evidence. However, further studies are needed, as some functional or pharmacokinetic data suggest a potential impact in certain situations.
RNPGx advises testing for the CYP2C19*17 allele, associated with ultrarapid metabolism in the homozygous state to reduce underexposure and increase the efficacy of mavacamten in this phenotypic group.
Possibly helpful tests for other alleles may be useful depending on clinical circumstances. For instance, in CYP2C19 poor metabolizer, testing for a deficient CYP3A4 allele (*20 or *22) may potentially impact the prediction of adverse effects or enhance echocardiographic monitoring; and genotyping for CYP3A5 alleles associated with a normal CYP3A phenotype may help explain inefficacy not accounted by the CYP2C19 ultrarapid metabolizer phenotype (CYP2C19 *17/*17). In cases of adverse drug reaction, associated with mavacamten overexposure, after CYP2C19*2 and *3 alleles genotyping, it may also be possible to test for other deleterious CYP2C19 alleles, either by implementing targeted strategies to identify the main rare alleles (see Table S2) or by using high‐throughput sequencing strategy. The CYP2C:GT haplotype can also be assessed based on previous studies involving other medications metabolized by CYP2C19.
FUNDING
No funding was received for this work.
CONFLICT OF INTEREST
The authors declared no competing interests for this work, and confirm that none of the authors participated in the clinical trials.
Supporting information
Table S1
Table S2.
References
- 1. Maron, B.J. , Gardin, J.M. , Flack, J.M. , Gidding, S.S. , Kurosaki, T.T. & Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation 92, 785–789 (1995). [DOI] [PubMed] [Google Scholar]
- 2. Maron, M.S. , Hellawell, J.L. , Lucove, J.C. , Farzaneh‐Far, R. & Olivotto, I. Occurrence of clinically diagnosed hypertrophic cardiomyopathy in the United States. Am. J. Cardiol. 117, 1651–1654 (2016). [DOI] [PubMed] [Google Scholar]
- 3. Veselka, J. , Anavekar, N.S. & Charron, P. Hypertrophic obstructive cardiomyopathy. Lancet 389, 1253–1267 (2017). [DOI] [PubMed] [Google Scholar]
- 4. Ho, C.Y. et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 138, 1387–1398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haute Autorité de Santé . Cardiomyopathie Hypertrophique (CMH) <https://www.has‐sante.fr/jcms/c_1100272/fr/cardiomyopathie‐hypertrophique‐cmh>. Accessed March 5, 2024.
- 6. Yang, Q. et al. Surgical septal myectomy outcome for obstructive hypertrophic cardiomyopathy after alcohol septal ablation. J. Thorac. Dis. 13, 1055–1065 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Camzyos | European Medicines Agency <https://www.ema.europa.eu/en/medicines/human/EPAR/camzyos#ema‐inpage‐item‐authorisation‐details>. Accessed January 28, 2024.
- 8. EMA‐Camyzos Summary of product characteristics.pdf <https://ec.europa.eu/health/documents/community‐register/2023/20230626159388/anx_159388_fr.pdf>. Accessed June 28, 2024.
- 9. Nag, S. , Gollapudi, S.K. , Del Rio, C.L. , Spudich, J.A. & McDowell, R. Mavacamten, a precision medicine for hypertrophic cardiomyopathy: from a motor protein to patients. Sci. Adv. 9, eabo7622 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heitner, S.B. et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: a clinical trial. Ann. Intern. Med. 170, 741–748 (2019). [DOI] [PubMed] [Google Scholar]
- 11. Olivotto, I. et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER‐HCM): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 396, 759–769 (2020). [DOI] [PubMed] [Google Scholar]
- 12. Grillo, M.P. et al. In vitro and in vivo pharmacokinetic characterization of mavacamten, a first‐in‐class small molecule allosteric modulator of beta cardiac myosin. Xenobiotica 49, 718–733 (2019). [DOI] [PubMed] [Google Scholar]
- 13. Gaedigk, A. , Casey, S.T. , Whirl‐Carrillo, M. , Miller, N.A. & Klein, T.E. Pharmacogene Variation Consortium: A Global Resource and Repository for Pharmacogene Variation. Clin. Pharmacol. Ther. 110, 542–545 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caudle, K.E. et al. Standardizing terms for clinical pharmacogenetic test results: consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet. Med. 19, 215–223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Relling, M.V. & Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the pharmacogenomics research network. Clin. Pharmacol. Ther. 89, 464–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McGurk, K.A. , Bilgehan, N. & Ware, J.S. Pharmacogenetic influences over mavacamten pharmacokinetics: considerations for the treatment of individuals with hypertrophic cardiomyopathy. Circulation 149, 1786–1788 (2024). https://pubmed.ncbi.nlm.nih.gov/38829931/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. FDA highlights of prescribing informations (2022).pdf <https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214998s000lbl.pdf>. Accessed June 28, 2024.
- 18. Merali, S. et al. Recommendation of mavacamten posology by model‐based analyses in adults with obstructive hypertrophic cardiomyopathy. CPT Pharmacometrics Syst. Pharmacol. 13, 1448–1461 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Picard, N. et al. Pharmacogenetics‐based personalized therapy: levels of evidence and recommendations from the French Network of Pharmacogenetics (RNPGx). Therapie 72, 185–192 (2017). [DOI] [PubMed] [Google Scholar]
- 20. Haute Autorité de Santé . CAMZYOS (mavacamten) – Cardiomyopathie hypertrophique obstructive <https://www.has‐sante.fr/jcms/p_3465194/fr/camzyos‐mavacamten‐cardiomyopathie‐hypertrophique‐obstructive>. Accessed January 10, 2024.
- 21. Tod, M. , Bourguignon, L. , Bleyzac, N. & Goutelle, S. A model for predicting the interindividual variability of drug‐drug interactions. AAPS J. 19, 497–509 (2017). [DOI] [PubMed] [Google Scholar]
- 22. Goutelle, S. & Tod, M. Quantitative methods for prediction of the effect of cytochrome P450 gene polymorphisms on substrate drug exposure. Clin. Pharmacokinet. 54, 319–320 (2015). [DOI] [PubMed] [Google Scholar]
- 23. Chiang, M. et al. Physiologically based pharmacokinetic modeling and simulation of Mavacamten exposure with drug‐drug interactions from CYP inducers and inhibitors by CYP2C19 phenotype. Clin. Pharmacol. Ther. 114, 922–932 (2023). [DOI] [PubMed] [Google Scholar]
- 24. Li, Q. , Liu, Y.N. , Chen, C. , Xu, R.A. , Xie, S. & Zhan, R. Effects of CYP2C19 inhibitors on mavacamten pharmacokinetics in rats based on UPLC‐MS/MS. Chem. Biol. Interact. 1, 110531 (2023). [DOI] [PubMed] [Google Scholar]
- 25. Perera, V. , Gretler, D.D. , Seroogy, J.D. , Chiang, M. , Palmisano, M. & Florea, V. Effects of omeprazole and verapamil on the pharmacokinetics, safety, and tolerability of mavacamten: two drug‐drug interaction studies in healthy participants. Clin. Pharmacol. Drug Dev. 12, 1241–1251 (2023). [DOI] [PubMed] [Google Scholar]
- 26. Zvyaga, T. et al. Evaluation of six proton pump inhibitors as inhibitors of various human cytochromes P450: focus on cytochrome P450 2C19. Drug Metab. Dispos. 40, 1698–1711 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Simon, T. et al. Clinical events as a function of proton pump inhibitor use, clopidogrel use, and cytochrome P450 2C19 genotype in a large nationwide cohort of acute myocardial infarction: results from the French registry of acute ST‐elevation and non‐ST‐elevation myocardial infarction (FAST‐MI) registry. Circulation 123, 474–482 (2011). [DOI] [PubMed] [Google Scholar]
- 28. Frelinger, A.L. et al. A randomized, 2‐period, crossover design study to assess the effects of dexlansoprazole, lansoprazole, esomeprazole, and omeprazole on the steady‐state pharmacokinetics and pharmacodynamics of clopidogrel in healthy volunteers. J. Am. Coll. Cardiol. 59, 1304–1311 (2012). [DOI] [PubMed] [Google Scholar]
- 29. Qi, F. , Zhu, L. , Li, N. , Ge, T. , Xu, G. & Liao, S. Influence of different proton pump inhibitors on the pharmacokinetics of voriconazole. Int. J. Antimicrob. Agents 49, 403–409 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Gautier‐Veyret, E. et al. Variability of voriconazole plasma concentrations after allogeneic hematopoietic stem cell transplantation: impact of cytochrome P450 polymorphisms and comedications on initial and subsequent trough levels. Antimicrob. Agents Chemother. 59, 2305–2314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Westphal, J.F. Macrolide‐induced clinically relevant drug interactions with cytochrome P‐450A (CYP) 3A4: an update focused on clarithromycin, azithromycin and dirithromycin. Br. J. Clin. Pharmacol. 50, 285–295 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lindquist, M. VigiBase, the WHO global ICSR database system: basic facts. Ther. Innov. Regul. Sci. 42, 409–419 (2008). [Google Scholar]
- 33. Stanke‐Labesque, F. , Gautier‐Veyret, E. , Chhun, S. & Guilhaumou, R. French Society of Pharmacology and Therapeutics. Inflammation is a major regulator of drug metabolizing enzymes and transporters: consequences for the personalization of drug treatment. Pharmacol. Ther. 215, 107627 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Veringa, A. et al. Voriconazole metabolism is influenced by severe inflammation: a prospective study. J. Antimicrob. Chemother. 72, 261–267 (2017). [DOI] [PubMed] [Google Scholar]
- 35. Gautier‐Veyret, E. et al. Optimization of voriconazole therapy for treatment of invasive aspergillosis: pharmacogenomics and inflammatory status need to be evaluated. Br. J. Clin. Pharmacol. 87, 2534–2541 (2021). [DOI] [PubMed] [Google Scholar]
- 36. Encalada Ventura, M.A. et al. Longitudinal analysis of the effect of inflammation on voriconazole trough concentrations. Antimicrob. Agents Chemother. 60, 2727–2731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gautier‐Veyret, E. et al. Pharmacogenetics may influence the impact of inflammation on voriconazole trough concentrations. Pharmacogenomics 18, 1119–1123 (2017). [DOI] [PubMed] [Google Scholar]
- 38. Botton, M.R. et al. PharmVar GeneFocus: CYP2C19. Clin. Pharmacol. Ther. 109, 352–366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee, C.R. et al. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin. Pharmacol. Ther. 112, 959–967 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quaranta, S. , Dupouey, J. , Colle, R. & Verstuyft, C. Pharmacogénétique des médicaments antidépresseurs: état des connaissances et des pratiques – recommandations du Réseau national de pharmacogénétique (RNPGx). Therapies 72, 301–309 (2017). [DOI] [PubMed] [Google Scholar]
- 41. Moriyama, B. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP2C19 and voriconazole therapy. Clin. Pharmacol. Ther. 102, 45–51 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chaudhry, A.S. et al. The CYP2C19 intron 2 branch point SNP is the ancestral polymorphism contributing to the poor metabolizer phenotype in livers with CYP2C19*35 and CYP2C19*2 alleles. Drug Metab. Dispos. 43, 1226–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pratt, V.M. et al. CYP3A4 and CYP3A5 genotyping recommendations: a joint consensus recommendation of the Association for Molecular Pathology, Clinical Pharmacogenetics Implementation Consortium, College of American Pathologists, Dutch Pharmacogenetics Working Group of the Royal Dutch Pharmacists Association, European Society for Pharmacogenomics and Personalized Therapy, and Pharmacogenomics Knowledgebase. J. Mol. Diagn. 25, 619–629 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodriguez‐Antona, C. et al. PharmVar GeneFocus: CYP3A5. Clin. Pharmacol. Ther. 112, 1159–1171 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Botton, M.R. et al. Structural variation at the CYP2C locus: characterization of deletion and duplication alleles. Hum. Mutat. 40, e37–e51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gaedigk, A. et al. CYP2C8, CYP2C9, and CYP2C19 characterization using next‐generation sequencing and haplotype analysis. J. Mol. Diagn. 24, 337–350 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beunk, L. et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene‐drug interaction between CYP2D6, CYP3A4 and CYP1A2 and antipsychotics. Eur. J. Hum. Genet. 32, 278–285 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Birdwell, K. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin. Pharmacol. Ther. 98, 19–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuehl, P. et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 27, 383–391 (2001). [DOI] [PubMed] [Google Scholar]
- 50. Kee, P.S. et al. Omeprazole treatment failure in gastroesophageal reflux disease and genetic variation at the CYP2C locus. Front. Genet. 13, 869160 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bråten, L.S. , Ingelman‐Sundberg, M. , Jukic, M.M. , Molden, E. & Kringen, M.K. Impact of the novel CYP2C:TG haplotype and CYP2B6 variants on sertraline exposure in a large patient population. Clin. Transl. Sci. 15, 2135–2145 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bråten, L.S. et al. A novel CYP2C‐haplotype associated with ultrarapid metabolism of escitalopram. Clin. Pharmacol. Ther. 110, 786–793 (2021). [DOI] [PubMed] [Google Scholar]
- 53. Zubiaur, P. et al. Impact of CYP2C:TG haplotype on CYP2C19 substrates clearance in vivo, protein content, and in vitro activity. Clin. Pharmacol. Ther. 114, 1033–1042 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ganoci, L. et al. Is CYP2C haplotype relevant for efficacy and bleeding risk in clopidogrel‐treated patients? Genes (Basel) 15, 607 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kwong, K.M. , Tam, C.C. , Chan, R. , Lee, S.W.L. , Ip, P. & Kwok, J. Comparison of single nucleotide polymorphism genotyping of CYP2C19 by loop‐mediated isothermal amplification and real‐time PCR melting curve analysis. Clin. Chim. Acta 478, 45–50 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2.