

Abstract

The effectiveness of metallocene catalysts in the cationic ring-opening polymerization (cationic ROP) of ε-caprolactone (CL) is influenced by the choice of metallocene/borate systems, particularly their bulkiness. Recent research examines this effect on the initiation and propagation stages of cationic ROP. We conducted a density functional theory study on the precatalyst activation of cationic CL ROP by zirconocene/borate catalysts, where four models of zirconocene precatalysts (Cp2ZrMe2 (a), (Me5Cp)CpZrMe2 (b), (Me5Cp)2ZrMe2 (c), and Ind2ZrMe2 (d)) were combined with boron cocatalysts B(C6F5)3 and [X+][B(C6F5)4–] (X+ = Ph3C+ or PhMe2NH+). We modeled and predicted their thermodynamic, steric, and electronic properties during ion-pair formation and separation. The calculated Gibbs energies of ion-pair formation correlated with the positive charge at the metal center and steric congestion of the catalyst. All catalyst precursors showed exothermic and exergonic insertion of CL, toluene solvent, and contact ion pairs; solvent-separated ion pairs were the preferred activation pathway. Catalyst c showed the most stable ion pair in precatalyst activation, with the lowest separation energy, aided by methyl group bulkiness and toluene solvent. We evaluated Cp′-based ligands using percent buried volume (%VBur) trends. Noncovalent interaction analysis indicated weak interactions at ion-pair contacts. This study enhances our understanding of cationic ROP and could aid in developing new polymerization catalysts for polyester synthesis.
1. Introduction
The extensive use of polymeric materials, or microplastics, significantly contributes to plastic pollution, harming marine and terrestrial ecosystems. In 2010, 4.8 to 12.7 million metric tons of plastic waste entered the ocean.1 This issue has spurred interest in creating sustainable, biodegradable polymeric materials.2 Poly(ε-caprolactone) (PCL), a commercially synthetic biodegradable plastic, is gaining attention due to its biocompatibility.3 PCLs have the potential to reduce plastic pollution and can be easily produced through ring-opening polymerization (ROP) of cyclic esters,4 offering an alternative to enzymatic hydrolysis.5
Research into living cationic ROP with cyclic esters is highly active.6 Numerous catalysts/initiators have been developed for this polymerization.7 Group 4 metallocene/borate systems are notable for synthesizing various functionalized polymers through cationic ROP of oxazolines,8 vinyl ethers,9 lactones,10 and cyclic carbonates.11 The nature of the metallocene polymerization catalysis depends on the reactivity at the active center of zirconocene alkyl cations. The [Cp2ZrMe]+ species, first isolated and characterized by Jordan et al., has become the archetypal cation for understanding metallocene-based polymerization catalysis.12,13 This “Jordan cation” established the fundamental principles of how these catalytically active species form and behave. The activation process involves converting a precatalyst Cp2ZrMe2 into [Cp2ZrMe]+ in two steps, where the performance depends on the choice of metallocene/borate systems (see Scheme 1). In step 1, an ion pair is formed with B(C6F5)3 and [X+][B(C6F5)4–] (X+ = Ph3C+ or PhMe2NH+) cocatalysts, resulting in ion pairs denoted as IPI and IPII, respectively (see Scheme 1). Step 2 involves dissociation of the ion pair to form the catalytically active species and the counteranions. The energy changes linked to these steps, ion-pair formation (ΔEipf) and separation (ΔEips), can be calculated using eqs 1–5 in Scheme 1. The effectiveness of metallocene catalysts in the ROP depends critically on the initial activation step, where the nature of the ion pair formed affects both catalyst stability and activity. Understanding this activation process is essential for controlling the polymerization rate, molecular weight distribution, and end-group fidelity in PCL synthesis. This study focuses on the activation step—a crucial process that determined catalyst availability for subsequent polymerization. The detailed mechanism of monomer coordination, ring opening, and chain growth has been addressed elsewhere.14
Scheme 1. Ion-Pair Formation and Separation Mechanism Occurring in the Precatalyst Activation of Zirconocene-Catalyzed Cationic ROP by Two Distinct Boron Cocatalysts, B(C6F5)3 and [X+][B(C6F5)4–] (X+ = Ph3C+ or PhMe2NH+) Cocatalysts (Denoted as IPI and IPII, Respectively).

Changes of energies for steps 1 and 2 (denoted as ΔEipf and ΔEips, respectively) are also indicated.
The metallocene/borate systems were first developed as olefin polymerization catalysts, and their activation chemistry, particularly the formation and properties of [Cp2ZrMe]+, has been thoroughly investigated both experimentally13 and computationally.15,16 Our recent studies explored the effects of various Cp′ ligands on the initiation and propagation of cationic ROP of cyclic lactones (CL) using zirconocene complex catalysts.14,17 These studies established key principles about ion-pair formation, stability, and steric–electronic effects that inform our current work on ROP applications. Here, we further investigated how these ligands influence precatalyst activation in the ROP through density functional theory (DFT) calculations and electronic and steric analyses. We examined four zirconocene precatalysts (a–d) (see Scheme 2) with two borate counteranions: [B(C6F5)4]− (I) and [MeB(C6F5)3]− (II), focusing on their intermolecular interactions during ion-pair formation and separation. DFT has been extensively used in similar studies18,19 to provide detailed insights into the cationic ROP process. Our results illustrate how different Cp′ ligands affect ion-pair formation and separation and identify the roles of toluene solvent, monomer, and precatalyst in catalyst activation, revealing weak interactions at the contact points between catalyst and cocatalyst through noncovalent interaction (NCI) analysis.
Scheme 2. Structures of Zirconocene Precatalysts (a–d) under Study.

2. Computational Details
DFT calculations
were performed using the Gaussian 09 program.20 The DFT methods have previously been applied
for activation of olefin polymerization.15,16,21 All structures were fully optimized without
any constraints in the gas phase and toluene using the dispersion-corrected
M06-2X functional22 in conjunction with
the def2-SVP basis set for all atoms with a 28-electron relativistic
effective core potential for Zr.23 The
basis sets were taken from the EMSL basis set exchange.24 This M06-2X/def2-SVP method is widely applied
for transition metal complexes,19,25 and the M06-2X functional
has been carefully validated and successfully applied in catalyst
activation studies26 and cationic ROP.27 This method yields ion-pair formation energy
(ΔEipf) that aligns well with DLPNO-CCSD(T)/def2-TZVP
single-point energy calculations28 (−28.1
vs −26.9 kcal/mol, respectively). Calculations using tight
DLPNO thresholds were carried out by Orca 5.0.3.29 Frequency analyses were performed to obtain Gibbs free
energies (T = 298.15 K, p = 1.013
bar) and to confirm that all located stationary points are minima
on the potential energy hypersurface. Molecular properties in toluene
solution were obtained by employing the universal implicit solvent
model based on density (SMD30) as implemented
in Gaussian 09, at the same level of theory as the geometry optimization.
Natural bond orbital (NBO) analysis31 was
used to obtain the charge distribution. The NCIweb server32 was used to visualize noncovalent interactions
(NCIs) within the ion pairs of the zirconocene catalysts and counteranions,
based on electron density (ρ) and reduced density gradient (RDG
or s), according to the equation:33 RDG = 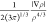 . Steric maps reported in this work were
generated using the SambVca 2.1 web application.34
. Steric maps reported in this work were
generated using the SambVca 2.1 web application.34
3. Results and Discussion
3.1. Structure and Stability of Ion Pairs
DFT calculations were performed on the ion-pair formation and separation of four zirconocene precatalysts (Scheme 2). Relative Gibbs free energies and enthalpies (ΔG and ΔH) for the zirconocene catalyst (a), using eqs 1–5, are plotted in Figure 1, while the energies for other catalysts are included in the Supporting Information (Figures S1–S3). Thermodynamic quantities of the two processes for all systems are reported in Table 1.
Figure 1.

Relative Gibbs free energies and enthalpies (ΔG and ΔH) of the ion-pair formation (eqs 1–2) and separation (eqs 3–5) in toluene and in the gas phase (values given in parentheses) calculated for the zirconocene catalyst (a) in different forms (e.g., Cp2ZrMe+, [Cp2ZrMe+][I], and [Cp2ZrMe+][II]).
Table 1. Geometric and Energetic Analysis for the Formation of Contact Ion Pairs (IPI and IPII)a.
| ion pair | ΔGipf (ΔHipf) (kcal/mol) | ΔGips (ΔHips) (kcal/mol) | Zr-Cp′ (Å) | Zr–B (Å) | Zr–Me(B) (Å) | Zr–F(B)b (Å) | ∠Cp′ZrCp′ (deg) |
|---|---|---|---|---|---|---|---|
| IPI-a | –11.0 (−28.2) | 29.8 (45.9) | 2.232 | 4.142 | 2.576 | 130.8 | |
| IPI-b | –13.2 (−30.4) | 27.8 (46.2) | 2.251 | 4.204 | 2.691 | 133.3 | |
| IPI-c | –17.1 (−34.6) | 26.9 (43.6) | 2.266 | 4.235 | 2.614 | 137.8 | |
| IPI-d | –9.2 (−27.1) | 26.6 (42.3) | 2.254 | 4.099 | 2.469 | 132.9 | |
| IPII-a | –36.8 (−39.1) | 23.7 (43.9) | 2.218 | 5.187 | 2.426/2.769 | 131.9 | |
| IPII-b | –36.5 (−41.2) | 23.7 (42.3) | 2.241 | 5.446 | 2.606/2.557 | 133.8 | |
| IPII-c | –38.8 (−44.0) | 21.1 (40.2) | 2.222 | 5.977 | 3.079/2.493 | 138.5 | |
| IPII-d | –33.0 (−36.2) | 23.0 (38.5) | 2.265 | 5.739 | 3.034/2.377 | 133.9 |
Enthalpies of ion-pair formation and separation are also included in parentheses.
Values are Zr–F distances calculated between the Zr atom and the F atoms at ortho and meta positions, respectively.
3.1.1. Formation of Ion Pairs
Generally, the geometries of the alkylzirconocenium cation fragments are similar (Figure 1), but the IPI and IPII complexes differ significantly in their geometry around Zr. As shown in Figure 2, the [MeB(C6F5)3]− anion forms stronger Zr–Me(B) interactions (2.469–2.691 Å), without Zr–F interactions, whereas the [B(C6F5)4]− anion chelates to the Zr center with Zr–F distances ranging from 2.416 to 3.079 Å for the ortho-F atom and 2.377–2.769 Å for the meta-F atom (Table 1). These findings align with previous studies indicating that IPs can adopt multiple structures featuring distinct Zr–F interactions.35,36 The optimized IPII structures also reveal that, compared to IPI, the distances between the Zr and B centers are noticeably longer (Figure 3). The Zr–B distances between the catalyst and cocatalyst range from 4.099 to 4.235 Å for IPI and from 5.187 to 5.977 Å for IPII. In the case of (Me5Cp)2ZrMe2 (c), larger distances are observed for both counteranions, while the shortest distance is noted for unsubstituted Cp ligand (a). Notably, IPI-d demonstrates a stronger interaction between the Zr atom and the methide group, evidenced by the shortest Zr–Me(B) (2.469 Å) and Zr–B (4.099 Å) distances compared to other IPs (Table 1 and Figure 2). Such variations in the Zr···B contact also reflect the steric hindrance of the ancillary ligand; the larger the cyclopentadienyl group, the longer the Zr···B distance.
Figure 2.

Toluene-optimized structures of the ion pairs [Cp′2ZrMe]+[MeB(C6F5)3]−IPI of four catalysts (a–d) formed in the precatalyst activation with B(C6F5)3. Values of the distances (Zr–Me(B) and Zr–B shown as orange and black dotted lines, respectively) of zirconium atoms (cyan) to the methylated group and boron (pink) atoms are given in Å. Ortho, meta, and para positions of F atoms are also indicated with o, m, and p, respectively.
Figure 3.

Toluene-optimized structures of the ion pairs [Cp′2ZrMe]+[B(C6F5)4]−IPII of four catalysts (a–d) formed in the precatalyst activation with B(C6F5)3. Values of the distances (Zr–F(B) and Zr–B shown as orange and black dotted lines, respectively) of zirconium atoms (cyan) to the methylated group and boron (pink) atoms are given in Å. Ortho, meta, and para positions of F atoms are also indicated with o, m, and p, respectively.
The structures where ortho- and meta-F atoms of the anion coordinate to the Zr center (IPII) are more thermodynamically stable than those of IPI. The computed formation free energies range from −11 to −36 kcal/mol (−28 to −39 kcal/mol in ΔHipf) (Figure 1), indicating that IP formation is thermodynamically favorable in solution. Among all IPs, IPI-c and IPII-c are the most stable. Notably, more negative ion-pair formation values (ΔGipf) for IPI correlate with the increasing number of methyl groups on the Cp ligand (ΔGipf = −11.0, −13.2, and −17.1 kcal/mol), closely related to the Cp′ZrCp′ angles and Zr–B distances (Table 1). A similar trend is seen for IPII. For both counteranions, the highest formation energies are obtained for IPI-d and IPII-d, at −9.2 and −33.0 kcal/mol, respectively, due to strong repulsive forces between the F atoms or the Me(B) group of the counteranion and the catalyst’s indene ring (see NCI plots below). It appears that the nature of the steric bulk (distributed vs localized) also influences the mode of ion-pair stabilization when comparing systems with extended π-conjugation and increased rigidity (see Results in the Supporting Information for (CpFlu)2ZrMe2 and (tBuCp)2ZrMe2 ). There is a notable difference between the computed ΔG and ΔH values for the formation of IPI and IPII ion pairs, highlighting the varying significance of entropy in the two processes. In the case of IPI, ΔG is much less negative (by about 20 kcal/mol) than ΔH due to the loss of translational and rotational degrees of freedom upon ion association. The number of particles decreases from 2 to 1, resulting in a large positive −TΔS term, and this decreases the thermodynamic driving force of the reaction. In contrast, in the case of IPII formation, ΔG and ΔH are closer as the number of particles is identical on the two sides of the reaction (2 vs 2), leading to a significantly smaller entropic effect. While our calculations demonstrate thermodynamically favorable ion pairs across all studied precatalysts, the process sensitivity to external conditions warrants consideration. The significant entropic contribution, especially in the case of IPI, suggests temperature dependence of the ion-pair stability through varying degrees of ion-pair separation and specific solvent–catalyst interactions. For example, more polar solvents would be expected to facilitate greater ion-pair separation compared with toluene, potentially affecting the activation process kinetics.
3.1.2. Separation of Ion Pairs
As shown in Table 1 and Figure 1, the complete dissociation of ion pairs is calculated to be endothermic and endergonic for all IPs (ΔHips > + 40 kcal/mol, ΔGips > + 20 kcal/mol), with the computed Gibbs free energies for separation in IPII ranging from 21.1 to 23.7 kcal/mol, which are about 5 kcal/mol lower than the corresponding energies (26.6–29.8 kcal/mol) obtained for IPI. These findings indicate that IPII is a weaker ion pair compared to IPI, and that ion-pair separation does not occur spontaneously, potentially requiring a molecule to facilitate this charge separation. A solvent-separated species has previously been detected experimentally in toluene solution.37,38 To support this process and identify potential species that may act as separators in ion-pair dissociation, three candidate species (precatalyst, CL monomer, and toluene solvent) were included at the ion-pair contact, and their geometric and electronic properties were evaluated using DFT calculations, as shown in Figure 4 and Table 2. The separation energy of the contact ion pair (ΔEss) by the separator (S) can be obtained using eq 6(15)
| 6 |
where A– = MeB(C6F5)3– or B(C6F5)4–.
Figure 4.

Optimized structures of the (a) monomer (CL)-separated ion pairs, MIP, (b) precatalyst-separated ion pairs, PIP, and (c) solvent (toluene)-separated ion pairs, SIP, in the presence of [MeB(C6F5)3]− (I) and [B(C6F5)4]− (II) counteranions. Distance values are given in Å. Coordination modes for each type of ion-pair separation are also indicated.
Table 2. Geometries, Gibbs Energies, and Enthalpies (kcal/mol) as well as NBO Charges for Zr and B Atoms Calculated for the Separation of Contact Ion Pairs (IPI) by CL, Precatalyst, and Solvent (Toluene), Which Are Denoted as MIP-I, PIP-I, and SIP–I, Respectivelya.
| competitive species | ΔG | ΔH | QZr (e) | QB (e) | Zr–F(B) (Å) | Zr–Cp′ (Å) | ∠Cp′ZrCp′ (deg) |
|---|---|---|---|---|---|---|---|
| MIP-I | –17.0 | –14.9 | 1.639 | 0.480 | 8.433 | 2.120 | 132.9 |
| (−14.0) | (−12.3) | (1.647) | (0.443) | (8.409) | (2.213) | (136.2) | |
| PIP-I | –17.0 | –21.3 | 1.467 | 0.487 | 5.324 | 2.221 | 133.8 |
| (−0.7) | (−7.5) | (1.446) | (0.443) | (7.267) | (2.212) | (135.2) | |
| SIP-I | –26.1 | –32.7 | 1.470 | 0.479 | 6.788 | 2.266 | 131.8 |
| (−26.3) | (−28.0) | (1.472) | (0.442) | (7.720) | (2.323) | (131.3) |
The corresponding values for the ion pairs IPII are also included in parentheses.
Table 2 shows that toluene has a pronounced effect on ion-pair separation (ΔGss = −26.1 kcal/mol) compared to CL and the precatalyst (ΔGss = −17 kcal/mol). This enhanced stabilization stems from specific cation–π interactions between Zr and toluene’s aromatic ring in SIP-I and SIP-II [3.170 vs 3.185 Å (Figure 4c)]. These interactions persist despite longer Zr···B distances in MIP-I and MIP-II (∼8.4 Å), compared to SIP-I and SIP-II (6.7 and 7.7 Å, respectively). The nature of the effects of solvent on ion-pair stability is expected to vary with solvent properties. Previous studies have shown that the ion-pair interaction is predominantly electrostatic, with solvation generally weakening cation–anion attraction.37,39 Aromatic solvents such as benzene or xylenes would likely exhibit similar stabilization through cation–π interactions, while more polar solvents such as THF or dichloromethane would stabilize separated ion pairs through different mechanisms. This solvent-dependent behavior is particularly relevant when considering bulkier ancillary ligands like Ind, which we found reduce ion-pair separation enthalpy due to steric effects. The data in Table 2 also indicate that the precatalyst is not a good separator as it tends to form a dimer species, [(Cp2ZrMe)2(μMe)]+, which reduces the catalyst performance in cationic ROP.27
3.1.3. Comparison with Experimental Studies on Ion-Pair Dynamics
Our computational results on ion-pair geometries and energetics align well with experimental observations of the dynamic behavior in metallocene-borate systems. The calculated Zr–B distances for [MeB(C6F5)3]− and [B(C6F5)4]− (4.099–4.235 and 5.187–5.977 Å, respectively) align with detailed structural and solution dynamics studies by Marks and co-workers showing distinct differences in ion-pair association between these anions.40 The stronger interaction we calculated for [MeB(C6F5)3]− supports experimental findings showing tighter ion pairs compared to [B(C6F5)4]− systems, which influence catalyst activity.35,41
The computed preference for solvent-separated species (ΔGss = −26.1 kcal/mol for toluene) provides theoretical support for experimental observations of the solution dynamics and η6-coordination of toluene. Recent NMR studies have demonstrated that aromatic solvents can coordinate to zirconocenium cations, significantly affecting the dynamics of these species.37 Our calculations reveal that this preference arises from favorable cation–π interactions between Zr and the aromatic ring (Zr–centroid distance of 3.17 Å). Investigations of arene coordination to group 4 metallocene cations have demonstrated the formation of stable [Cp*MMe2(η6-arene)]+ complexes (M = Ti, Zr, and Hf) with the [MeB(C6F5)3]− counteranion.38 These experimental findings support our calculated favorable energetics for solvent separation and the predicted η6-coordination mode of toluene.
3.2. Electronic and Steric Feature Analysis
To evaluate the nature of the interaction between the zirconocene catalysts and their counteranions, we conducted analyses of various electronic properties, including natural bond orbital (NBO), frontier molecular orbitals (FMOs), steric maps, and NCI.
3.2.1. NBO Charge
The electronic properties of these catalysts show a complex relationship with their reactivity. NBO analysis reveals that methyl substitution on the Cp ligand increases electron density at the Zr center through two mechanisms: direct electron donation and altered ring-metal bonding. This electronic enrichment has several important consequences for catalyst behavior:
-
1.
Ion-pair character: Higher Zr positive charge in methylated systems (QZr increasing from 1.535e to 1.706e; Table 3) strengthens interaction with counteranions. However, this is moderated by increased steric separation, consistent with established principles that weaker coordinating anions generally lead to higher catalyst activity.42
-
2.
Electronic-steric synergy: Electron-donating methyl groups enhance metal-Cp bonding strengths (see Zr-Cp′ in Table 1), partially compensating for reduced orbital overlap caused by steric congestion. The combined effect maintains catalyst stability while allowing necessary ion-pair separation.
-
3.
Reactivity implications: More electron-rich metal centers show modified Lewis acidity, which affects both counteranion coordination strength and monomer activation. Optimal activity typically occurs with intermediate electron density, balancing activation ability with stability.
Table 3. Electronic and Steric Analysis for the Formation of Contact Ion Pairs (IPI and IPII).
| ion pair | QZr (e) | QB (e) | HOMO (eV) | LUMO (eV) | Egap (eV) | Δ (attractive-steric clash) | % VBur, cat | % VBur, cocat |
|---|---|---|---|---|---|---|---|---|
| IPI-a | 1.535 | 0.519 | –7.996 | –1.457 | 6.539 | 0.381 | 62.9 | 19.2 |
| IPI-b | 1.634 | 0.509 | –7.922 | –1.241 | 6.681 | 0.193 | 67.9 | 18.8 |
| IPI-c | 1.706 | 0.518 | –7.543 | –1.152 | 6.391 | 0.182 | 71.8 | 17.6 |
| IPI-d | 1.601 | 0.522 | –7.294 | –1.547 | 5.747 | –1.634 | 66.6 | 18.9 |
| IPII-a | 1.589 | 0.441 | –7.938 | –1.364 | 6.575 | –1.569 | 63.4 | 23.1 |
| IPII-b | 1.664 | 0.441 | –7.850 | –1.215 | 6.635 | –1.767 | 68.5 | 20.7 |
| IPII-c | 1.785 | 0.441 | –7.633 | –1.479 | 6.153 | –1.561 | 73.2 | 16.5 |
| IPII-d | 1.700 | 0.442 | –7.333 | –1.699 | 5.634 | –3.579 | 68.1 | 20.1 |
These electronic effects align with extensive experimental studies showing that counteranion coordination strength significantly influences polymerization activity.39,43 Our computational results suggest that ligand electronic properties can be used to fine-tune this coordination strength, complementing the established effects of the steric bulk.
3.2.2. Frontier Molecular Orbital (FMO) Analysis
The HOMO–LUMO energies and their energy gaps (Egap) were evaluated for the ion pairs in Table 3 to understand their stability. The LUMO energy of IPI-a is −1.457 eV, increasing with sterically crowded Cp′ ligands: IPI-b (−1.241 eV) and IPI-c (−1.152 eV). Meanwhile, the presence of the Ind ligand lowers the HOMO–LUMO energy gap by 0.5–0.6 eV, resulting in gaps of 5.747 eV in IPI-d and 5.634 eV in IPII-d. Higher LUMO energy corresponds to greater ion-pair complex stability, while lower LUMO energy indicates reduced stability.
3.2.3. Steric Map Analysis
Percent buried volume (% VBur) is a key tool for analyzing steric properties in organometallic chemistry.44 We used this % VBur descriptor to quantify the steric bulkiness of a Cp′ ligand coordinated to the metal center. Alkylzirconocenium cations in each IP were assessed using SambVca 2.1,34 and steric maps were calculated using a sphere radius of 3.5 Å centered on the Zr atom, as shown in Figure 5. The % VBur values for the Cp′ ligands across different IPs (denoted as % VBur,cat) are collected in Table 3. Analyzing % VBur,cat values (catalyst: IPI, IPII) reveals an increase in steric bulk as follows: Cp2ZrMe2 (a: 62.9, 63.4) < (Me5Cp)CpZrMe2(b: 67.9, 68.5) < (Me5Cp)2ZrMe2 (c: 71.8, 73.2). Overall, %VBur,cat values are slightly higher for IPII than for IPI (Table 3), with catalyst c being the most sterically congested. Notably, catalysts b and d had similar % VBur,cat values (68), indicating that differences between IPI and IPII are mainly due to the steric effects of the specific ligands.
Figure 5.

Steric maps of Cp′2ZrMe+ structures (% VBur,cat) in the ion-pair complexes, IPI (left) and IPII (right), of catalysts a–d (labeled as (a–d), respectively). A sphere radius of 3.5 Å centered on the Zr atom is used for each contour map.
We also measured the steric parameters for the counteranions. This analysis was conducted by comparing the buried volume difference between the IP complexes and their alkylzirconocenium cation counterparts, calculated at a 6.0 Å radius around the metal center (Figures S4 and S5), which is assumed to represent the Zr···B distances of all studied IPs. The percent buried volumes for the counteranion fragment (% VBur,cocat) are summarized in Table 3. The results indicate that the IPs for catalyst a have the highest buried volume of the anions, whereas those for catalyst c exhibit the lowest % VBur,cocat. Thus, the % VBur,cocat values inversely correlate with the calculated % VBur,cat values.
3.2.4. Noncovalent Interactions
Our NCI analysis14 reveals a complex network of interactions between the zirconocene catalysts (a–d) and both counteranions ([MeB(C6F5)3]− and [B(C6F5)4]−). The 2D and 3D NCI plots (Figures 6 and S6) map these interactions through RDG values (−0.06 to +0.06) and low-gradient isosurfaces (0.3 Å cutoff), where blue, green, and red regions indicate attractive, van der Waals (vdW), and repulsive interactions, respectively.
Figure 6.

3D iso-surfaces (left) and 2D scatters (right) from NCI analysis for the four ion pairs [Cp′2ZrMe]+[MeB(C6F5)3]−: (a) IPI-a, (b) IPI-b, (c) IPI-c, and (d) IPI-d. The 0.3 Å cutoff is used for each iso-surface. Three specific interactions (① Zr–Me(B), ② Cp′-Me(B), and ③ Cp′-C6F5(B)) at the contact ion pairs were indicated. Additional vdW interactions between the two Cp′ rings were assigned as ④. Steric repulsion between the Me(B) group and the indene ligand is also shown with red lines.
The stability of each ion pair is governed by three categories of noncovalent interactions:
-
1.
Primary metal-anion interactions Zr–Me(B)/Zr–F(B): Strong attractive interactions (blue regions, −0.06 to −0.03) that are essential for initial catalyst activation.
-
2.
Secondary ligand-anion contacts Cp′-Me(B), and Cp′-C6F5(B): Moderate van der Waals interactions (green regions, −0.02 to +0.02) that enhance ion-pair stability.
-
3.
Intramolecular stabilization (Cp′–Cp′): Additional vdW interactions between Cp′ rings in methylated systems
The interaction patterns vary systematically with ligand structure: catalyst a (unsubstituted Cp) primarily shows metal-anion interactions with minimal secondary contacts. Catalysts b and c (methylated) exhibit increasing vdW stabilization from Cp′-Me(B) contacts, with c showing the strongest combination of primary and secondary attractive forces. Catalyst d (Ind) shows unique repulsive interactions (red regions, λ2 > + 0.04) due to indene rigidity. The counteranions exhibit distinct interaction modes: [MeB(C6F5)3]-IPI shows concentrated interactions through the Me– bridge, while [B(C6F5)4]-IPII has a more distributed interaction network through multiple F– contacts. These findings provide key insights for catalyst design.
3.2.5. Electronic–Steric Interplay
The methyl substituents on Cp ligands influence the catalyst properties through both steric and electronic mechanisms. The electron-donating nature of methyl groups increases electron density at the metal center, as evidenced by the trend in NBO charges at Zr (QZr = 1.535e, 1.634e, and 1.706e for catalysts a, b, and c, respectively; Table 3). This electronic enrichment strengthens the donor capability of the Cp′ ligands compared to unsubstituted Cp, affecting the metal-counteranion interaction strength. Simultaneously, the increasing steric bulk of methylated Cp ligands (reflected in %VBur values increasing from 62.9 to 71.8, Figure 5) forces greater Zr–B separation, as seen in the systematic increase in the Zr–B distances (Table 1). This steric–electronic interplay manifests in several ways:
-
1.
Ion-pair stability: While methyl substitution increases electron density at Zr, potentially strengthening ion-pair interaction, the dominant steric effects result in longer Zr–B distances and weaker overall ion-pair binding.
-
2.
Counteranion interaction: The electronic enrichment of the metal center reduces its Lewis acidity, while steric congestion limits the counteranion approach. This is particularly evident in IPI-c, where despite having the highest electron density at Zr, it shows optimal ion-pair stability due to balanced steric effects.
-
3.
Activation barriers: The enhanced electron density at Zr partially compensates for the increased steric hindrance, maintaining favorable activation energetics despite greater spatial separation of the ion pairs.
Clearly, the ion-pair stability depends on both the electronic and steric properties. This complex interplay suggests that optimal catalyst design requires careful balancing of electronic and steric factors rather than maximizing either independently.
4. Conclusions
We present a detailed DFT investigation of zirconocene/borate catalyst activation for cationic ROP of ε-caprolactone, examining [MeB(C6F5)3]− and [B(C6F5)4]− counteranions. Our analysis reveals how structural and electronic factors at the activation stage influence subsequent polymerization behavior. Ion-pair formation shows systematic variation with the ligand structure. The increasing −ΔGipf through the series IP-a, IP-b, and IP-c correlates with both steric bulk (% VBur) and electronic effects. These effects extend to other ligand architectures, as demonstrated by additional studies on fluorenyl- and tert-butyl-substituted systems. NCI analysis identifies key stabilizing interactions that affect both activation and the subsequent ROP process: primary metal-anion contacts essential for controlled polymerization, secondary ligand-anion interactions that influence reaction pocket geometry, and intramolecular contacts that stabilize catalyst structure during the monomer approach.
For ROP catalysis, several critical features emerge:
-
1.
Ion-pair characteristics directly influence monomer activation: IPII systems show easier dissociation than IPI, suggesting higher potential polymerization activity. Solvent-separated species, particularly with toluene, provide an optimal pathway for generating active catalysts while maintaining control.
-
2.
Ligand electronic effects impact ion-pairing reactivity for polymerization: Electron-donating methyl groups of the Cp ring modify metal center Lewis acidity, affecting both counteranion and monomer activation. The degree of methylation offers a way to tune catalyst reactivity while maintaining stability.
-
3.
Noncovalent interactions influence ion-pairing stability for polymerization behavior: Secondary stabilizing contacts help define monomer approach trajectories, and steric bulk from methylation creates controlled reaction spaces while maintaining necessary flexibility.
This mechanistic understanding of catalyst activation should facilitate the development of more efficient and selective catalysts for biodegradable polyester synthesis through ROP, particularly for the controlled synthesis of high-molecular-weight PCL.
Acknowledgments
Financial support of the University of Phayao and the Thailand Science Research and Innovation Fund (Fundamental Fund 2025, grant no. 5026/2567) is gratefully acknowledged. J.J. thanks the School of Science, University of Phayao, for grant no. PBTSC67019. Computer time and software are partly facilitated by the National e-Science Infrastructure Consortium of Thailand (www.e-science.in.th). J.O. thanks the financial support of the National Research Development and Innovation Office (NKFIH) through grant K146661. M.L. acknowledges the support of the Research Council of Finland, decision 357509.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c08377.
Summary of thermodynamic parameters for ion-pair formation and separation for different zirconocene catalysts, calculated at the M06-2X/def2-SVP level of theory in toluene; relative Gibbs free energies and enthalpies of ion-pair formation and separation for zirconocene catalysts in various forms, including gas-phase values; steric maps of ion pairs and alkylzirconocenium cation fragments for the precatalysts; NCI analysis results, including 3D isosurfaces and 2D scatters for the ion pairs; and additional results (geometries, steric map, and NCI) for other ion pairs with different Cp′ ligands, CpFluZrMe2 and (tBuCp)2ZrMe2 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jambeck J. R.; Geyer R.; Wilcox C.; Siegler T. R.; Perryman M.; Andrady A.; Narayan R.; Law K. L. Plastic waste inputs from land into the ocean. Science 2015, 347 (6223), 768–771. 10.1126/science.1260352. [DOI] [PubMed] [Google Scholar]
- Samir A.; Ashour F. H.; Hakim A. A. A.; Bassyouni M. Recent advances in biodegradable polymers for sustainable applications. npj Mater. Degrad. 2022, 6 (1), 68. 10.1038/s41529-022-00277-7. [DOI] [Google Scholar]
- Mangaraj S.; Yadav A.; Bal L. M.; Dash S.; Mahanti N. K. Application of biodegradable polymers in food packaging industry: A comprehensive review. J. Package Technol. Res. 2019, 3, 77–96. 10.1007/s41783-018-0049-y. [DOI] [Google Scholar]
- Wang Y.; Wang X.; Zhang W.; Sun W.-H. Progress of ring-opening polymerization of cyclic esters catalyzed by iron compounds. Organometallics 2023, 42 (14), 1680–1692. 10.1021/acs.organomet.3c00028. [DOI] [Google Scholar]; a Nifant’ev I.; Ivchenko P. Coordination ring-opening polymerization of cyclic esters: a critical overview of DFT modeling and visualization of the reaction mechanisms. Molecules 2019, 24, 4117. 10.3390/molecules24224117. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Meelua W.; Wanjai T.; Chokbunpiam T.; Jitonnom J. Mechanism of ring-opening polymerization of l-lactide by lanthanide aryloxide: A theoretical study on the effect of the aryloxide ligands on the process. Int. J. Quantum Chem. 2023, 123 (16), e27134 10.1002/qua.27134. [DOI] [Google Scholar]; DOI: https://doi.org/10.1002/qua.27134; c Jitonnom J.; Molloy R.; Punyodom W.; Meelua W. Theoretical studies on aluminum trialkoxide-initiated lactone ring-opening polymerizations: Roles of alkoxide substituent and monomer ring structure. Comput. Theor. Chem. 2016, 1097, 25–32. 10.1016/j.comptc.2016.10.009. [DOI] [Google Scholar]; d Sattayanon C.; Sontising W.; Jitonnom J.; Meepowpan P.; Punyodom W.; Kungwan N. Theoretical study on the mechanism and kinetics of ring-opening polymerization of cyclic esters initiated by tin(II) n-butoxide. Comput. Theor. Chem. 2014, 1044, 29–35. 10.1016/j.comptc.2014.06.008. [DOI] [Google Scholar]
- Engel J.; Cordellier A.; Huang L.; Kara S. Enzymatic ring-opening polymerization of lactones: traditional approaches and alternative strategies. ChemCatChem 2019, 11 (20), 4983–4997. 10.1002/cctc.201900976. [DOI] [Google Scholar]; DOI: https://doi.org/10.1002/cctc.201900976
- Xu Y.; Wang L.; Chen C.; Huang P.; Dai H.; Jiang W.; Zhou Y. Living cationic polymerization of ε-caprolactone catalyzed by a metal-free lewis acid of trityl tetrafluoroborate. Macromolecules 2023, 56 (2), 501–509. 10.1021/acs.macromol.2c02056. [DOI] [Google Scholar]
- Kaluzynski K.; Pretula J.; Lewinski P.; Kaźmierski S.; Penczek S. Catalysis in polymerization of cyclic esters. Catalyst and initiator in one molecule. Polymerization of ε-caprolactone. J. Catal. 2020, 392, 97–107. 10.1016/j.jcat.2020.09.026. [DOI] [Google Scholar]
- Kourti M.-E.; Fega E.; Pitsikalis M. Block copolymers based on 2-methyl- and 2-phenyl-oxazoline by metallocene-mediated cationic ring-opening polymerization: synthesis and characterization. Polym. Chem. 2016, 7 (16), 2821–2835. 10.1039/C6PY00405A. [DOI] [Google Scholar]; a Kourti M.-E.; Vougioukalakis G. C.; Hadjichristidis N.; Pitsikalis M. Metallocene-mediated cationic ring-opening polymerization of 2-methyl- and 2-phenyl-oxazoline. J. Polym. Sci. A Polym. Chem. 2011, 49 (11), 2520–2527. 10.1002/pola.24679. [DOI] [Google Scholar]
- Batagianni E.; Marathianos A.; Koraki A.; Maroudas A.-P.; Pitsikalis M. Metallocene-mediated cationic polymerization of vinyl ethers: Kinetics of polymerization and synthesis and characterization of statistical copolymers. J. Macromol. Sci., Part A: Pure Appl. Chem. 2016, 53 (3), 140–151. 10.1080/10601325.2016.1132908. [DOI] [Google Scholar]
- Hayakawa M.; Mitani M.; Yamada T.; Mukaiyama T. Living ring-opening polymerization of lactones using cationic zirconocene complex catalysts. Macromol. Chem. Phys. 1997, 198 (5), 1305–1317. 10.1002/macp.1997.021980502. [DOI] [Google Scholar]; a Kostakis K.; Mourmouris S.; Karanikolopoulos G.; Pitsikalis M.; Hadjichristidis N. Ring-opening polymerization of lactones using zirconocene catalytic systems: Block copolymerization with methyl methacrylate. J. Polym. Sci. A Polym. Chem. 2007, 45 (16), 3524–3537. 10.1002/pola.22099. [DOI] [Google Scholar]
- Hayakawa M.; Mitani M.; Yamada T.; Mukaiyama T. Living ring-opening polymerization of cyclic carbonate using cationic zirconocene complex as catalyst. Macromol. Rapid Commun. 1996, 17 (12), 865–870. 10.1002/marc.1996.030171204. [DOI] [Google Scholar]; DOI: https://doi.org/10.1002/marc.1996.030171204
- Jordan R. F.; Dasher W. E.; Echols S. F. Reactive cationic dicyclopentadienyl zirconium(IV) complexes. J. Am. Chem. Soc. 1986, 108 (7), 1718–1719. 10.1021/ja00267a068. [DOI] [Google Scholar]; a Jordan R. F.; LaPointe R. E.; Bradley P. K.; Baenziger N. Synthesis and chemistry of cationic alkyl, alkenyl, and allyl complexes derived from the soluble, cationic hydride (C5H4Me)2Zr(H)(THF)+. Organometallics 1989, 8 (12), 2892–2903. 10.1021/om00114a026. [DOI] [Google Scholar]
- Jordan R. F.Chemistry of cationic dicyclopentadienyl Group 4 metal-alkyl complexe. In Advances in Organometallic Chemistry, Stone F. G. A., West R., Eds.; Vol. 32; Academic Press, 1991; pp 325–387. [Google Scholar]
- Meelua W.; Linnolahti M.; Jitonnom J. Mechanism of cationic ring-opening polymerisation of ε-caprolactone using metallocene/borate catalytic systems: a DFT and NCI study on chain initiation, propagation and termination. RSC Adv. 2024, 14 (17), 11715–11727. 10.1039/D4RA01178C. [DOI] [PMC free article] [PubMed] [Google Scholar]; 10.1039/D4RA01178C
- Chan M. S. W.; Vanka K.; Pye C. C.; Ziegler T. Density functional study on activation and ion-pair formation in Group IV metallocene and related olefin polymerization catalysts. Organometallics 1999, 18 (22), 4624–4636. 10.1021/om9903285. [DOI] [Google Scholar]
- Vanka K.; Chan M. S. W.; Pye C. C.; Ziegler T. A density functional study of ion-pair formation and dissociation in the reaction between boron- and aluminum-based Lewis acids with (1,2-Me2Cp)2ZrMe2. Organometallics 2000, 19 (10), 1841–1849. 10.1021/om990830p. [DOI] [Google Scholar]
- Meelua W.; Wanjai T.; Jitonnom J. Computational evaluation of zirconocene catalysts for epsilon-caprolactone cationic ring-opening polymerization. Sci. Rep. 2024, 14 (1), 3952. 10.1038/s41598-024-54157-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jitonnom J.; Meelua W. Cationic ring-opening polymerization of cyclic carbonates and lactones by group 4 metallocenes: A theoretical study on mechanism and ring-strain effects. J. Theor. Comput. Chem. 2017, 16 (1), 1750003. 10.1142/S0219633617500031. [DOI] [Google Scholar]; a Sassmannshausen J. Computational studies of the unusual water adduct [Cp2TiMe(OH2)](+): the roles of the solvent and the counterion. Dalton Trans. 2014, 43 (29), 11195–11201. 10.1039/C4DT00310A. [DOI] [PubMed] [Google Scholar]; b Jitonnom J.; Meelua W. Data on electronic structures for the study of ligand effects on the zirconocene-mediated trimethylene carbonate polymerization. Data Brief 2018, 20, 1867–1869. 10.1016/j.dib.2018.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine A.; Coussens B. B.; Hirvi J. T.; Berthoud A.; Friederichs N.; Severn J. R.; Linnolahti M. Effect of ligand structure on olefin polymerization by a metallocene/borate catalyst: a computational study. Organometallics 2015, 34 (11), 2415–2421. 10.1021/om501185x. [DOI] [Google Scholar]
- Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, 2009.
- Karttunen V. A.; Linnolahti M.; Turunen A.; Pakkanen T. A.; Severn J. R.; Maaranen J.; Kokko E.; Pitkänen P. The influence of the ligand structure on activation of hafnocene polymerization catalysts: A theoretical study. J. Organomet. Chem. 2008, 693 (1), 155–163. 10.1016/j.jorganchem.2007.10.037. [DOI] [Google Scholar]; a Vanka K.; Chan M. S. W.; Pye C. C.; Ziegler T. Exploring the activation of olefin polymerisation catalysts with density functional theory. Macromol. Symp. 2001, 173 (1), 163–178. . [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120 (1), 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Andrae D.; Häußermann U.; Dolg M.; Stoll H.; Preuß H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77 (2), 123–141. 10.1007/BF01114537. [DOI] [Google Scholar]
- Schuchardt K. L.; Didier B. T.; Elsethagen T.; Sun L.; Gurumoorthi V.; Chase J.; Li J.; Windus T. L. Basis set exchange: a community database for computational sciences. J. Chem. Inf. Model. 2007, 47 (3), 1045–1052. 10.1021/ci600510j. [DOI] [PubMed] [Google Scholar]
- Goossens H.; Catak S.; Glassner M.; de la Rosa V. R.; Monnery B. D.; De Proft F.; Van Speybroeck V.; Hoogenboom R. Cationic ring-opening polymerization of 2-propyl-2-oxazolines: Understanding structural effects on polymerization behavior based on molecular modeling. ACS Macro Lett. 2013, 2 (8), 651–654. 10.1021/mz400293y. [DOI] [PubMed] [Google Scholar]
- Collins S.; Linnolahti M. Activation of Substituted Metallocene Catalysts Using Methylaluminoxane. ChemCatChem 2022, 14 (7), e202101918 10.1002/cctc.202101918. [DOI] [Google Scholar]; DOI: https://doi.org/10.1002/cctc.202101918; a Linnolahti M.; Collins S. Thermodynamics of metallocene catalyst activation: alignment of theory and experiment. Dalton Trans. 2022, 51 (29), 11152–11162. 10.1039/D2DT01711C. [DOI] [PubMed] [Google Scholar]; 10.1039/D2DT01711C
- Meelua W.; Keawkla N.; Oláh J.; Jitonnom J. DFT study of formation and properties of dinuclear zirconocene cations: Effects of ligand structure, solvent, and metal on the dimerization process. J. Organomet. Chem. 2020, 905, 121024. 10.1016/j.jorganchem.2019.121024. [DOI] [Google Scholar]
- Riplinger C.; Sandhoefer B.; Hansen A.; Neese F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139 (13), 134101. 10.1063/1.4821834. [DOI] [PubMed] [Google Scholar]
- Neese F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2022, 12 (5), e1606 10.1002/wcms.1606. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113 (18), 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Hirshfeld F. L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44 (2), 129–138. 10.1007/BF00549096. [DOI] [Google Scholar]
- Novoa T.; Laplaza R.; Peccati F.; Fuster F.; Contreras-García J. The NCIWEB server: a novel implementation of the noncovalent interactions index for biomolecular systems. J. Chem. Inf. Model. 2023, 63 (15), 4483–4489. 10.1021/acs.jcim.3c00271. [DOI] [PubMed] [Google Scholar]
- Johnson E. R.; Keinan S.; Mori-Sánchez P.; Contreras-García J.; Cohen A. J.; Yang W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132 (18), 6498–6506. 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]; a Laplaza R.; Peccati F.; A. Boto R.; Quan C.; Carbone A.; Piquemal J.-P.; Maday Y.; Contreras-García J. NCIPLOT and the analysis of noncovalent interactions using the reduced density gradient. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11 (2), e1497 10.1002/wcms.1497. [DOI] [Google Scholar]
- Falivene L.; Cao Z.; Petta A.; Serra L.; Poater A.; Oliva R.; Scarano V.; Cavallo L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11 (10), 872–879. 10.1038/s41557-019-0319-5. [DOI] [PubMed] [Google Scholar]
- Zaccaria F.; Sian L.; Zuccaccia C.; Macchioni A.. Chapter One - Ion pairing in transition metal catalyzed olefin polymerization. In Advances in Organometallic Chemistry; Pérez P. J., Ed.; Academic Press, 2020; Vol. 73, pp 1–78. Ed.; Vol. [Google Scholar]
- Correa A.; Cavallo L. Dynamic properties of metallocenium ion pairs in solution by atomistic simulations. J. Am. Chem. Soc. 2006, 128 (33), 10952–10959. 10.1021/ja062407v. [DOI] [PubMed] [Google Scholar]
- Sian L.; Dall’Anese A.; Macchioni A.; Tensi L.; Busico V.; Cipullo R.; Goryunov G. P.; Uborsky D.; Voskoboynikov A. Z.; Ehm C.; et al. Role of solvent coordination on the structure and dynamics of ansa-zirconocenium ion pairs in aromatic hydrocarbons. Organometallics 2022, 41 (5), 547–560. 10.1021/acs.organomet.1c00653. [DOI] [Google Scholar]
- Gillis D. J.; Quyoum R.; Tudoret M.-J.; Wang Q.; Jeremic D.; Roszak A. W.; Baird M. C. Synthesis and characterization of the series of d0 arene complexes [Cp*MMe2(η6-arene)][MeB(C6F5)3] (M = Ti, Zr, Hf). Organometallics 1996, 15 (16), 3600–3605. 10.1021/om960174i. [DOI] [Google Scholar]; a Sarsfield M. J.; Ewart S. W.; Tremblay T. L.; Roszak A. W.; Baird M. C. Synthesis of a series of new, highly electrophilic, monocyclopentadienyltitanium olefin polymerization initiators. J. Chem. Soc., Dalton Trans. 1997, (18), 3097–3104. 10.1039/A703112B. [DOI] [Google Scholar]; 10.1039/A703112B
- Chen E. Y.-X.; Marks T. J. Cocatalysts for metal-catalyzed olefin polymerization: activators, activation processes, and structure-activity relationships. Chem. Rev. 2000, 100 (4), 1391–1434. 10.1021/cr980462j. [DOI] [PubMed] [Google Scholar]
- Yang X.; Stern C. L.; Marks T. J. Cationic zirconocene olefin polymerization catalysts based on the organo-Lewis acid tris(pentafluorophenyl)borane. A synthetic, structural, solution dynamic, and polymerization catalytic study. J. Am. Chem. Soc. 1994, 116 (22), 10015–10031. 10.1021/ja00101a022. [DOI] [Google Scholar]
- Bochmann M.; Lancaster S. J. Monomer-dimer equilibria in homo- and heterodinuclear cationic alkylzirconium complexes and their role in polymerization catalysis. Angew. Chem., Int. Ed. Engl. 1994, 33 (15–16), 1634–1637. 10.1002/anie.199416341. [DOI] [Google Scholar]
- Yang X.; Stern C.; Marks T. J. Models for organometallic molecule-support complexes. Very large counterion modulation of cationic actinide alkyl reactivity. Organometallics 1991, 10 (4), 840–842. 10.1021/om00050a008. [DOI] [Google Scholar]; a Jia L.; Yang X.; Stern C. L.; Marks T. J. Cationic metallocene polymerization catalysts based on tetrakis(pentafluorophenyl)borate and its derivatives. probing the limits of anion ″noncoordination″ via a synthetic, solution dynamic, structural, and catalytic olefin polymerization study. Organometallics 1997, 16 (5), 842–857. 10.1021/om960880j. [DOI] [Google Scholar]
- Chien J. C. W.; Song W.; Rausch M. D. Polymerization of propylene by zirconocenium catalysts with different counter-ions. J. Polym. Sci., Part A: Polym. Chem. 1994, 32 (12), 2387–2393. 10.1002/pola.1994.080321219. [DOI] [Google Scholar]; a Matsui S.; Mitani M.; Saito J.; Tohi Y.; Makio H.; Matsukawa N.; Takagi Y.; Tsuru K.; Nitabaru M.; Nakano T.; et al. A family of zirconium complexes having two phenoxy-imine chelate ligands for olefin polymerization. J. Am. Chem. Soc. 2001, 123 (28), 6847–6856. 10.1021/ja0032780. [DOI] [Google Scholar]; b Zhou J.; Lancaster S. J.; Walker D. A.; Beck S.; Thornton-Pett M.; Bochmann M. Synthesis, structures, and reactivity of weakly coordinating anions with delocalized borate structure: The assessment of anion effects in metallocene polymerization catalysts. J. Am. Chem. Soc. 2001, 123 (2), 223–237. 10.1021/ja002820h. [DOI] [PubMed] [Google Scholar]; c Ioku A.; Hasan T.; Shiono T.; Ikeda T. Effect of cocatalyst on propene polymerization with [t-BuNSiMe2(C5Me4)]TiMe2. Macromol. Chem. Phys. 2002, 203 (4), 748–755. . [DOI] [Google Scholar]
- Falivene L.; Cavallo L.; Talarico G. Buried volume analysis for propene polymerization catalysis promoted by group 4 metals: A tool for molecular mass prediction. ACS Catal. 2015, 5 (11), 6815–6822. 10.1021/acscatal.5b01363. [DOI] [Google Scholar]; a Clavier H.; Nolan S. P. Percent buried volume for phosphine and N-heterocyclic carbene ligands: steric properties in organometallic chemistry. Chem. Commun. 2010, 46 (6), 841–861. 10.1039/b922984a. [DOI] [PubMed] [Google Scholar]; 10.1039/B922984A
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.