

Abstract
Biallelic variants in phosphatidylinositol glycan anchor biosynthesis, class G (PIGG) cause hypotonia, intellectual disability, seizures, and cerebellar features. We present 8 patients from 6 families with a childhood‐onset motor neuropathy and neurophysiology demonstrating variable motor conduction block and temporal dispersion. All individuals had a childhood onset tremor, 5 of 8 had cerebellar involvement, and 6 of 8 had childhood febrile seizures. All individuals have biallelic PIGG variants, including the previously reported pathogenic variant Trp505*, plus 6 novel variants. Null enzyme activity is demonstrated via PIGO/PIGG double knockout system for Val339Gly and Gly19Glu, and residual activity for Trp505* due to read‐through. Emm negative blood group status was confirmed in 1 family. PIGG should be considered in unsolved motor neuropathy. ANN NEUROL 2025;97:388–396
Phosphatidylinositol glycan anchor biosynthesis, class G (PIGG) is one of 22 phosphatidylinositol glycan (PIG) genes involved in the biosynthesis of glycosylphosphatidylinositol (GPI). GPI is a glycolipid that anchors over 150 proteins to the cell membrane, which in turn play a critical role in neurogenesis. 1 The core glycan structure of the glycolipid GPI consists of 3 mannoses, all modified with ethanolamine‐phosphate (EtN‐P). PIGG is responsible for the enzyme facilitating addition of EtN‐P to the second mannose. Contrary to previous understanding that the EtN‐P on the third mannose was the bridge to the GPI‐anchored protein (GPI‐AP), it has recently been shown that selected GPI‐APs are bound by EtN‐P on the second mannose, which sheds mechanistic insight into how variants in PIGG might cause disease. Biallelic variants in PIG genes cause inherited GPI deficiency (IGD); a group of disorders associated with intellectual disability (ID), seizures, and facial dysmorphism. 2 These features, in addition to cerebellar atrophy with associated ataxia and nystagmus, have recently been reported with recessive PIGG variants. 3 The red blood cell (RBC) antigen Emm, which was unidentified until recently, has been proven to be free GPI, 4 and biallelic PIGG variants independently identified as causing Emm‐negative blood group, with or without an associated neurodevelopmental syndrome. 5 We describe 8 cases from 6 families carrying biallelic variants in PIGG, displaying a distinct neuropathy syndrome, expanding the known phenotype.
Methods
Patient Selection and Genetic Testing
Families were recruited in the United Kingdom, Ireland, Cyprus, The Netherlands, and Serbia with informed consent obtained from all patients according to local institutional requirements. Patients were clinically assessed by neuromuscular/neurogenetic experts. Genetic testing was performed with either whole exome sequencing (WES) or whole genome sequencing (WGS), on a clinical or research basis in line with local practice (Supplementary Table S1). Virtual panels were applied to WES/WGS data to exclude known causes of neuropathy and ID, if applicable. Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) criteria. 6
Functional Analysis of PIGG Variants
As previously reported, PIGO knockout (PIGO KO) cells show partial loss of surface GPI‐APs, which is completely removed by further knockout of PIGG. 7 Introducing the PIGG gene into PIGO/PIGG double knockout (DKO) cells restores the expression of GPI‐APs to the level of a PIGO single knockout, allowing the activity of PIGG variants to be analyzed by flow cytometry. It has previously been shown that decay‐accelerating factor (DAF) and cell surface Fc receptor CD16 are sensitive to the partially reduced activity of PIGG. Therefore, to measure the PIGG variant activity, PIGO/PIGG DKO HEK293 cells (permanently expressing CD16) were transfected with the strong SRα promoter (pME) or weaker thymidine kinase promoter (pTK) driven wild‐type (WT) or mutant PIGG‐glutathione S‐transferase (PIGG‐GST) plasmids. 3 To determine transfection efficiency, luciferase expression plasmid was co‐transfected with PIGG‐GST plasmids. Restoration of the surface expression of DAF and CD16 was analyzed 2 days later by staining cells with anti‐DAF antibody (clone IA10) or anti‐CD16 antibody (3G8 Biolegend) followed by phycoerythrin labeled anti‐mouse IgG and analyzed by flow cytometry. An isotype control antibody, matching the class of the test antibody but not targeting any antigen (DAF, mouse IgG2a; CD16, and mouse IgG1), was used to confirm fluorescence is due to specific antibody binding. The protein expression of each PIGG‐GST variant was then analyzed by Western blotting with anti‐GST antibody (anti‐goat GST; GE Healthcare) using the cell lysate of each transfectant. To quantify PIGG‐GST levels, band intensities of PIGG‐GST were divided by the band intensities of GAPDH (loading control) and by luciferase activities (transfection efficiency).
Emm Blood Group Testing
Emm blood group antigen testing was performed as previously described. 5 The presence of the red cell Emm antigen was determined with a hemagglutination assay (indirect antiglobulin test in tubes with polyethylene glycol as enhancer). The anti‐Emm used was a polyclonal human antiserum, with anti‐Emm antibodies of the IgG class and produced by an unrelated Emm negative female patient.
Results
Clinical Description
Eight patients from 6 non‐consanguineous families were identified with biallelic variants in PIGG. Clinical characteristics are summarized in Table 1 and Supplementary Table S1. Two of 8 patients were male with mean age at assessment of 28.3 years. The age of onset of neuromuscular symptoms ranged from 4 months to early teens; all but one individual presented with lower limb symptoms. All had a postural tremor (mean age of onset 7.1 years, Supplementary Video S1) and in 3 cases this preceded the neuromuscular symptoms. Three‐quarters (6/8) of the patients had febrile seizures, all resolving by the age of 6 years, and none went on to develop epilepsy. There was variable, mild ID (3/8). Cerebellar signs were variably present: nystagmus (3/8; Supplementary Video S2) and ataxia (3/8). Individual 5:I was the only patient with developmental delay and dysmorphism.
Table 1.
Clinical Features of PIGG Families
| Family | 1 | 2 | 3 | 4 | 5 | 6 | ||
|---|---|---|---|---|---|---|---|---|
| Individual | I | II | I | I | I | II | I | I |
| Variant 1 | p.(Trp505*) | p.(Trp678*) | p.(Trp505*) | p.(Trp505*) | c.2735+2T>C | p.Asp876ArgfsTer111 | ||
| Variant 2 | Homozygous | p.(Gly41*) | Homozygous | p.(Val339Gly) p.(Gly19Glu) | p.(Gly278Arg) | Homozygous | ||
| Neuropathy phenotype | dHMN | dHMN | dHMN | dHMN | dHMN | dHMN | HMN | dHMN |
| Motor CB or TD | No | CB/TD | CB/TD | CB | TD and CB/TD | CB and TD | TD | CB and CB/TD |
| Ethnicity | White British | White British | Irish | Irish | Dutch | Dutch | Cyprus | White Serbian |
| Sex | M | F | F | F | F | M | F | F |
| Age at NM symptom (tremor) onset, yr | 11 (3) | Teens (9) | Early teens (15) | 3 (6) | Early childhood (4) | 12 (4) | 4 mo (?) | 2 (9) |
| Age at assessment (current), yr | 48 (57) | 43 (52) | 19 (27) | 19 (19) | 22 (23) | 20 (21) | 12 (27) | 43 (43) |
| Presenting NM symptom | Sprained ankles | Tripping | Tripping/falls | Tripping | Leg pain during exercise | Spontaneous movements in calves | Hypotonia | Frequent falls |
| CMTES (age, yr) | 5 (53) | 4 (47) | 5 (19) | 3 (19) | NA | NA | NA | 7 (43) |
| Lower limb weakness (MRC grade) | Distal (4+/5) | Distal (4+/5) | Distal (4/5) | Distal (4+/5) | Distal (4+/5) | Distal (4/5) | Distal (4+/5) proximal (4/5) | Distal (3/5) |
| Upper limb weakness | Nil | Nil | Nil | Nil | Nil | Nil | Distal (4/5) | Distal (4/5) |
| Nystagmus | No | Gaze‐evoked torsional | Gaze‐evoked | No | No | No | Gaze evoked | No |
| Strabismus | No | Yes | No | No | No | No | Yes | No |
| Ataxia | No | No | Finger nose ataxia | Mild heel shin ataxia | No | No | Mild gait ataxia | No |
| Postural tremor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| ID /cognitive impairment | No | No | Mild ID | No | Learning problems | No | Mild ID | No |
| Childhood febrile seizures (until, yr) | No | Yes, 1 episode | Yes (4) | Yes (4) | Yes (6) | Yes (4) | Yes (18 mo) | No |
| Cerebellar atrophy | Mild superior vermis | No | Yes | No | No | Unknown | No | Unknown |
| Nerve thickening (modality) | Yes (MRI thigh) | Yes (MRI spine) | Yes (MRI spine) | Yes (MRI spine) | Yes (Nerve US) | No | No | Unknown |
CB = conduction block; CMTES = Charcot–Marie‐Tooth Examination Score; CS = conduction slowing; (d)HMN = (distal) hereditary motor neuropathy; F = female; ID = intellectual disability; LL = lower limb; M = male; mo = months; MRC = medical research council; MRI = magnetic resonance imaging; NA = not applicable; NM = neuromuscular; TD = temporal dispersion; US = ultrasound.
Note: CD/TD signifies significant amplitude decrease but limitations of the study preclude delineation between CB versus TD.
The neuropathy was generally a mild, minimally progressive, distal, motor neuropathy, with only 4 of 8 patients having minor sensory symptoms or signs. Six of 8 patients had a foot deformity (Fig1A–F) and subtle upper motor neuron signs were seen in 3 of 8 patients. Individual 4:II showed striking spontaneous muscle activity after exercise, but not at rest, although further characterization with electromyography (EMG) was not possible (Supplementary Video S3).
Figure 1.
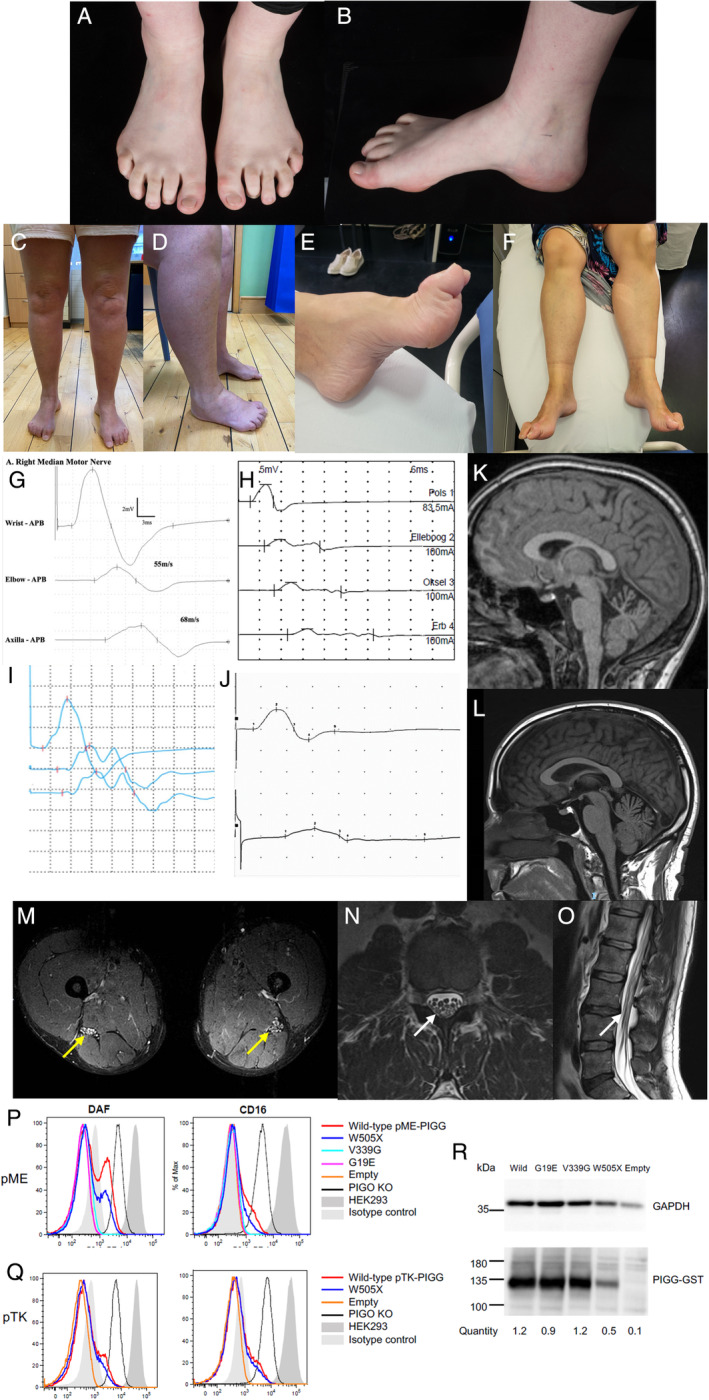
(A–F) Photography of lower limbs showing foot deformity and distal wasting (A and B patient 2:I, C and D patient 1:II, E and F patient 6:I); (G–J) Waveforms of motor nerve conduction studies showing conduction block and temporal dispersion (G patient 2:1 right median, H patient 4:I right median, I patient 5:I right ulnar, J patient 6:I left median); (K–L) MRI of the brain, sagittal T1 views showing prominent (K patient 2:I) and mild (L patient 1:I) superior vermian atrophy; (M) MRI of the thigh of patient 1:I (T2 TIRM axial view) shows thickening of sciatic nerve bilaterally returning high signal (yellow arrows); (N, O) MRI of the lumbar spine of patient 1:I shows thickened intradural roots within the cauda equina (N axial T2, O sagittal T2, white arrows); (P–R) PIGO/PIGG DKO HEK293 cells (permanently expressing CD16) were transfected with the strong promoter (pME, P) or weaker promoter (pTK, Q) driven wild‐type or mutant PIGG. Two days later, fluorescence‐activated cell sorting (FACS) analysis (P, Q) and Western Blotting (R) were performed. FACS analysis – X‐axis is relative cell number, Y‐axis is fluorescent intensity of phycoerythrin. Western blots – quantity was calculated by the band intensities of PIGG‐GST normalized with the band intensities of GAPDH (loading control) and with luciferase activity (transfection control). MRI = magnetic resonance imaging. [Color figure can be viewed at www.annalsofneurology.org]
Neurophysiology (Table 2) showed a motor neuropathy with normal sensory conduction in all individuals. The studies of 7 of 8 patients demonstrated variable motor conduction block (CB), temporal dispersion, and sometimes a combination of the two (Fig1G–J and Supplementary Fig S1), mostly in the forearm in the median and ulnar nerves. This is assessed by reduced motor amplitudes to proximal stimulation with either a fall in motor response area (motor CB) or prolongation of motor response (dispersion) or both. There was also some minor motor slowing seen generally but not within the demyelinating range. The only convincing conduction slowing was the outlying median velocity of 36.3 m/s in the forearm of individual 6:I. Where performed, imaging confirmed cerebellar atrophy in 2 of 6 individuals (Fig1K, L) and nerve thickening in 5 of 7 individuals (magnetic resonance imaging [MRI] or nerve ultrasound; Fig1M–O and Supplementary S1). Individual 5:I spontaneously improved with time, which was confirmed by serial neurophysiological studies (Supplementary Table S2).
Table 2.
Upper Limb Motor Nerve Conduction Studies
| Family | 1 | 2 | 3 | 4 | 5 | 6 | ||
|---|---|---|---|---|---|---|---|---|
| Individual | I | II | I | I | I | II | I | I |
| Age at study | 48 | 43 | 19 | 12 | 21 | 19 | 24 | 43 |
| Summary | LD motor neuropathy | LD motor neuropathy with median CB/TD | LD motor neuropathy with median and ulnar mixed CB/TD | LD motor neuropathy with ulnar CB | Motor neuropathy with median TD and mixed CB/TD in ulnar | Motor neuropathy with ulnar CB and median TD | LD motor neuropathy with ulnar TD | LD motor neuropathy with intermediate velocity in median and median CB and ulnar CB/TD |
| Median nerve | ||||||||
| Median CMAP (wrist) mV | 4.2 | 7.4 | 6.1 | 5.8 | 8.8 | 8.0 | 5.5 | 4.6 |
| Median CMAP (elbow) mV | 3.3 | 3.8 | 1.4 | 4.1 | 3.4 | 5.1 | 3.6 | 1.9 |
| Median DML ms | 4.7 | 3.9 | 3.1 | 3.5 | 3.4 | 4.8 | 3.5 | 3.5 |
| Median NCV (wrist‐elbow) m/s | 48 | 47 | 53 | 54 | 49 | 52 | 50 | 36.3 |
| Median F latency (ms) | 35 | 30 | 36 | ND | 32 | 34 | 33.4 | ND |
| Ulnar nerve | ||||||||
| Ulnar CMAP (wrist) mV | 5.9 | 5.8 | 4.1 | 4.3 | 5.7 | 6.2 | 4.8 | 2.7 |
| Ulnar CMAP (below elbow) mV | 3.7 | 3.9 | 0.5 | 2.6 | 2.9 | 3.2 | 2.3 | 1.1 |
| Ulnar CMAP (above elbow) mV | 3.1 | 3.6 | 0.9 | 2.6 | 2.8 | 3.2 | 2.1 | ND |
| Ulnar DML ms | 3.7 | 4.4 | 2.9 | 3.3 | 3 | 2.8 | 3.1 | 3.75 |
| Ulnar NCV (wrist‐below elbow) m/s | 55 | 53 | 67 | 42 | 49 | 61 | 53 | 47.3 |
| Ulnar NCV (around elbow) m/s | 50 | 56 | 67 | 110 | 57 | 44 | 85 | ND |
| Ulnar F latency ms | 52 | 30 | 40 | ND | 29 | 43 | ND | ND |
Note: All nerves studied on the right except patients 1:I, median nerve of 5:I and 6:I.
CB = conduction block; CMAP = compound motor action potential; DML = distal motor latency; LD = length‐dependent; NCV = nerve conduction velocity; ND = not done; TD = temporal dispersion; UL = upper limb.
Genetics
Six variants in PIGG were detected (see Table 1, variant classification Supplementary Table S3) including the previously reported c.1515G>A p.(Trp505*) (pathogenic), 3 , 8 c.832G>A p.(Gly278Arg) (variant of uncertain significance [VUS]), 3 , 9 and the novel c.121G>T p.(Gly41*) (pathogenic), c.2034G>A p.(Trp678*) (pathogenic), c.2735+2T>C (likely pathogenic), c.2625dup p.(Asp876ArgfsTer111) (likely pathogenic), and the pair of variants in cis c.56G>A p.(Gly19Glu) and c.1016T>G p.(Val339Gly) (both likely pathogenic). No other relevant variants were detected in any individuals; variants unrelated to the disease detected in individuals 2:I and 3:I are detailed in Supplementary Table S3.
Functional Studies and Emm Blood Group Testing
Three variants were tested in the PIGO/PIGG DKO system. Restoration of the GPI‐AP expression on PIGO/PIGG DKO cells by transfection with WT or variant PIGG cDNA was compared (Fig1P strong promoter, and Fig1Q weak promoter), and PIGG variants’ expression analyzed by Western blotting (Fig1R). As previously shown, transfection of WT PIGG shows decreased activity when C‐terminally tagged compared with non‐tagged PIGG in the PIGO KO 3 (red compared with black lines, Fig1P, Q). However, Val339Gly and Gly19Glu variants had null enzymatic activity even if they were transfected with the strong promoter driven construct (Fig1P turquoise and magenta lines), but they expressed protein levels similar to WT PIGG (Fig1R). The nonsense Trp505* variant showed decreased activity compared to WT PIGG driven by either the weak or strong promoter (Fig1P, Q blue compared with red lines) but retained residual activity because of partial expression of full‐length protein (Fig1R).
In the 2 members of family 4 (both compound heterozygous for Trp505* in trans with Val339Gly and Gly19Glu), there was no Emm antigen expression detectable on the red cells of the patients, confirming Emm negative blood group status (Supplementary Table S4).
Discussion
We report the first series of patients with biallelic variants in PIGG and a motor neuropathy associated with prominent tremor, febrile seizures, and variable cerebellar involvement (present in 3/8 patients, comparable to a previous study) but without ID in the majority. 3 Prior studies have reported hypotonia and diminished deep tendon reflexes (DTRs), suggesting a motor neuropathy, but without confirmatory neurophysiology. 2 , 3 The neuropathy, although relatively mild, is the unifying feature in this cohort. The prominence of early‐onset postural tremor seen in 4 cases without cerebellar signs, favors a neurogenic origin, supported by all cases of tremor reported by Tremblay‐Laganière et al (5/21) having either diminished DTRs or hypotonia. 3 A notable feature of the neuropathy is the prominent motor CB and temporal dispersion, with no major slowing of motor conduction in the segments with CB/dispersion. The presence of these features at non‐compression sites typically suggests an inflammatory etiology although CB/dispersion is a feature of some inherited neuropathies. 10 More unusual is motor CB or dispersion occurring in a motor predominant, inherited neuropathy, only reported rarely (SORD, PLEKHG5, and SIGMAR1). 11 , 12 , 13 The improvement of individual 5:I over time, for which the mechanism is not understood, again contributes to PIGG‐neuropathy acting as an inflammatory mimic.
The allele frequency of Trp505* in population databases (1719/1614208 alleles, heterozygous frequency 1.06 × 10−3, plus 2 homozygotes, in GnomADv4.0.0) in the context of a rare disease, merits discussion. Although this is compatible with a rare recessive disorder, the corresponding disease prevalence is expected to be higher. 14 An explanation for this discordance is the mild and variable phenotypes previously reported in individuals homozygous for this variant; isolated febrile seizures 3 and autism. 8 Similarly, the patients in our cohort (individuals 1:I, 1:II, and 3:I) with homozygous Trp505* have a relatively mild phenotype without the classical features of IGD. This is corroborated by the residual PIGG enzyme activity demonstrated in PIGO/PIGG DKO HEK293 cells transfected with the Trp505* mutant; we have confirmed the previously hypothesized residual protein product via likely read through of the nonsense codon. 3 , 15 Considering the population frequency and the above evidence, it is therefore likely that Trp505* is a hypomorphic allele, as seen in other IGD disorders, 16 and homozygotes may have minimal symptoms or do not manifest. Contrastingly both the Val339Gly and Gly19Glu variants (seen in cis in individuals 4:I and 4:II) show no enzyme activity and have population allele frequencies in the order of 0.5 to 1 x 10−5 in keeping with a complete loss‐of‐function allele.
The role of GPI and GPI‐APs in neurogenesis is clearly evidenced by the characteristic neurological features of IGD caused by almost 20 of the PIG and associated PGAP (post‐GPI attachment to proteins) families of genes. 1 The function of GPI as an anchor for dozens of cell surface proteins has led to work investigating the effect of the disease causing PIG genes on specific proteins, with disease mechanisms hypothesized. 17 , 18 , 19 , 20 However, to our knowledge, the exact disease mechanisms of the IGDs has not been elucidated.
All but one of the genes producing an IGD syndrome report associated hypotonia, but apart from 4 cases with variants in PIGB described with both axonal and demyelinating polyneuropathies (without accompanying neurophysiological data) 19 a neuropathy has not been explicitly identified in IGD.
Two broad hypotheses can be considered regarding the pathogenesis of PIGG‐related neuropathy and neuropathy in the broader context of this family of genes. First, that GPI is fundamental to peripheral neurogenesis and all IGD syndromes contain a neuropathy as part of their presentation due to aberrant GPI, and the reason this had not been previously demonstrated is that in many of the patients with IGD and severe central nervous system abnormalities or early death, this has not been investigated. Evidence as to which GPI‐AP, or combination of GPI‐APs, is implicated in disease is lacking, but the role of vitamin B6 has previously been postulated; particularly in view of the reported pyridoxine‐responsive seizures in some patients with IGD due to variants in PIGO and PIGS. 20 , 21 Murakami et al noted that the GPI‐AP alkaline phosphate converts the active form of vitamin B6 (pyridoxal 5’‐phosphate [PLP]) to pyridoxal allowing transport across the blood–brain‐barrier. Analogies can be drawn with both the recessive mutations in PDXK, which cause a B6‐responsive inherited motor and sensory neuropathy due to reduced enzymatic conversion of pyridoxal to PLP 22 , and the neuropathy caused by nutritional B6 deficiency. 23 Given the theoretical therapeutic implications, further work to confirm or refute the involvement of alkaline phosphate, and therefore B6, in the neuropathy of patients with IGD, is critical given the reported peripheral neuropathy caused by excess pyridoxine consumption. 23
Alternatively, the role of glial‐cell‐line‐derived neural growth factor (GDNF) as a survival factor for central and peripheral neurons could be considered for IGD‐related neuropathy. The cellular responses to GDNF have been shown to require the cell surface receptor GDNFR‐α which is a GPI‐AP. 24 Extrapolating, reduced surface expression of GDNFR‐α due to a PIG gene defect could render GDNF ineffective and result in a peripheral (and central) neuropathy.
The component of GPI that binds to the cell membrane is the phospholipid phosphatidylinositol. Independent of GPI, this moiety exists in 7 phosphorylation states, termed phosphoinositides, each controlling numerous cellular processes. Defects in these phosphorylation pathways are implicated in numerous neurological disorders, many of which phenotypically overlap with IGD syndromes. 25 Mutations in some phosphoinositide metabolism genes (FIG4, PTEN, MTMR2, and SBF1) also cause neuropathies with conduction slowing and/or CB, 26 , 27 , 28 suggesting a more fundamental role of GPI in the pathophysiology of this inherited neuropathy, independent of the GPI‐APs that are affected.
The above arguments, however, do not account for the specific phenotype seen in PIGG‐related neuropathy. The second hypothesis is that defects in PIGG have specific pathological effects on peripheral nerve causing this unusual neuropathy phenotype. Work by Ishida et al has furthered understanding of the role of PIGG in cell‐surface protein anchoring. They challenged the established notion that GPI‐APs are bound to the third mannose of GPI. Through a series of experiments, they showed that the EtN‐P on the second mannose, previously thought to be discarded after the GPI‐AP was anchored, was retained and itself formed the bridge to some selected GPI‐APs. 7 Comparing the expression levels of various GPI‐APs in PIGG KO HEK293 cells with those rescued by transient co‐transfection with PIGG cDNA, a number of GPI‐APs were PIGG‐dependent. The largest dependence was seen in NTNG2 (recessive variants in NTNG2 also cause a neurodegenerative disorder), 29 but importantly CNTN1 was among the most PIGG‐dependent. This is particularly relevant given the known acquired autoimmune paranodopathy caused by antibodies against CNTN1. 30 The paranodopathy causes an “acquired” neurophysiological picture with conduction slowing, dispersion and CB, but histopathology had demonstrated that the pathology (and the resultant neurophysiological features) lies in disruption of structures at the Node of Ranvier, and not in the myelin. 31 The neurophysiological findings in our PIGG‐neuropathy cohort, I demonstrating CB and dispersion in an otherwise predominantly axonal neuropathy, could theoretically place the pathology at the node/paranode, and in conjunction with evidence that CNTN1 is PIGG‐dependent, allow speculation that variants in PIGG cause a genetic paranodopathy. Although nonspecific, the consistency of the early onset neuropathic tremor in out cohort mirrors the frequent tremor seen in CNTN1‐related paranodopathy. 32 Last, CD59 is another GPI‐AP and biallelic variants in CD59 cause an inherited immune‐mediated neuropathy. Although not the most PIGG‐dependent GPI‐AP, 7 parallels can be drawn between this condition and the neurophysiological features in our cohort, particularly the spontaneous improvement of individual 5:I. 33
The importance of GPI in human health and disease, is illustrated by the fact that in addition to the neurological phenotype, PIGG has been demonstrated to define the rare blood group system Emm. 5 Duval et al have shown that the Emm antigen is not, like other blood groups, a GPI‐AP but free, unlinked, GPI. 4 The epitope for Emm was shown to be the EtN‐P on the second and third mannose groups; PIGG facilitates the addition of the second mannose and individuals with biallelic PIGG variants have shown absent Emm expression on RBC, that is, Emm negativity. 4 , 5 , 34 Patients with IGD and variants in PIGO (the gene facilitating the addition of EtN‐P to the third mannose) showed decreased but not absent Emm expression on RBCs, whereas those with PIGN and PIGA variants had normal of upregulated Emm levels. 4 In our study, the negative Emm blood group status of the siblings of family 4 support the literature that PIGG causes both neurological and hematological manifestations, and acts as further evidence for the pathogenicity of Trp505* in trans with Val339Gly and Gly19Glu.
In conclusion, our series of patients with biallelic variants in PIGG broadens the phenotype of PIGG‐related disease to include a motor‐predominant neuropathy associated with tremor, motor CB, and dispersion, that can neurophysiologically mimic acquired disease, without major ID. PIGG should be considered in all unsolved cases of inherited motor‐predominant neuropathy. Further clinical studies, including neurophysiology, are needed to characterize the hypotonia seen in other PIG‐related disorders to delineate the presence, or absence, of neuropathy, and the neurophysiological phenotype. More work is needed to further understand the mechanism of PIGG‐related neuropathy. Application of existing methods to demonstrate reduced PIGG activity or expression for our unstudied novel variants would add support to their pathogenicity, but are unlikely to aid mechanistic understanding. Ideally, studies would investigate nerve histopathology of affected individuals, tissue‐specific gene expression of GPI‐APs, and immunological reactivity to anti‐CNTN1 (and other) antibodies of the paranode of PIGG deficient neurons, or other cell lines, challenging the hypothesis of a genetic paranodopathy. Alternatively, disease models in patient‐derived induced pluripotent stem cell‐derived motor neurons, or animal models, could shed light on pathological mechanisms.
Author Contributions
C.J.R., A.O'C., P.M.C., S.M., and M.M.R. contributed to the conception and design of the study. C.J.R., A.O'C., N.E.V., W.vR., E.Z.P., S.P., P.C.L., M.S., E.vB., V.I., B.H., J.C.M., J.B., Y.M., M.L., S.M.M., and M.M.R. contributed to the acquisition and analysis of data. C.J.R., A.O'C., Y.M., and M.M.R. contributed to drafting the text or preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Video S1. Tremor in individual 1:II.
Video S2. Nystagmus in individual 1:II.
Video S3. Spontaneous muscle activity after exercise in individual 4:II.
Figure S1. Further examples of conduction block and temporal dispersion, MRI images showing nerve root thickening, nerve ultrasound demonstrating patchy nerve thickening.
Figure S2. Uncropped Western Blots.
Table S1. Additional clinical features.
Table S2. Serial nerve conduction studies in individual 5:I.
Table S3. Variant classification.
Table S4. Hemagglutination results EMM antigen typing.
Acknowledgments
C.J.R. and M.M.R. are grateful to the Medical Research Council (MRC MR/S005021/1) and the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712 and 1UOINS109403‐01 and R21TROO3034) and M.M.R. also to the Muscular Dystrophy Association (MDA510281) and the Charcot Marie Tooth Association (CMTA) for their support. This research was also supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. M.L. is grateful to the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712). This work was also supported by a grant from the Ministry of Health, Labour, and Welfare, and a grant from the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development (AMED; grants 23FC1033, JP22ek0109614, and JP23bm1223019 to Y.M.). S.M.M. is a member of the European Reference Network for Rare Neurological Diseases—Project ID No. 739510. W.vR. is supported by funding provided by the Dutch Research Council (NWO; VENI scheme grant 09150161810018) and Prinses Beatrix Spierfonds (neuromuscular fellowship grant W.F19‐03). Part of this research was made possible through access to data in the National Genomic Research Library, which is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The National Genomic Research Library holds data provided by patients and collected by the NHS as part of their care and data collected as part of their participation in research. The National Genomic Research Library is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The authors are grateful to Dr Stephan Goedee, University Medical Center Utrecht, for providing nerve ultrasound images.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Kinoshita T. Biosynthesis and biology of mammalian GPI‐anchored proteins. Open Biol 2020;10. 10.1098/rsob.190290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Makrythanasis P, Kato M, Zaki MS, et al. Pathogenic variants in PIGG cause intellectual disability with seizures and Hypotonia. Am J Hum Genet 2016;98:615–626. 10.1016/j.ajhg.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tremblay‐Laganière C, Maroofian R, Nguyen TTM, et al. PIGG variant pathogenicity assessment reveals characteristic features within 19 families. Genet Med 2021;23:1873–1881. 10.1038/s41436-021-01215-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duval R, Nicolas G, Willemetz A, et al. Inherited glycosylphosphatidylinositol defects cause the rare Emm‐negative blood phenotype and developmental disorders. Blood 2021;137:3660–3669. 10.1182/blood.2020009810. [DOI] [PubMed] [Google Scholar]
- 5. Lane WJ, Aeschlimann J, Vege S, et al. PIGG defines the Emm blood group system. Sci Rep 2021;11:1–11. 10.1038/s41598-021-98090-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ishida M, Maki Y, Ninomiya A, et al. Ethanolamine‐phosphate on the second mannose is a preferential bridge for some GPI‐anchored proteins. EMBO Rep 2022;23:1–16. 10.15252/embr.202154352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arteche‐López A, Rodríguez MJG, Calvin MTS, et al. Towards a change in the diagnostic algorithm of autism spectrum disorders: evidence supporting whole exome sequencing as a first‐tier test. Genes (Basel) 2021;12:1–16. 10.3390/genes12040560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stranneheim H, Lagerstedt‐Robinson K, Magnusson M, et al. Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med 2021;13:1–15. 10.1186/s13073-021-00855-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rajabally YA, Adams D, Latour P, Attarian S. Hereditary and inflammatory neuropathies: a review of reported associations, mimics and misdiagnoses. J Neurol Neurosurg Psychiatry 2016;87:1051–1060. 10.1136/jnnp-2015-310835. [DOI] [PubMed] [Google Scholar]
- 11. Record CJ, Pipis M, Blake J, et al. Unusual upper limb features in SORD neuropathy. J Peripher Nerv Syst 2022; 27:175–177. 10.1111/jns.12492. [DOI] [PubMed] [Google Scholar]
- 12. Villar‐Quiles RN, Le VT, Leonard‐Louis S, et al. Leukoencephalopathy and conduction blocks in PLEKHG5‐associated intermediate CMT disease. Neuromuscul Disord 2021;31:756–764. 10.1016/j.nmd.2021.06.004. [DOI] [PubMed] [Google Scholar]
- 13. Frezatti RSS, Tomaselli PJ, Figueiredo FB, et al. Conduction block and temporal dispersion in a SIGMAR1‐related neuropathy. J Peripher Nerv Syst 2022;27:316–319. 10.1111/jns.12517. [DOI] [PubMed] [Google Scholar]
- 14. Pipis M, Rossor AM, Laura M, Reilly MM. Next‐generation sequencing in Charcot–Marie–tooth disease: opportunities and challenges. Nat Rev Neurol 2019;15:644–656. 10.1038/s41582-019-0254-5. [DOI] [PubMed] [Google Scholar]
- 15. Dunn JG, Foo CK, Belletier NG, et al. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. Elife 2013;2013:1–32. 10.7554/eLife.01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Almeida AM, Murakami Y, Layton DM, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 2006;12:846–851. 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 17. Johnstone DL, Nguyen TTM, Zambonin J, et al. Early infantile epileptic encephalopathy due to biallelic pathogenic variants in PIGQ: report of seven new subjects and review of the literature. J Inherit Metab Dis 2020;43:1321–1332. 10.1002/jimd.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tremblay‐Laganière C, Kaiyrzhanov R, Maroofian R, et al. PIGH deficiency can be associated with severe neurodevelopmental and skeletal manifestations. Clin Genet 2021;99:313–317. 10.1111/cge.13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murakami Y, Nguyen TTM, Baratang N, et al. Mutations in PIGB cause an inherited GPI biosynthesis defect with an axonal neuropathy and metabolic abnormality in severe cases. Am J Hum Genet 2019;105:384–394. 10.1016/j.ajhg.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Efthymiou S, Dutra‐Clarke M, Maroofian R, et al. Expanding the phenotype of PIGS‐associated early onset epileptic developmental encephalopathy. Epilepsia 2021;62:e35–e41. 10.1111/epi.16801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuki I, Takahashi Y, Okazaki S, et al. Vitamin B 6 –responsive epilepsy due to inherited GPI deficiency. Neurology 2013;81:1467–1469. 10.1212/WNL.0b013e3182a8411a. [DOI] [PubMed] [Google Scholar]
- 22. Chelban V, Wilson MP, Warman Chardon J, et al. PDXK mutations cause polyneuropathy responsive to pyridoxal 5′‐phosphate supplementation. Ann Neurol 2019;86:225–240. 10.1002/ana.25524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kramarz C, Murphy E, Reilly MM, Rossor AM. Nutritional peripheral neuropathies. J Neurol Neurosurg Psychiatry 2024;95:61–72. 10.1136/jnnp-2022-329849. [DOI] [PubMed] [Google Scholar]
- 24. Treanor JJS, Goodman L, De Sauvage F, et al. Characterization of a multicomponent receptor for GDNF. Nature 1996;382:80–83. 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- 25. Volpatti JR, Al‐Maawali A, Smith L, et al. The expanding spectrum of neurological disorders of phosphoinositide metabolism. Dis Model Mech 2019;12. 10.1242/dmm.038174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Azzedine H, Bolino A, Taïeb T, et al. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot‐Marie‐tooth disease associated with early‐onset glaucoma. Am J Hum Genet 2003;72:1141–1153. 10.1086/375034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nicholson G, Lenk GM, Reddel SW, et al. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain 2011;134:1959–1971. 10.1093/brain/awr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bansagi B, Phan V, Baker MR, et al. Multifocal demyelinating motor neuropathy and hamartoma syndrome associated with a de novo PTEN mutation. Neurology 2018;90:E1842–E1848. 10.1212/WNL.0000000000005566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dias CM, Punetha J, Zheng C, et al. Homozygous missense variants in NTNG2, encoding a presynaptic netrin‐G2 adhesion protein, Lead to a distinct neurodevelopmental disorder. Am J Hum Genet 2019;105:1048–1056. 10.1016/j.ajhg.2019.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Querol L, Nogales‐Gadea G, Rojas‐Garcia R, et al. Antibodies to contactin‐1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2013;73:370–380. 10.1002/ana.23794. [DOI] [PubMed] [Google Scholar]
- 31. Doppler K, Appeltshauser L, Wilhelmi K, et al. Destruction of paranodal architecture in inflammatory neuropathy with anti‐contactin‐1 autoantibodies. J Neurol Neurosurg Psychiatry 2015;86:720–728. 10.1136/jnnp-2014-309916. [DOI] [PubMed] [Google Scholar]
- 32. Zhao M, Chen G, Li S, et al. Recurrent CNTN1 antibody‐positive nodopathy: a case report and literature review. Front Immunol 2024;15:1–8. 10.3389/fimmu.2024.1368487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haliloglu G, Maluenda J, Sayinbatur B, et al. Early‐onset chronic axonal neuropathy, strokes, and hemolysis: inherited CD59 deficiency. Neurology 2015;84:1220–1224. 10.1212/WNL.0000000000001391. [DOI] [PubMed] [Google Scholar]
- 34. Shah RJ, Senjaliya SB, Harimoorthy V, et al. Anti‐Emm, a rare specificity to the high‐incidence antigen Emm in an Indian patient defining the new blood group system EMM (ISBT042). Asian J Transfus Sci 2021;15:223–225. 10.4103/ajts.ajts_59_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Tremor in individual 1:II.
Video S2. Nystagmus in individual 1:II.
Video S3. Spontaneous muscle activity after exercise in individual 4:II.
Figure S1. Further examples of conduction block and temporal dispersion, MRI images showing nerve root thickening, nerve ultrasound demonstrating patchy nerve thickening.
Figure S2. Uncropped Western Blots.
Table S1. Additional clinical features.
Table S2. Serial nerve conduction studies in individual 5:I.
Table S3. Variant classification.
Table S4. Hemagglutination results EMM antigen typing.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.