

ABSTRACT
Epstein‐Barr virus (EBV) is a ubiquitous human ɣ‐herpesvirus implicated in various malignancies, including Burkitt's lymphoma and gastric carcinomas. In most EBV‐associated cancers, the viral genome is maintained as an extrachromosomal episome by the EBV nuclear antigen‐1 (EBNA1). EBNA1 is considered to be a highly stable protein that interacts with the ubiquitin‐specific protease 7 (USP7). Here, we show that pharmacological inhibitors and small interfering RNA (siRNA) targeting USP7 reduce EBNA1 protein levels in a proteosome‐dependent manner. Proteomic analysis revealed that USP7 inhibitor GNE6776 altered the EBNA1 protein interactome, including disrupting USP7 association with EBNA1. GNE6776 also inhibited EBNA1 binding to EBV oriP DNA and reduced viral episome copy number. Transcriptomic studies revealed that USP7 inhibition affected chromosome segregation and mitotic cell division pathways in EBV+ cells. Finally, we show that GNE6776 selectively inhibited EBV+ gastric and lymphoid cell proliferation in cell culture and slowed EBV+ tumor growth in mouse xenograft models. These findings suggest that USP7 inhibitors perturb EBNA1 stability and function and may be exploited to treat EBV latent infection and tumorigenesis.
Keywords: EBNA1, EBV, gastric cancer, inhibitor, USP7
1. Introduction
Epstein‐Barr virus (EBV) is an oncogenic human gamma‐herpesvirus that infects nearly 90% of the world's population [1, 2]. Globally, 1.5% of human malignancies can be attributed to EBV [3, 4]. EBV has been linked to multiple types of malignancies, including, but not limited to, nasopharyngeal carcinoma (NPC), gastric carcinoma (GC), Burkitt's lymphoma (BL), and Hodgkin's lymphoma (HL) [2]. EBV cancers are thought to depend on the transforming activities of numerous viral genes and long‐term persistence of the viral genome in transformed cells [5, 6].
All EBV+ tumors express EBV‐encoded nuclear antigen‐1 (EBNA1), which is an essential multifunctional sequence‐specific DNA‐binding protein that regulates both viral and host gene expression and the persistence of the viral DNA within the host nucleus [7, 8]. Accordingly, EBNA1 is a potential target for inhibiting tumor growth in EBV‐associated cancers. The importance of EBNA1 is highlighted by the fact that EBNA1 is the only protein consistently expressed in all EBV+ tumors and all latency types [9]. During latent infection, EBV requires EBNA1 to facilitate viral DNA replication and maintenance of the EBV episomal genome [10, 11]. These functions are dependent on the interaction between EBNA1 and the latent origin of replication (oriP) [7]. EBNA1 binding to oriP facilitates EBV DNA replication, episomal maintenance, and transcriptional activation of other viral genes [10, 11]. EBNA1 autoregulates its own transcription through binding to the BamHI Q promoter (Qp), thus tightly controlling the expression levels of EBNA1 during latent infection [12]. Therefore, disruption of either EBNA1 expression or the capacity to bind to viral DNA can deregulate its expression and EBV persistence [13, 14].
The ubiquitin‐specific protease‐7 (USP7), also known as herpes‐associated ubiquitin‐specific protease, is a deubiquitinating enzyme (DUB) involved in the regulation of cellular protein turnover via the ubiquitin proteasome system (UPS) (reviewed in [15, 16]). Much like all DUBs, USP7 can stabilize proteins and rescue them from turnover by removing ubiquitin moieties from ubiquitinated substrates [15, 16]. USP7 interacts with many cellular proteins implicated in cancer, including p53, MDM2, DNMT1, and UHRF1, where it regulates ubiquitination and proteosome‐dependent degradation of target proteins (reviewed in [17, 18]). USP7 also interacts with many viral regulatory proteins. The herpes simplex virus 1 (HSV) tegument protein ICP0, which has E3 ubiquitin ligase activity, binds USP7 and targets it for proteosome‐dependent degradation [18, 19, 20]. In contrast, LANA, the Kaposi's sarcoma herpesvirus (KSHV) orthologue of EBNA1, interacts with USP7 to regulate the LANA‐dependent DNA replication function [21]. The physical interaction of USP7 with EBNA1 has been characterized extensively [22, 23, 24, 25]. A partial crystal structure of the interaction between USP7 and EBNA1 reveals a USP7‐binding domain on EBNA1 within the 395‐450 residues [23]. Further analyses indicated that USP7 facilitates the recruitment of EBNA1 to oriP [22]. However, whether USP7 deubiquitinase activity affects EBNA1 stability and/or function in EBV genome persistence remains unclear.
Several small molecule inhibitors of USP7 have been developed and characterized in molecular mechanistic detail [26, 27, 28, 29, 30]. Here, we use several USP7‐specific inhibitors to investigate the enzymatic role of USP7 in regulating EBNA1 protein stability and function in EBV episome maintenance. We have extended the current understanding of the role of USP7 binding to EBNA1 by providing evidence that USP7 is necessary to sustain EBNA1 protein levels and EBV episomal maintenance. Our findings demonstrate that inhibiting the catalytic activity of USP7 impairs both the EBNA1 protein stability and function in EBV episome. Moreover, our study reveals that USP7 inhibitors selectively disrupt the proliferation of EBV+ cells in tissue culture and mouse xenograft models, underscoring the potential of these inhibitors as a targeted therapy for EBV+ cancer cells.
2. Materials and Methods
2.1. Cell Lines and Reagents
AGS (gastric cancer, EBV−), YCCEL1 (gastric cancer, EBV+, latency I), SNU719 (gastric cancer, EBV+, latency I), Raji (Burkitt lymphoma, EBV+, latency III), and BJAB (B‐cell lymphoma, EBV‐) were grown in RPMI‐164 medium (Gibco) supplemented with 15% fetal bovine serum (FBS) (Gibco), 50 U/mL penicillin and streptomycin (Corning), 10% l‐glutamine (Corning), and 10% Glutamax (Gibco). HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 50 U/mL penicillin and streptomycin. All cells were maintained in an incubator at 37°C with 5% CO2.
2.2. Western Blot
Western blots were performed as previously described [31]. Briefly, cells were collected and lysed in radioimmunoprecipitation assay (RIPA) buffer (0.1% sodium dodecyl sulfate, 150 mM NaCl, 0.5% deoxycholic acid, 1.0% IgePal, 50 mM Tris at pH 8.0). Total protein concentration was quantified with Pierce BCA Protein Assay Kit according to the manufacturer's protocol (Thermo Scientific). Equal amounts of protein were loaded onto a 4%–16% gradient SDS‐polyacrylamide gel (Invitrogen), separated by electrophoresis, and transferred onto a nitrocellulose membrane (Bio‐Rad). Membranes were blocked with 5% milk in 1× Tris‐buffered saline‐Tween20, then immunoblotted with antibodies against proteins, as indicated in the figures and imaged on Amersham Imager 680. The following antibodies were used: mouse monoclonal anti‐FLAG‐M2‐peroxidase (Sigma‐Aldrich, A8592), rabbit polyclonal anti‐USP7 (Invitrogen, PA534911), mouse monoclonal anti‐USP7 (Invitrogen, MA531515), rabbit polyclonal anti‐EBNA1 (Pocono Rabbit Farm), and anti‐β‐actin‐peroxidase (Sigma Aldrich, A3854). Quantitation of gel bands were performed using ImageJ v1.54k.
2.3. Digital Droplet PCR
Digital droplet PCR was performed as previously described [32]. Briefly, DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen), and then DNA concentration was calculated using the NanoDrop2000 Spectrophotometer (ThermoFisher). DNA was digested and emulsified with droplet generator oil (Bio‐Rad) using the QX‐100 droplet generator and amplified using GeneAmp 9700 thermocycler (Applied Biosystems). The droplets were quantified using the QX200 droplet reader (Bio‐Rad).
2.4. Drug Treatments
For cell culture, GNE6776 (MedChem Express‐ Cat: HY‐107986), XL177A (MedChem Express‐ Cat: HY‐138794), and (R)‐FT671 (Aobious‐ Cat: AOB12773) were resuspended in DMSO to make a stock solution and then diluted in fresh media to reach the final working concentration. A titration was performed on EBV+ gastric cells (YCCEL1) and B cells (Raji) to determine the concentration and duration of drug treatment, which was then analyzed in triplicate for statistical analysis. EBV‐ cells received the same drug treatment conditions as EBV+ cells to determine if there was selectivity. Cell comparisons of Raji versus BJAB, or YCCEL1 versus AGS were selected based on similar proliferation rates. Cells were replenished with new media containing the same concentration of drugs or DMSO every day until the specified time as indicated.
For EBNA1 stability assays using cycloheximide, transfected cells were treated with GNE6776 at 24 h posttransfection. Following 24 h posttreatment of GNE6776, the media was replenished with new media containing either only GNE6776 or in combination with cycloheximide (ThermoFisher‐ Cat:J66004. XF).
For mouse studies, compounds were weighed and mixed with 0.5% methylcellulose and 0.2% Tween‐80 in PBS. After distribution into treatment and vehicle groups to normalize fluorescence (SNU719 and BJAB NIR) or luciferase signal (Raji and AGS) based on IVIS measurements (D0), treatment was administered by oral gavage twice a day (bis in die, b.i.d.) at a dose of 25 or 50 mg/kg body weight. The vehicle control contained formulation reagents without GNE6776.
2.5. Pulse‐Field Gel Electrophoresis
Samples were treated as indicated, then pelleted and washed with 1× PBS. The pellets were resuspended in 2% low‐melt agarose (Bio‐Rad) and cast in CHEF Reusable Plug Mold (Bio‐Rad). The agarose‐encased samples were digested in 1 mg/mL proteinase K overnight in 1% SDS with 0.2 M EDTA. Pulse‐field gel electrophoresis was performed as previously described [33].
2.6. Southern Blot
The Southern blot was performed as described [33]. Briefly, the gel was transferred onto a nylon membrane overnight and UV‐fixed (UV Stratalinker 2400). A α‐32P‐labeled probe specific for the EBV WP region or cellular α‐satellite repeat DNA (5’‐tttcttttgatagtgcagttttgaaacattctttttaaaaaatctgcagt‐3’) was used to hybridize to the DNA on the membrane then visualized with a Typhoon 9410 variable‐mode imager (GE Healthcare Life Sciences).
2.7. Transfections and RNA Interference
To generate transiently expressing FLAG‐tagged EBNA1, cells were seeded onto a 6‐well plate and then transfected using the plasmids as indicated in the figures with OptiMEM (Gibco) and lipofectamine 2000 (Invitrogen) according to the manufacturer. siRNA‐mediated knockdown was performed as previously described [31]. Plasmids for tranfection included pCMV‐FLAG‐EBNA1(WTdGA)‐oriP‐Hygro (N2624) and pCMV‐FLAG‐ΔEBNA1(Δ395‐450dGA)‐oriP‐Hygro (N3763). Specific ON‐TARGETplus Smartpool siRNA against USP7 (AAGCGUCCCUUUAGCAUUA, GCAUAGUGAUAAACCUGUA, UAAGGACCCUGCAAAUUAU, GUAAAGAAGUAGACUAUCG) or ON‐TARGETplus nontargeting pool (UGGUUUACAAUGUCGACUAA, UGGUUUACAUGUUGUGUGA, UGGUUUACAUGUUUUCUGA, UGGUUUACAUGUUUUCCUA) (Horizon Discovery).
2.8. Immunoprecipitation
Cells were lysed with 1% Triton X‐100, 1× protease inhibitor (Sigma Aldrich), 1 mM PMSF, 1 mM Na3VO4, 10 mM NaF, 150 mM NaCl, 1× phosphatase inhibitor, and 0.25 units/uL benzonase nuclease at 4°C for 2 h. Samples were then centrifuged at 4°C for 5 min at 20,000 × g, and the supernatant was transferred to a new tube for protein quantification. Proteins of interest were immunoprecipitated, as indicated in the figures.
2.9. Chromatin Immunoprecipitation
ChIP analyses were performed as previously reported [33]. Briefly, HEK293T cells were transfected with wild‐type pCMV‐FLAG‐EBNA1‐oriP or pCMV‐Empty Vector using lipofectamine 2000. Cells were then collected after 72 h posttransfection for ChIP analysis.
2.10. Liquid Chromatography‐Tandem Mass Spectrometry (LC‐MS/MS) Analyses and Data Processing
HEK293T cells were transfected with pCMV‐FLAG‐EBNA1 containing oriP and prepared for immunoprecipitation. Immunoprecipitated samples were run into an SDS‐gel for a short distance, and the entire stained gel lanes were excised and digested with trypsin. Tryptic peptides were analyzed using data‐dependent acquisition (DDA) on a Q Exactive Plus mass spectrometer (ThermoFisher Scientific) coupled to a Vanquish Neo UHPLC system (ThermoFisher Scientific). DDA data were searched against a UniProt human protein database (August 2023) and a common contaminant database using MaxQuant 1.6.3.3 [34]. Consensus identification lists were generated with false discovery rates set at 1% for protein and peptide identifications.
2.11. Gene Ontology Analysis
Genes identified through mass spectrometry were analyzed using ShinyGo 0.80, software developed and maintained by the Xijin Ge lab at South Dakota State University [35].
2.12. Resazurin Assay
Cells were seeded at 5 × 103 per well into a 96‐well plate. The cells were grown in the presence or absence of the indicated inhibitors, with each condition performed in quadruplicates. Puromycin was used as the positive control. Cells were then collected at various time points as indicated. Resazurin was added to each well on the last day, and the plates were processed using the PerkinElmer EnVision Xcite multilabel plate reader.
2.13. RNA‐Seq
Samples were collected, and RNA was isolated using RNAeasy (Qiagen) according to the manufacturer's protocol, including DNase I treatment. RNA‐seq library and data processing were performed as described previously [36]. Briefly, QuantSeq (Lexogen) was used to generate the library, which was then sequenced on NextSeq. 500 (Illumina). STAR was used to align reads to the human GRCh38 and EBV NC_007605.1 genomes simultaneously, and read counts were estimated using RSEM v1.2.12 software [37, 38]. Genes with low read counts (less than 10) in all samples were removed and differential gene expression was analyzed using DESeq. 2 [39]. Significance was calculated using Wald's test and p‐values were adjusted for multiple testing using the Benjamini‐Hochberg method. Shrinkage of fold change estimates were performed using DESeq. 2 lfcShrink function with type = “ashr.” Pathway analysis was performed using ShinyGO 0.8 database.
2.14. Mouse Xenograft Studies
All mice in this study were managed based on Animal Care (AAALAC) per the NIH Office of Laboratory Animal Welfare: “PHS Policy on the Humane Care and Use of Research Animals.” National and institutional guidelines for the care and use of laboratory animals were followed. All procedures were approved by The Wistar Institute Institutional Animal Care and Use Committee (IACUC) in compliance with the Animal Welfare Act (AWA) and PHS Policy on Humane Care and Use of Laboratory Animals.
Both male and female NSG mice (NOD.Cg‐Prkdc scid Il2rgtm1Wjl/SzJ) bred in‐house at The Wistar Institute under protocol number 201281 were used for the xenograft studies. All mice were fed sterile food and water ad libitum and enrolled in their respective studies at 8 weeks of age. The mice were housed in micro‐isolator cages in a designated, specific pathogen‐free facility at The Wistar Institute (Philadelphia, PA). For visualization by the In Vivo Imaging System Spectrum CT (IVIS, Revvity Inc.), cells were transduced to express iRP713 NIR (BJAB and SNU719) or mCherry‐eLuciferase (Raji and AGS). Mice were engrafted with a cell suspension (> 98% viability) of 1 × 106 for B cells (Raji and BJAB) and 5 × 106 cells for gastric cancer cells (SNU719 and AGS) resuspended in 1× PBS (pH 7.4) and mixed with 20% of ice‐cold Matrigel (Corning Life Sciences). Cells were injected subcutaneously into the flank of each mouse (BJAB‐NIR left flank/Raji mCherry‐eLuciferase right flank or SNU719 NIR left flank/AGS mCherry‐eLuciferase right flank). The mice were monitored daily and weighed three times per week. The experimental endpoints were reached when the tumor volume of at least one of the tumor pairs reached 1000 mm3 in the vehicle control group. This was to avoid exceeding the humane endpoint of 2000 mm3 total tumor volume. For the BJAB/RAJI pair, this was treatment Day 17; for the SNU719/AGS pair, this was treatment Day 24.
Mice were euthanized via CO2 administration following the American Association for Accreditation of Laboratory Animal Care (AAALAC) euthanasia guidelines.
All tumors were measured by caliper, and tumor volume was calculated as follows: tumor volume = [length (L) × width (W)2]/2.
2.15. Quantitation and Visualization of Tumor Growth Using the Xenogen IVIS Bioluminescent Imaging System
Groups were normalized by bioluminescent imaging using the Spectrum IVIS CT Bioluminescent Imaging System (Perkin‐Elmer; Waltham, MA) to ensure the average flux (photons/sec) was equivalent across groups and to monitor cell growth throughout the study. Before imaging, mice were injected intraperitoneally with d‐luciferin (Gold Biotechnology) with a dose of 7.5 mg/kg in a dose volume of 10 mL/kg body weight for 15 min. The optimal interval between luciferin injection and bioluminescent imaging was determined by performing an initial kinetic curve for the different cell lines in mice. Mice were immobilized using isoflurane before and during imaging.
2.16. Flow Cytometry
2.16.1. Analysis of Cell Cycle Kinetics
After treatment, cells were permeabilized with cold, 70% ethanol and resuspended in PBS containing PI (10 mg/mL) and RNAse A solution (100 µg/mL) for 30 min before proceeding to acquisition on a BD FACSymphony A5 SE (BD Biosciences; Bedford, MA) Cell cycle profiles were analyzed using FloJo software (Ashland, OR).
2.16.2. Ki67 Analysis by Flow Cytometry
Tumor sections were homogenized and filtered through a 45 μM filter, followed by a 35 μM filter filtration. Cells were washed twice with PBS and then fixed with 70% cold ethanol for 1 h at −20°C. Cells were subjected to live/dead stain (Zombie, Biolegend, San Diego, CA) for 30 min, then washed three times with cell staining buffer (PBS with 5% fetal bovine serum and 0.09% sodium azide) before subjecting to fixation and permeabilization (Cytofix/Cytoperm, BD Biosciecnes; Bedford, MA). Fixed and permeabilized cell suspensions were then mixed with an antibody to Ki67 conjugated with Alexa 488 (Biolegend, San Diego, CA) and incubated at room temperature for 30 min. Cells were washed three times with cell staining buffer and flow cytometry was performed on BD FACSymphony A5 SE (BD Biosciences; Bedford, MA). Data were analyzed using FloJo software (Ashland, OR). The sequential gating strategy included a. cell‐size (FSC‐A/SSC‐A), b. doublet discrimination (FSC‐A vs. FSC‐H), c. live dead screen (Zombie), and d. selection of tumor cells from the ex vivo homogenate (mCherry for AGS and Raji, AF700 for SNU719‐NIR and BJAB‐NIR) before the mean fluorescence intensity of Ki67 (AF488) was determined.
3. Results
3.1. USP7 Enzymatic Activity Is Important for Maintaining EBNA1 Protein Levels
To investigate the potential role of USP7 deubiquitinase activity in the regulation of EBNA1, we tested the effects of a well‐characterized USP7‐specific pharmacological inhibitor (GNE6776) on EBNA1 protein expression levels in several EBV+ cells. GNE6776 prevents USP7 deubiquitinase activity by non‐covalently targeting USP7 within 12 angstroms of the catalytic pocket to prevent ubiquitin binding [26]. We found that treatment with GNE6776 on EBV+ B cells (Raji) for 96 h and EBV+ gastric cells (YCCEL1) for 48 h reduced EBNA1 protein levels by ~35% and ~76%, respectively, as measured by Western blot (Figure 1A–D). Next, we tested other USP7 inhibitors and another EBV+ cell line to ensure the initial findings were not an artifact of a singular drug nor cell‐line specific. XL177A covalently binds within 5 angstroms of the catalytic pocket, while (R)‐FT671 non‐covalently binds within 4.7 angstroms of the catalytic domain of USP7 [28, 29, 30]. EBNA1 protein levels in SNU719 were consistently decreased relative to cellular β‐actin by ~52% with (R)‐FT671 and ~47% by XL177A (Figure 1E–H). Since EBNA1 protein levels may be regulated indirectly by effects on viral transcription or episome copy number, we tested the effects of GNE6776 on EBNA1 protein stability in EBV− AGS cells transiently transfected with FLAG‐EBNA1 expression vector. We observed a ~79% loss of FLAG‐EBNA1 relative to cellular β‐actin in transfected 293 T cells in response to treatment with GNE6776 (Figure 1I–J).
Figure 1.
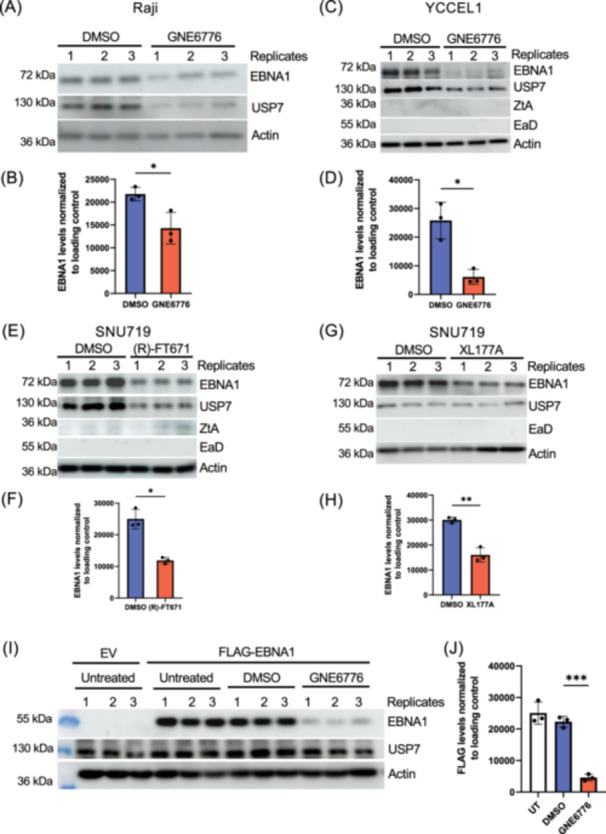
USP7 inhibitors reduce EBNA1 protein levels. (A) Raji cells were treated with DMSO or GNE6776 (40 μM) for 96 h and then assayed by Western blot for EBNA1, USP7, or loading control β‐Actin (actin). Molecular weight markers are indicated to the left. (B) Quantification of EBNA1 levels relative to actin for three biological replicates shown in panel A. (C) YCCEL1 cells were treated with DMSO or GNE6776 (20 μM) for 48 h and then assayed by Western blot for EBNA1, USP7, Zta, Ea‐D or loading control β‐Actin. (D) Quantification of EBNA1 levels relative to actin for 3 biological replicates shown in panel C. (E) SNU719 cells was treated with DMSO or (R)‐FT671 (30 μM) for 96 h and then assayed by Western blot for EBNA1, USP7, Zta, Ea‐D or loading control β‐Actin. (F) Quantification of EBNA1 levels relative to actin for 3 biological replicates shown in panel E. (G) SNU719 cells were treated with DMSO or XL177A (30 μM) for 6 days and then assayed by Western blot for EBNA1, USP7, Ea‐D or loading control β‐Actin. (H) Quantification of EBNA1 levels relative to actin for three biological replicates shown in panel E. (I) AGS cells were transfected with the empty vector (EV) or vector expressing Flag‐tagged EBNA1. At 24 h posttransfection, the media was replaced with either normal media containing DMSO or 20 µM of GNE6776 for 48 h. (J) Densitometry of FLAG‐EBNA1 levels normalized to β‐actin for three replicates shown in panel I. The error bar represents the standard deviation mean (sdm). ***p < 0.0005 **p < 0.01, *p < 0.0.05 using a two‐tailed student t‐test.
We next asked if GNE6776‐induced loss of EBNA1 protein was dependent on proteosome degradation (Figure 2A,B). We tested the effects of the proteasome inhibitor MG132 on EBNA1 protein levels in the absence or presence of GNE6776. We found that MG132 had little effect on EBNA1 protein levels in the absence of GNE6776, but partially rescued ( ~ 74%) EBNA1 protein levels when combined with GNE6776 (Figure 2A,B). Control experiments indicated that MG132 increased total ubiquitin levels, which were further increased when combined with GNE6776 (Figure 2A, middle panel). To determine whether GNE6776 affected EBNA1 protein stability, we blocked protein synthesis by addition of cycloheximide, and assayed EBNA1 protein decay rates in 293T‐FLAG‐EBNA1‐transfected cells. We found that GNE6776 reduced EBNA1 protein levels significantly more (~36%) in the presence of cycloheximide relative to DMSO treatment (Figure 2C,D). This suggests that USP7 inhibition impairs the half‐life of EBNA1 and the effects of USP7 inhibitors are likely to affect EBNA1 protein stability.
Figure 2.
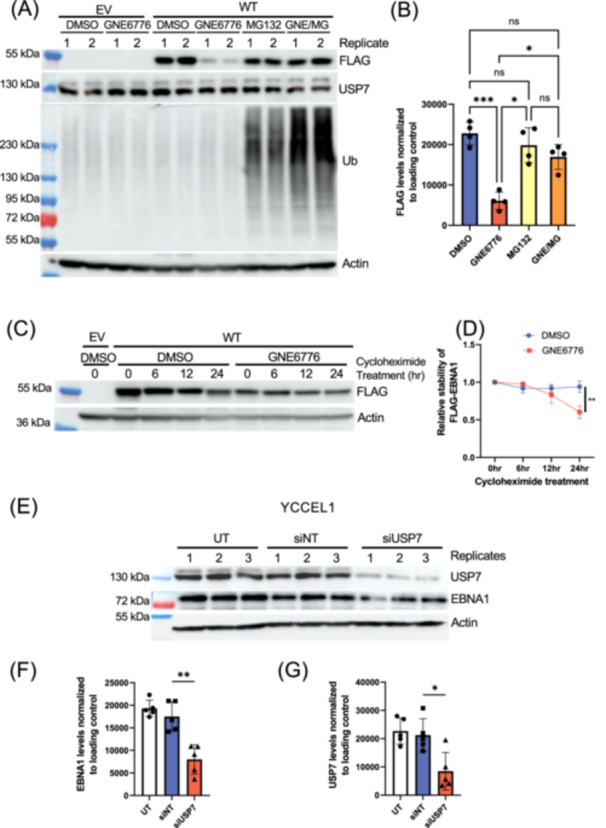
EBNA1 protein levels is destabilized by GNE6776 and knockdown of USP7. (A) A western blot representative of the analysis on AGS cells transfected with either EV or WT and treated with either DMSO, GNE6776, MG132, or combination of GNE6776 and MG132 (GNE/MG). Blots were probed for FLAG, USP7, ubiquitin (Ub), or loading control β‐actin. (B) Densitometry of FLAG‐EBNA1 levels normalized to the loading control n n = 4 *p < 0.002 ***p < 0.0005 by one‐way ANOVA. (C) Western blot analysis of cells transfected with either EV or WT and treated with DMSO or GNE6776 in the absence or presence of cycloheximide at various durations. (D) Densitometry of the time course analysis as shown in panel C. **p < 0.007 by 2‐way ANOVA. (E) YCCEL1 cells were untreated (UT) or transfected with either a nontargeting siRNA (siNT) or a siRNA pool specific against USP7 (siUSP7). Cells were collected for western blot and analyzed for USP7, EBNA1, and β‐Actin. Quantification of EBNA1 (F) and USP7 (G) relative to β‐actin from (E). *p < 0.02, **p < 0.002 by two‐tailed t‐test.
To confirm that the observations were not a product of off‐target effects by the inhbitors, we performed siRNA knockdown specifically against USP7 in EBV+ gastric cell line YCCEL1. These cells were chosen since they are highly transfectable and could achieve ~60% reduction in USP7. This knock‐down of USP7 correlated with a ~51% depletion of EBNA1 at 3 days posttransfection (Figure 2E–G). We further validated these findings in HEK293T cells stably expressing EBV through a bacmid and observed that ~53% USP7 reduction resulted in ~46% EBNA1 reduction (Figure S1A–C). In contrast, deletion of the USP7 interaction domain (EBNA1∆395‐450) did not affect the response of EBNA1 to GNE6776 (Figure S1D,E). These results suggest that USP7 enzymatic activity, and not USP7 binding to EBNA1 aa395‐450, is responsible for the stabilization of EBNA1.
3.2. USP7 Inhibitors Alter EBNA1 Protein Interactome
EBNA1 is known to interact with several cellular proteins, including USP7 [33, 40, 41]. To determine if USP7 inhibitors alter these interactions, we performed EBNA1 immunoprecipitation followed by LC‐MS/MS analysis of cells treated with GNE6776 or DMSO control. We found significant changes in many of EBNA1's interaction partners after GNE6776 treatment (Figure 3A). For example, CK2α1 and CK2α2, which have a known interaction with EBNA1 [42] are reduced by ~100‐fold and ~47.9‐fold, respectively. We also found that USP7 interaction with EBNA1 was decreased significantly (~171‐fold) (Figure S2). This was validated by the IP‐Western blot (Figure 3B). While most EBNA1 interactions were reduced by GNE6776 treatment, some interactions, were significantly increased, such as those with STK38L, SEMG1, and ACLY. Gene Ontology analysis identified molecular functions associated with mRNA splicing and RNA metabolism for EBNA1‐interacting proteins reduced by GNE6776 (Figure 3C). In contrast, proteins increased for EBNA1 interaction were associated with processes relating to degranulation and exocytosis (Figure S3A,B). A network diagram of the biological processes impacted by GNE6776 displays two pathways that GNE6776 impairs‐ posttranscriptional regulation and chromatin regulatory processes, including telomeric DNA binding and helicase activity (Figure 3D). These findings show that GNE6776 alters the EBNA1 protein interactome with distinct functional associations.
Figure 3.
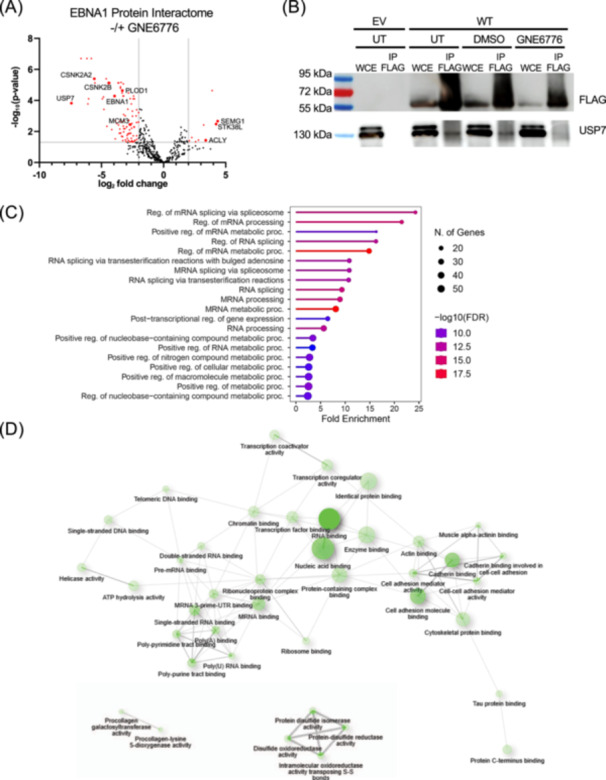
USP7 inhibitor perturbed EBNA1 protein‐interactome. (A) 293 T cells expressing FLAG‐EBNA1 protein were treated with DMSO or 20 µM GNE6776, followed by FLAG‐immunopurification and tandem LC‐MS/MS analysis to identify EBNA1‐associated proteins. The volcano plot shows DMSO and GNE6776 treatment differences using log2 fold change in protein abundance (x axis) and ‐Log10 p value. (B) Co‐immunoprecipitation of AGS cells transfected with EV or FLAG‐EBNA1 untreated or treated with DMSO or GNE6776. Whole‐cell extract (WCE) or FLAG‐IP samples were assayed by Western blot for FLAG or USP7. (C) Gene set enrichment analyses of downregulated protein interactions on samples after 48 h of GNE6776 treatment compared to DMSO based on ShinyGO 0.8 analysis of the gene's biological processes. (D) Network diagram of the molecular functions significantly affected by GNE6776 treatment based on EBNA1 interacting proteins downregulated by drug treatment. The FDR cutoff was 0.05.
3.2.1. USP7 Inhibitors Affect EBNA1 Chromatin Binding and Episome Maintenance Function
Although USP7 reduces EBNA1 protein levels, it is not yet known whether these levels reflect the DNA bound fraction nor the relative ratio of EBNA1 to viral DNA. To investigate these questions, we assayed EBNA1 and USP7 binding to EBV genome by chromatin immunoprecipitation (ChIP) in SNU719 cells (Figure 4A,B). GNE6776 treatment led to a ~61% decrease in EBNA1 binding to FR and ~48% decrease at Qp (Figure 4A). It has been previously reported that USP7 can bind the FR region [22]. Our data are consistent with these findings, but we further demonstrated that USP7 can also interact with the Qp and OriLyt regions (Figure 4B). Treatment with GNE776 significantly impaired USP7's interaction with these regulatory regions, to an extent far exceeding that of protein depletion (Figure 1I). This suggests that USP7 inhibitors disrupt chromatin interactions of EBNA1 and USP7 with viral DNA. To assess the effects of USP7 inhibition on EBNA1 functional activity we assayed the EBNA1‐dependent DNA replication of an oriP plasmid in transiently transfected 293 T cells (Figure 4C,D). GNE6776 reduced oriP‐dependent DNA replication by ~50%. Next, we tested the effects of USP7 inhibitors on EBV episome maintenance in EBV‐associated gastric carcinoma cell lines (Figure 4E–I). Treatment of SNU719 cells with GNE6776 for 6 days led to a ~60% reduction in viral episomal DNA levels as measured by Southern blot analysis of pulse‐field gel electrophoresis (PFGE) (Figure 4E,F). We observed a similar loss of EBV DNA copy number in another EBVaGC cell line (YCCEL1) as measured by digital droplet PCR (ddPCR) (Figure 4G). Other USP7 inhibitors X177A (Figure 4H) or (R)‐FT6771 (Figure 4I) in SNU719 cells revealed similar significant loss of EBV DNA content relative to cellular DNA using ddPCR. Together, these results indicate that USP7's deubiquitinase activity promotes EBNA1 and USP7 chromatin binding, oriP‐dependent DNA replication and viral episome maintenance.
Figure 4.
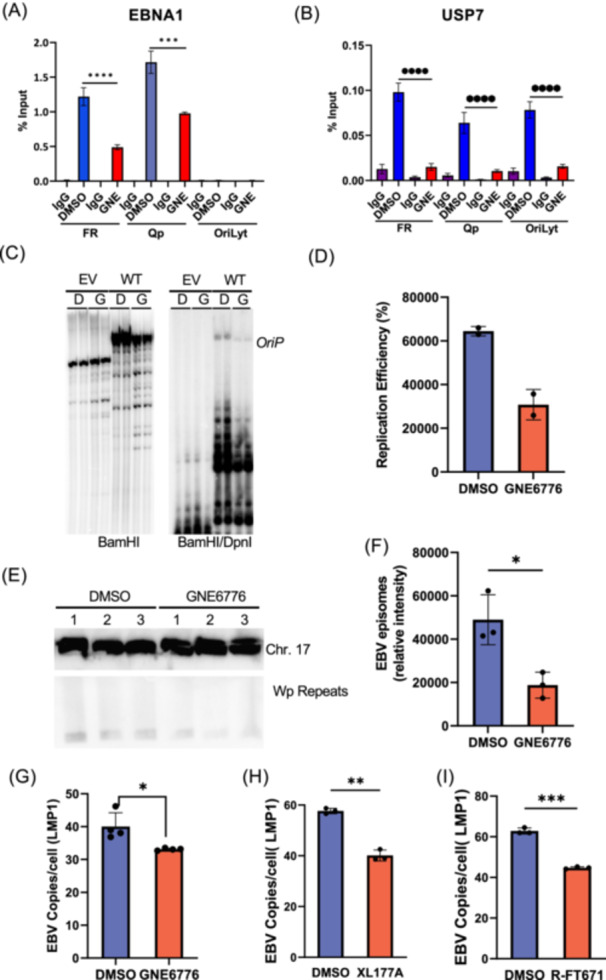
GNE6776 inhibits EBNA1 DNA binding and EBV episome maintenance. (A) SNU719 cells were treated with DMSO or GNE6776 (20 μM) for 6 days and then assayed by ChIP‐qPCR with IgG control or EBNA1 antibody and assayed by qPCR with primers amplifying FR, Qp, or OriLyt regions of EBV genome. Bars represent the sdm of 3 biological replicates. (B) ChIP‐qPCR as described in panel A, except using antibodies for USP7 and IgG control. ****p <0.0001, ***p <0.001 using student two‐tailed t‐test. (C) A replication assay in 293 T cells transfected with either EV or WT EBNA1 and then treated with either DMSO or GNE6776. (n = 2) (D) Quantitation of the replication efficiency of panel E. (E) SNU719 cells treated with DMSO or GNE6776 followed by pulse‐field gel electrophoresis (PFGE) and Southern blot probed for EBV genome (Wp repeats, lower panel) or cellular loading control Chr 17 probe (top panel). (F) Quantification of EBV episome copy number averaging 3 biological replicates shown in panel C. Error bars are sdm. *p < 0.05 student two‐tailed t‐test. (G) Digital droplet PCR (ddPCR) analysis of EBV DNA copy number in YCCEL1 cells treated with DMSO or GNE6776 using a probe for LMP1 gene relative to cellular RPP30. (H) ddPCR analysis of EBV DNA copy number in SNU719 cells treated with DMSO or XL177A. (I) Same as in panels E and F, except comparing DMSO with (R)‐FT671. *p < 0.05, **p < 0.005, ***p < 0.0009 student two‐tailed t‐test.
3.3. Inhibiting USP7 Selectively Blocks EBV+ Cancer Cell Growth
Given the negative impact of GNE6776 on EBNA1 stability and function, we next tested if GNE6776 selectively inhibits EBV+ cancer cell proliferation. We compared EBV‐ and EBV+ gastric carcinoma (AGS and SNU719, respectively) and B cell lines (BJAB and Raji, respectively) based on their matched proliferation rates. We measured changes in the metabolic activity of cells grown in the presence or absence of GNE6776 with a resazurin assay. The metabolic activity of SNU719 and Raji cells in the presence of GNE6776 was inhibited by nearly 40% and 100%, respectively (Figure 5A). In contrast, EBV‐ cells BJAB and AGS displayed minor metabolic inhibition in the presence of GNE6776 compared to the negative control (DMSO) (Figure 5A). We further validated the initial findings with other USP7 inhibitors (R)‐FT671 and XL177A and observed similar selective inhibition in EBV+ cells (Figure S4A,B). Cell cycle analyses confirmed that EBV+ cells were selectively impaired by the presence of GNE6776 compared to EBV‐ cells (Figure 5B). While GNE6776 had no impact on BJAB, AGS displayed an increase in the S phase population (Figure 5A and Figure S4A) Only EBV+ cells displayed significantly reduction in S and G2/M population in the presence of GNE6776 compared to EBV‐ cells (Figure 5B). Collectively, these findings suggest that EBV+ cancer cells are selectively vulnerable to inhibition of USP7's enzymatic activity.
Figure 5.
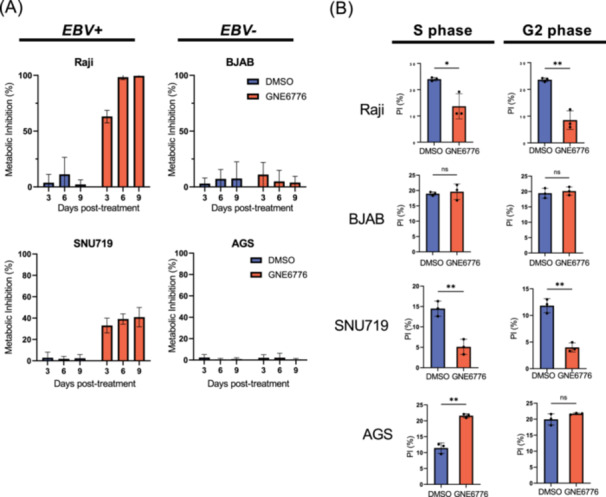
Inhibiting USP7 selectively blocks EBV+ cancer cell growth. (A) Raji, BJAB, SNU719, and AGS cells were grown in the presence of either DMSO or GNE6776 and then assessed for their metabolic activity using resazurin at 3, 6, and 9 days posttreatment. (B) Raji, BJAB, SNU719, and AGS cells were treated with either DMSO (blue) or GNE6776 (red) for either 4 days (BJAB and Raji) or 6 days (AGS and SNU719) and then fixed and stained with propidium iodide for cell cycle analysis shown as percent in S or G2 phase. n = 3 *p < 0.03, **p < 0.004 by two‐tailed t‐test.
To further our analyses, we utilized comparative transcriptomics to interrogate the systemic impact of GNE6776 on EBV− versus EBV+ cancer cells using AGS and SNU719, respectively. Principal component analysis of gene expression revealed separation by cell line along PC1 and GNE6776 treatment along PC2, indicating significant differences between each group (Figure 6A). Next, we performed a hierarchical clustering on the differentially expressed genes based on the interaction between condition and cell lines and found signature genes impacted by GNE6776 found in EBV+ cells only (Figure 6B and Figure S5). There were 86 genes unique to SNU719 that showed de‐regulated gene expression after GNE6776 treatment (Figure 6C). In SNU719 cells, 24 genes were downregulated and 71 were upregulated after treatment, many of which were found in chromatin segregation during cell cycle. We performed a gene set enrichment analysis on the genes that were negatively impaired by GNE6776 specifically in SNU719 cells and discovered that most were involved in chromosome segregation and cell cycle/growth control (Figure 6D,E). In contrast, several of the upregulated genes were involved in response to stress and cell death (Figure 6D,F). While both AGS‐ and SNU719‐treated cells display reduced gene expression for cell growth (Figure 6E and Figure S6B), only SNU719 displayed a significant reduction in proliferation (Figure 5), which further suggests USP7 inhibition has an EBV‐specific effect.
Figure 6.
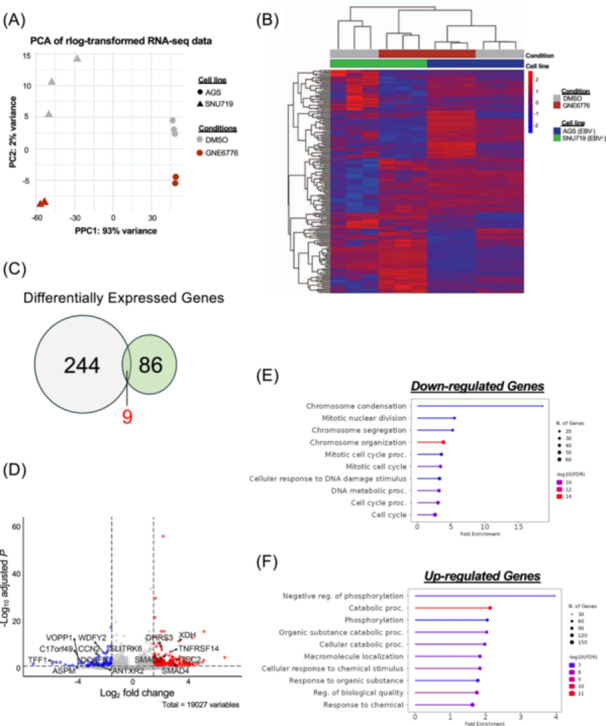
RNAseq transcriptomic analysis of GNE6776 treatment of EBV+ versus EBV− GC cells. (A) PCA analysis of RNAseq from AGS and SNU719 cells treated with DMSO or GNE6776. (B) Heat map ordered by hierarchical clustering of differentially expressed genes (DEGs, padj < 0.05 and fold change > 1.5) after treatment. (C) Venn diagram of DEGs for AGS (gray) or SNU719 (green) in response to GNE6776 treatment. (D) Volcano plot analyzing DEGs unique to EBV+ SNU719. (E and F) Gene set enrichment analysis for downregulated genes and upregulated genes (panel E) specific to SNU719 after GNE6776 treatment. The FDR cutoff was 0.05.
3.4. Treatment With USP7 Inhibitors Significantly Impairs Tumorigenesis in EBV+ Xenografts
The selective inhibition of EBV+ cell growth in cell culture (Figure 5) prompted us to test whether USP7 inhibitors could block EBV+ tumor cell growth in mouse xenograft models. We compared the effects of GNE6776 on the tumor growth EBV+ and EBV− cell lines. We compared EBV+ Raji to EBV‐ BJAB as a matched pair of Burkitt's lymphoma cell lines, and EBV+ SNU719 to EBV‐ AGS as a matched pair of GC cell lines based on their similar proliferation rates. Overall, we found that GNE6776 selectively inhibited Raji‐tumor growth compared to BJAB at 25 and 50 mg/kg and selectively inhibited SNU719 relative to AGS at 50 mg/kg as demonstrated by both tumor inhibition (Figure 7A–D) and Ki67 intracellular staining of tumor cells (Figure 7E). BJAB cells showed a slight reduction in tumor growth at higher concentration (50 mg/kg) but this effect was significantly less than that observed for EBV+ Raji cell tumors. We also measured body and organ weights to assess the overall toxicity of GNE6776 (Figure S7A–C). The weight of the mice and survival curves of GNE6776‐treated mice were comparable to those of vehicle‐treated mice, even at higher doses (Figure S7B,D). Liver and kidney weights were not impacted by GNE6776 treatment, although spleen weights were elevated at higher doses (Figure S7A,C). It is possible that B‐cells may be more sensitive to an extended GNE6776 treatment at higher doses and do not require as high a concentration of GNE6776 compared to gastric cells. These findings show that GNE6776 is well tolerated in mouse models and can selectively inhibit EBV+ relative to the growth matched to EBV− tumors in vivo.
Figure 7.
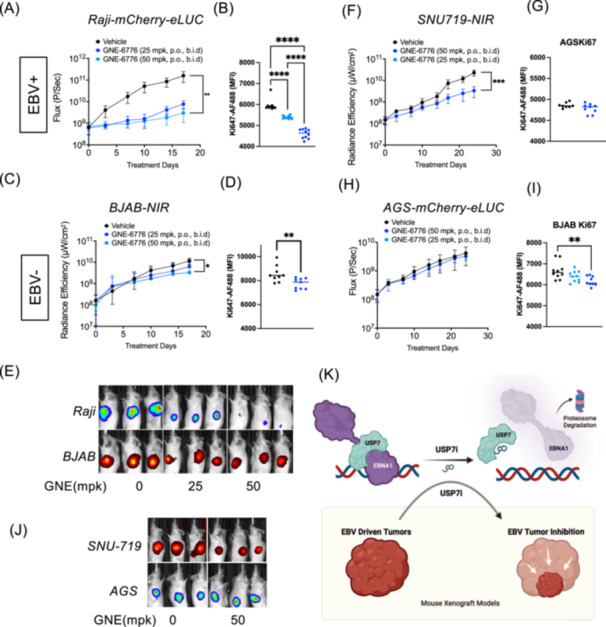
GNE6776 selectively inhibits EBV+ tumor growth in mouse xenograft models. (A–E) Mice injected with 1 × 106 Raji cells expressing mCherry‐eLuc (top panel) and 1 × 106 BJAB cells expressing NIR (bottom panel) into opposing flanks were then treated daily with either GNE6776 at 25 mpk or 50 mpk or with vehicle control by oral administration (po/bid) and assayed by IVIS bioluminescence imaging for tumor growth (A and C) and quantification of Ki67 expression in recovered fluorescent‐positive tumor cells by flow cytometry (B and D), and representative mouse images (E). (F–J) Mice injected with 5 × 106 SNU719 cells expressing NIR (top panel) and 5 × 106 AGS cells expressing mCherry‐eLuc (bottom panel) into opposing flanks were then treated daily (po/bid) with either GNE6776 at 50 mpk or with vehicle control and assayed by IVIS bioluminescence imaging (panels F and H) for tumor growth or Ki67 staining (G and I), or representative images (J). *p < 0.05, ***p < 0.001, ****p < 0.0005 by student two‐tailed t‐test. (K) Model of USP7 inhibitor effects on EBNA1 stability and function in suppressing EBV+ cancer cell growth.
4. Discussion
EBNA1 is a unique viral‐encoded protein required for EBV genome persistence and is expressed in all EBV‐associated malignancies, which makes it an excellent candidate for therapeutic intervention. Previous studies have identified USP7 as an EBNA1 interacting protein and characterized the effects of EBNA1 on USP7 activities, including p53 and PML [24, 25, 43, 44]. These previous studies also revealed that EBNA1 could be ubiquitinated in vitro and that purified USP7 could deubiquitinate EBNA1 [24]. They also showed that siRNA depletion of USP7 reduced EBNA1 DNA binding at FR and its transcriptional activation function [45]. However, the effects of pharmacological inhibitors of USP7 on EBNA1 stability and function, as well as EBV tumor cell growth, have not been reported previously.
Here, we show that USP7 inhibitors destabilize EBNA1 protein in a proteosome‐dependent manner (Figures 1 and 2). We also show that USP7 inhibitors alter EBNA1 protein interactome, including the loss of association with USP7 (Figure 3). USP7 inhibitors reduced EBNA1 and USP7 binding to the EBV oriP and compromised oriP‐dependent DNA replication and viral episome maintenance in EBV+ cancer cell lines (Figure 4). These findings suggest that USP7 DUB activity stabilizes and regulates EBNA1 protein interactions required for functional activity at oriP (Figure 7K). Consistent with this idea, we found USP7 inhibitors altered EBNA1 interaction with several cell cycle regulatory factors, including loss of MCM3, RCC1, and PARP1, and gain of STK38 and STK38L (Figure S2). Proteomic analysis identified cell proliferation and transcriptomic analysis identified chromosome condensation (Figure 6) as major pathways affected by USP7 inhibitors in EBV+ cells. Thus, USP7 inhibitors affect EBNA1 interactions with targets important for cell cycle control and episome maintenance function.
Both EBNA1 and USP7 interact with many proteins that impact DNA replication and cell cycle control [46]. Previous studies demonstrated that EBNA1 regulates USP7 interactions with MDM2 and p53 to control of cell cycle [47]. EBNA1 has been shown to interact with USP7's TRAF‐like domain (residues 53‐208), while USP7 inhibitors generally target the catalytic core (residues 208‐560) [26, 28, 29, 30] (reviewed in [15]). At present, it is unclear whether the USP7 inhibitors occlude or alter the conformation of the binding site between EBNA1 and USP7. The USP7‐EBNA1 is also known to form a ternary complex with casein kinase 2 (CK2) and have higher association with each other within promyelocytic leukemia (PML) nuclear bodies (NB) [42]. Our proteomics analysis revealed a ~100‐fold decrease interaction between CK2 and EBNA1 (Figure S2B). Thus, it is possible that USP7 interacts weakly with EBNA1 through additional domains or that USP7 inhibitors affect other interacting partners such as impacts the CK2‐USP7‐EBNA1 ternary complex and/or PML NB regulation to further inhibit the cell cycle and EBV‐driven tumor growth.
USP7 has been implicated in several types of cancer [17] and is also known to interact with many other viral proteins, including the KSHV LANA, the orthologue of EBNA1 [21]. The USP7 binding motif in LANA shares some similarity to the EBNA1 binding motif, and its deletion led to a slight increase in LANA DNA replication activity [21]. While USP7 inhibition can effectively obstruct EBV‐tumorigenesis and replication, future experiments are warranted to determine if other oncogenic ɣ‐herpesvirus such as KSHV are also sensitive to USP7 disruption.
There are several limitations to our study. We compare EBV+ and EBV− cell lines containing different genetic backgrounds and limit our study to only a few EBV latency types. Some of these different cell types and backgrounds may account for differences in the extent of USP7 inhibitor activity on EBNA1 stability and function. Future experiments should be conducted using a more extensive representation of EBV latency I, II, and III cell types to assess the potential of USP7 therapeutic intervention in all EBV‐driven cancers. We focused primarily on the properties of GNE6776, but other inhibitors may prove more efficacious in vivo. While our data suggests that destabilization of EBNA1 contributes to an EBV‐specific inhibition, it is also likely that other USP7 targets contribute to this effect. USP7 inhibitor (XL177A) was found to suppress growth in bone marrow cells through p53 signaling [29]. Our transcriptomic analysis identified a few p53 targets more upregulated than EBV− cells treated with GNE6776 such as SESN2, BTG2, and PINK1. EBV encodes both a miRNA (EBV‐miR‐BHRF1‐1) [48] and another latent protein (EBNA3C) [49] that can both destabilize p53, but it is not known if these are USP7‐dependent. Much more work is required to determine which USP7 targets are most consequential for growth inhibition, and which USP7 inhibitors could be sufficiently safe and effective for advancement to clinical trials. In conclusion, our findings indicate that pharmacological inhibition of USP7 impairs EBNA1 function and EBV‐driven cell growth, and that USP7 is an attractive candidate for EBV‐targeted therapy.
Author Contributions
Christopher Chen: conceptualization, funding acquisition, investigation, methodology, visualization, data curation, writing–original draft, review and editing. Kush Addepalli: investigation. Samantha Soldan: investigation, methodology, visualization, formal analysis. Sarah Preston‐Alp: investigation, visualization, formal analysis, data curation. Leonardo Munoz: investigation, visualization, formal analysis. Rishi J. Patel: investigation. Coltin Albitz: investigation. Hsin Yao Tang: investigation, methodology, formal analysis, visualization, writing–original draft. Paul Lieberman: supervision, conceptualization, funding acquisition, investigation, methodology, visualization, writing–original draft, review and editing.
Supporting information
Supporting information.
Supporting information.
Acknowledgments
We want to thank the members of the Wistar Cancer Center Cores in Proteomics, Genomics, Animal Facility, and Flow Cytometry for their excellent technical support. This work was supported by R01 CA259171, P01 CA2690243, R01 AI53508 (PML), P30 Cancer Center Support Grant P30 CA010815 (D. Altieri), T32 CA009171 to CC. The funders provided salary support for PML, SSS, LJCM (NIH DE017336, AI53508, CA140652, CA093606, CA2059171‐02S1, HYT (R50 CA221838), and CC (T32 CA009171). The funders had no role in the study design, publication decision, data collection and analysis, or manuscript preparation.
Data Availability Statement
The data that support the findings of this study are openly available in NCBI GEO at https://www.ncbi.nlm.nih.gov/geo/.
References
- 1. Young L. S., Yap L. F., and Murray P. G., “Epstein‐Barr Virus: More Than 50 Years Old and Still Providing Surprises,” Nature Reviews Cancer 16, no. 12 (2016): 789–802. [DOI] [PubMed] [Google Scholar]
- 2. Soldan S. S., Messick T. E., and Lieberman P. M., “Therapeutic Approaches to Epstein‐Barr Virus Cancers,” Current Opinion in Virology 56 (2022): 101260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wong Y., Meehan M. T., Burrows S. R., Doolan D. L., and Miles J. J., “Estimating the Global Burden of Epstein‐Barr Virus‐Related Cancers,” Journal of Cancer Research and Clinical Oncology 148, no. 1 (2022): 31–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shannon‐Lowe C. and Rickinson A., “The Global Landscape of EBV‐Associated Tumors,” Frontiers in Oncology 9 (2019): 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thorley‐Lawson D. A., “EBV Persistence‐‐Introducing the Virus,” Current Topics in Microbiology and Immunology 390, no. 1 (2015): 151–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thorley‐Lawson D. A. and Allday M. J., “The Curious Case of the Tumour Virus: 50 Years of Burkitt's Lymphoma,” Nature Reviews Microbiology 6, no. 12 (2008): 913–924. [DOI] [PubMed] [Google Scholar]
- 7. Frappier L., “Ebna1,” Current Topics in Microbiology and Immunology 391 (2015): 3–34. [DOI] [PubMed] [Google Scholar]
- 8. De Leo A., Calderon A., and Lieberman P. M., “Control of Viral Latency by Episome Maintenance Proteins,” Trends in Microbiology 28, no. 2 (2020): 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Young L. S., Arrand J. R., and Murray P. G., “Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis,” in EBV Gene Expression and Regulation Arvin A., Campadelli‐Fiume G., Mocarski E., et al. (Cambridge, 2007). [PubMed] [Google Scholar]
- 10. Yates J. L., Warren N., and Sugden B., “Stable Replication of Plasmids Derived From Epstein‐Barr Virus in Various Mammalian Cells,” Nature 313, no. 6005 (1985): 812–815. [DOI] [PubMed] [Google Scholar]
- 11. Altmann M., Pich D., Ruiss R., Wang J., Sugden B., and Hammerschmidt W., “Transcriptional Activation by EBV Nuclear Antigen 1 Is Essential for the Expression of Ebv's Transforming Genes,” Proceedings of the National Academy of Sciences 103, no. 38 (2006): 14188–14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshioka M., Crum M. M., and Sample J. T., “Autorepression of Epstein‐Barr Virus Nuclear Antigen 1 Expression by Inhibition of Pre‐Mrna Processing,” Journal of Virology 82, no. 4 (2008): 1679–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin Q. and Flemington E. K., “siRNAs Against the Epstein Barr Virus Latency Replication Factor, EBNA1, Inhibit Its Function and Growth of Ebv‐Dependent Tumor Cells,” Virology 346, no. 2 (2006): 385–393. [DOI] [PubMed] [Google Scholar]
- 14. Hong M., Murai Y., Kutsuna T., et al., “Suppression of Epstein‐Barr Nuclear Antigen 1 (EBNA1) by Rna Interference Inhibits Proliferation of Ebv‐Positive Burkitt's Lymphoma Cells,” Journal of Cancer Research and Clinical Oncology 132, no. 1 (2006): 1–8. [DOI] [PubMed] [Google Scholar]
- 15. Pozhidaeva A. and Bezsonova I., “USP7: Structure, Substrate Specificity, and Inhibition,” DNA Repair 76 (2019): 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Z., Kang W., You Y., et al., “USP7: Novel Drug Target in Cancer Therapy,” Frontiers in Pharmacology 10 (2019): 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saha G., Roy S., Basu M., and Ghosh M. K., “USP7 ‐ A Crucial Regulator of Cancer Hallmarks,” Biochimica et Biophysica Acta (BBA) ‐ Reviews on Cancer 1878, no. 3 (2023): 188903. [DOI] [PubMed] [Google Scholar]
- 18. Bojagora A. and Saridakis V., “USP7 Manipulation by Viral Proteins,” Virus Research 286 (2020): 198076. [DOI] [PubMed] [Google Scholar]
- 19. Pfoh R., Lacdao I. K., Georges A. A., et al., “Crystal Structure of USP7 Ubiquitin‐Like Domains With an ICP0 Peptide Reveals a Novel Mechanism Used by Viral and Cellular Proteins to Target USP7,” PLoS Pathogens 11, no. 6 (2015): e1004950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boutell C., Canning M., Orr A., and Everett R. D., “Reciprocal Activities Between Herpes Simplex Virus Type 1 Regulatory Protein ICP0, a Ubiquitin E3 Ligase, and Ubiquitin‐Specific Protease Usp7,” Journal of Virology 79, no. 19 (2005): 12342–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jäger W., Santag S., Weidner‐Glunde M., et al., “The Ubiquitin‐Specific Protease USP7 Modulates the Replication of Kaposi's Sarcoma‐Associated Herpesvirus Latent Episomal Dna,” Journal of Virology 86, no. 12 (2012): 6745–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sarkari F., Sanchez‐Alcaraz T., Wang S., Holowaty M. N., Sheng Y., and Frappier L., “EBNA1‐Mediated Recruitment of a Histone H2B Deubiquitylating Complex to the Epstein‐Barr Virus Latent Origin of Dna Replication,” PLoS Pathogens 5, no. 10 (2009): e1000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saridakis V., Sheng Y., Sarkari F., et al., “Structure of the p53 Binding Domain of HAUSP/USP7 Bound to Epstein‐Barr Nuclear Antigen 1,” Molecular Cell 18, no. 1 (2005): 25–36. [DOI] [PubMed] [Google Scholar]
- 24. Holowaty M. N., Sheng Y., Nguyen T., Arrowsmith C., and Frappier L., “Protein Interaction Domains of the Ubiquitin‐Specific Protease, USP7/HAUSP,” Journal of Biological Chemistry 278, no. 48 (2003): 47753–47761. [DOI] [PubMed] [Google Scholar]
- 25. Holowaty M. N., Zeghouf M., Wu H., et al., “Protein Profiling With Epstein‐Barr Nuclear Antigen‐1 Reveals an Interaction With the Herpesvirus‐Associated Ubiquitin‐Specific Protease HAUSP/USP7,” Journal of Biological Chemistry 278, no. 32 (2003): 29987–29994. [DOI] [PubMed] [Google Scholar]
- 26. Di Lello P., Pastor R., Murray J. M., et al., “Discovery of Small‐Molecule Inhibitors of Ubiquitin Specific Protease 7 (USP7) Using Integrated NMR and In Silico Techniques,” Journal of Medicinal Chemistry 60, no. 24 (2017): 10056–10070. [DOI] [PubMed] [Google Scholar]
- 27. Kategaya L., Di Lello P., Rougé L., et al., “USP7 Small‐Molecule Inhibitors Interfere With Ubiquitin Binding,” Nature 550, no. 7677 (2017): 534–538. [DOI] [PubMed] [Google Scholar]
- 28. Turnbull A. P., Ioannidis S., Krajewski W. W., et al., “Molecular Basis of USP7 Inhibition by Selective Small‐Molecule Inhibitors,” Nature 550, no. 7677 (2017): 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schauer N. J., Liu X., Magin R. S., et al., “Selective USP7 Inhibition Elicits Cancer Cell Killing Through a p53‐dependent Mechanism,” Scientific Reports 10, no. 1 (2020): 5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lamberto I., Liu X., Seo H. S., et al., “Structure‐Guided Development of a Potent and Selective Non‐Covalent Active‐Site Inhibitor of USP7,” Cell Chemical Biology 24, no. 12 (2017): 1490–1500.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen C. and Bridge E., “DNA‐PK Phosphorylation at Ser2056 During Adenovirus E4 Mutant Infection is Promoted by Viral DNA Replication and Independent of the MRN Complex,” Virology 565 (2022): 82–95. [DOI] [PubMed] [Google Scholar]
- 32. Monaco M. C. G., Soldan S. S., Su C., et al., “EBNA1 Inhibitors Block Proliferation of Spontaneous Lymphoblastoid Cell Lines From Patients With Multiple Sclerosis and Healthy Controls,” Neurology Neuroimmunology & Neuroinflammation 10, no. 5 (2023): e200149, 10.1212/NXI.0000000000200149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dheekollu J., Wiedmer A., Soldan S. S., et al., “Regulation of EBNA1 Protein Stability and DNA Replication Activity by PLOD1 Lysine Hydroxylase,” PLoS Pathogens 19, no. 6 (2023): e1010478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cox J. and Mann M., “MaxQuant Enables High Peptide Identification Rates, Individualized P.P.B.‐Range Mass Accuracies and Proteome‐Wide Protein Quantification,” Nature Biotechnology 26, no. 12 (2008): 1367–1372. [DOI] [PubMed] [Google Scholar]
- 35. Ge S. X., Jung D., and Yao R., “Shinygo: A Graphical Gene‐Set Enrichment Tool for Animals and Plants,” Bioinformatics 36, no. 8 (2020): 2628–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Preston‐Alp S., Caruso L. B., Su C., et al., “Decitabine Disrupts EBV Genomic Epiallele DNA Methylation Patterns Around CTCF Binding Sites to Increase Chromatin Accessibility and Lytic Transcription in Gastric Cancer,” mBio 14, no. 5 (2023): e0039623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dobin A., Davis C. A., Schlesinger F., et al., “Star: Ultrafast Universal RNA‐Seq Aligner,” Bioinformatics 29, no. 1 (2013): 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li B. and Dewey C. N., “Rsem: Accurate Transcript Quantification From RNA‐Seq Data With or Without a Reference Genome,” BMC Bioinformatics 12 (2011): 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Love M. I., Huber W., and Anders S., “Moderated Estimation of Fold Change and Dispersion for RNA‐Seq Data With Deseq. 2,” Genome Biology 15, no. 12 (2014): 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Malik‐Soni N. and Frappier L., “Proteomic Profiling of EBNA1‐Host Protein Interactions in Latent and Lytic Epstein‐Barr Virus Infections,” Journal of Virology 86, no. 12 (2012): 6999–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Frappier L., “The Epstein‐Barr Virus EBNA1 Protein,” Scientifica 2012 (2012): 438204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sivachandran N., Cao J. Y., and Frappier L., “Epstein‐Barr Virus Nuclear Antigen 1 Hijacks the Host Kinase CK2 to Disrupt PML Nuclear Bodies,” Journal of Virology 84, no. 21 (2010): 11113–11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Holowaty M. N. and Frappier L., “HAUSP/USP7 as an Epstein‐Barr Virus Target,” Biochemical Society Transactions 32, no. 5 (2004): 731–732. [DOI] [PubMed] [Google Scholar]
- 44. Frappier L., “Viral Disruption of Promyelocytic Leukemia (PML) Nuclear Bodies by Hijacking Host Pml Regulators,” Virulence 2, no. 1 (2011): 58–62. [DOI] [PubMed] [Google Scholar]
- 45. Sarkari F., Wheaton K., La Delfa A., et al., “Ubiquitin‐Specific Protease 7 Is a Regulator of Ubiquitin‐Conjugating Enzyme Ube2e1,” Journal of Biological Chemistry 288, no. 23 (2013): 16975–16985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jagannathan M., Nguyen T., Gallo D., et al., “A Role for USP7 in DNA Replication,” Molecular and Cellular Biology 34, no. 1 (2014): 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sheng Y., Saridakis V., Sarkari F., et al., “Molecular Recognition of p53 and MDM2 by USP7/HAUSP,” Nature Structural & Molecular Biology 13, no. 3 (2006): 285–291. [DOI] [PubMed] [Google Scholar]
- 48. Xu D. M., Kong Y. L., Wang L., et al., “EBV‐miR‐BHRF1‐1 Targets p53 Gene: Potential Role in Epstein‐Barr Virus Associated Chronic Lymphocytic Leukemia,” Cancer Research and Treatment 52, no. 2 (2020): 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yi F., Saha A., Murakami M., et al., “Epstein‐Barr Virus Nuclear Antigen 3C Targets p53 and Modulates Its Transcriptional and Apoptotic Activities,” Virology 388, no. 2 (2009): 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are openly available in NCBI GEO at https://www.ncbi.nlm.nih.gov/geo/.