

Abstract
A deletion mutation ΔK210 in cardiac troponin T (cTnT) was recently found to cause familial dilated cardiomyopathy (DCM). To explore the effect of this mutation on cardiac muscle contraction under physiological conditions, we determined the Ca2+-activated force generation in permeabilized rabbit cardiac muscle fibers into which the mutant and wild-type cTnTs were incorporated by using our TnT exchange technique. The free Ca2+ concentrations required for the force generation were higher in the mutant cTnT-exchanged fibers than in the wild-type cTnT-exchanged ones, with no statistically significant differences in maximal force-generating capability and cooperativity. Exchanging the mutant cTnT into isolated cardiac myofibrils also increased the free Ca2+ concentrations required for the activation of ATPase. In contrast, a deletion mutation ΔE160 in cTnT that causes familial hypertrophic cardiomyopathy (HCM) decreased the free Ca2+ concentrations required for force generation, just as in the case of the other HCM-causing mutations in cTnT. The results indicate that cTnT mutations found in the two distinct forms of cardiomyopathy (i.e., HCM and DCM) change the Ca2+ sensitivity of cardiac muscle contraction in opposite directions. The present study strongly suggests that Ca2+ desensitization of force generation in sarcomere is a primary mechanism for the pathogenesis of DCM associated with the deletion mutation ΔK210 in cTnT.
Contraction of the vertebrate-striated muscles (i.e., skeletal and cardiac muscles) is regulated by Ca2+ through its binding to a specific regulatory protein complex, troponin (Tn), which is distributed at regular intervals along the entire thin filament (1, 2). Tn is a complex of three different proteins, troponin T (TnT; tropomyosin-binding component), troponin I (TnI; inhibitory component), and troponin C (TnC; Ca2+-binding component). On Ca2+ binding to TnC, a Ca2+-induced interaction of TnC with TnI relieves the inhibitory action of TnI exerted on the thin filament and enables the myosin head to cyclically interact with actin in the thin filament and generate force. The Ca2+ sensitivity of muscle contraction is determined by the Ca2+-binding affinity of TnC, which is dynamically altered through interaction with TnI and TnT in the myofilament lattice (3–8).
Mutations in genes for cardiac troponin T (cTnT) and cardiac troponin I (cTnI) have been found to cause familial hypertrophic cardiomyopathy (HCM), an autosomal dominant heart disease characterized by asymmetrical ventricular hypertrophy with a high incidence of sudden death in young adults (9). We have already examined the effects of eight HCM-linked cTnT mutations (I79N, R92Q, ΔE160, E244D, R278C, and two truncated mutants produced by a splice donor site mutation Int15G1→A) and six HCM-linked cTnI mutations (R145G, R145Q, R162W, ΔK183, G203S, and K206Q) on the contractile functions of cardiac muscle by using a technique for exchanging the exogenous Tn complex into skinned muscle fibers and isolated myofibrils. We found that Ca2+ sensitization in cardiac muscle contractility is a common effect caused by HCM-linked mutations in cTnT and cTnI (10–16). Several groups also have reported Ca2+-sensitizing effects of the HCM-linked mutations in cTnT and cTnI. Tobacman et al. (17) reported that ΔE160 cTnT mutant increased the Ca2+ sensitivity of acto-S1 MgATPase activity. Szczesna et al. (18) reconstituted I79N, R92Q, F110I, and R278C cTnT mutants into skinned cardiac muscle fibers and reported that these mutations increased Ca2+ sensitivity. Redwood et al. (19) also reported that one of the two truncated cTnT mutants had a Ca2+-sensitizing effect on acto-S1 MgATPase activity. Recently, Miller et al. (20) and Chandra et al. (21) reported that transgenic mice expressing I79N and R92Q cTnT mutants did exhibit increased Ca2+ sensitivity in skinned muscle fibers. Elliott et al. (22) reported that R145G and R162W mutations in cTnI increased the Ca2+ sensitivity of actin-tropomyosin-activated myosin S-1 ATPase activity, and James et al. (23) reported that transgenic mice expressing R145G (R146G in the mouse sequence) cTnI mutant exhibit increased Ca2+ sensitivity in skinned muscle fibers. These studies strongly suggest that Ca2+ sensitization of force generation in sarcomere is a primary mechanism for the pathogenesis of HCM with the mutations in Tn subunits (24, 25). Recently, however, a novel mutation (ΔK210) in the cTnT gene was found to cause a quite different form of cardiomyopathy, dilated cardiomyopathy (DCM), which is characterized by cardiac dilation and reduced systolic function leading to heart failure with high mortality (26). In the present study, an attempt was made to directly exchange the recombinant human ΔK210 cTnT into membrane-permeabilized (skinned) rabbit cardiac muscle fibers. This technique overcomes the potentially significant complications caused by compensatory mechanisms expected to occur when transgenesis is used in a whole animal. The study revealed that the functional consequence of the mutation ΔK210 in cTnT is a decrease in the Ca2+ sensitivity of cardiac muscle contraction. The results strongly suggest that the primary mechanism for the pathogenesis of DCM associated with the mutation ΔK210 is a deficiency of force generation by sarcomere in cardiac muscle, in contrast to the enhancement of force generation in HCM associated with the other mutations in cTnT.
Materials and Methods
Mutagenesis of Recombinant Human cTnT.
The cloning and mutagenesis of human cTnT cDNA were carried out as described previously (14). To generate the ΔK210 and ΔE160 cDNAs, mutageneses were first carried out by PCR according to the GeneSOEing method described by Horton (27) by using the following oligonucleotide primers: 5′-GGT GGT GGA AGC GTA CGA AGA GG-3′ (18F, SplI site is underlined), 5′-CCT CTC AGC CAG AAT CTT CTT CTT TTC CCG (645R), 5′-AAG AAG ATT CTG GCT GAG AGG AGG AAG GTG (622F), and 5′-GCT GCA GGA TCC TAT TTC CAG CGC CCG G (878 R, BamHI site is underlined) for ΔK210; 5′-GGT GGT GGA AGC GTA CGA AGA GG-3′ (18F, SplI site is underlined), 5′-CCT GTT CTC CTC CTC TCG TCG AGC CCT CTC (495R), 5′-CGA CGA GAG GAG GAG AAC AGG AGG AAG GCT (472F), and 5′-GCT GCA GGA TCC TAT TTC CAG CGC CCG G (878 R, BamHI site is underlined) for ΔE160. The obtained PCR products digested by SplI and BamHI were ligated to a NcoI linker made from oligonucleotides 5′-CAT GTC TGA CAT CGA AGA AGT GGT GGA AGA and 5′-GTA CTC TTC CAC CAC TTC TTC GAT GTC AGA and constructed into the pET-3d vector. The complete nucleotide sequences of the mutant cTnT cDNAs were confirmed by DNA sequencing.
Preparation of Skinned Fibers and Force Measurements.
Rabbit cardiac skinned muscle fibers were prepared from the left ventricular trabeculae of young male albino rabbits (≈3 mo old) as described (10). In brief, small bundles (0.5–1 mm wide and 5–7 mm long) of trabeculae tied to glass capillary tubes were skinned with relaxing solution containing 0.5% Brij-58 for 30 min at 25°C and were stored up to 3 wk at −20°C in relaxing solution containing 50% glycerol. A small fiber (≈120 μm in diameter) dissected from the stock-skinned trabecula was mounted in a thermostatically controlled chamber with a capacity of 0.2 ml. Fiber length between hooks was ≈1 mm, and the resting sarcomere length was set to 2.3 μm by using laser diffraction. The force generated by skinned muscle fibers was measured at 25°C with a strain gauge, UL-2GR (Minebea, Japan). The relaxing solution consisted of (in mM) 50 Mops/KOH (pH 7.0), 100 KCl, 6 MgCl2, 5 ATP, 4 EGTA, 0.5 DTT, and 10 creatine phosphate, as well as 35 units/ml creatine kinase. Activating solutions with desired free Ca2+ concentrations were prepared by adding appropriate amounts of CaCl2, calculated as described (28), to the relaxing solution.
ATPase Activity Measurement.
Porcine cardiac myofibrils were isolated from left ventricular muscle, and their ATPase activity was measured in a reaction mixture (150 μl) that consisted of 90 mM KCl, 5 mM MgCl2, 20 mM Mops/KOH (pH 7.0), 1 mM Ca2+-EGTA, 4 mM ATP, and 45 μg of myofibrils, as described (13).
Purification of Proteins.
Expression and purification of the recombinant human cTnTs were performed as described previously (10). The wild-type and mutant cTnTs were eluted at 0.3–0.4 M NaCl on a fast-performance liquid chromatography (FPLC) ion-exchange column, RESOURCE Q (6 ml) (Amersham Pharmacia Biotech), with a linear gradient of 0–0.5 M NaCl in the presence of 20 mM Tris⋅HCl (pH 8.0)/6 M urea/5 mM trans-1, 2-cyclohexanediamine-N,N,N′,N′-tetraacetic acid/and 15 mM 2-mercaptoethanol. Rabbit native cTnT, cTnI, and cTnC were prepared from the left ventricular myocardium of young male albino rabbits (≈3 mo old) according to the method of Tsukui and Ebashi (29) by using FPLC ion-exchange columns, RESOURCE Q and S (6 ml), and Mono Q and S HR 5/5 (Amersham Pharmacia Biotech). The purified proteins used in the present study are shown in Fig. 1.
Figure 1.

SDS/PAGE of recombinant human cTnT and native rabbit cardiac Tn three components. Lane 1, rabbit skinned cardiac muscle; lane 2, recombinant human wild-type cTnT; lane 3, recombinant human ΔK210 cTnT; lane 4, native rabbit cTnT; lane 5, native rabbit cTnI, and lane 6, native rabbit cTnC. The gel was stained with Coomassie brilliant blue R-250.
TnT Exchange in Skinned Fibers.
cTnT exchange in the skinned fibers and isolated myofibrils was performed by the method described previously (30–32).
SDS/PAGE Analysis.
SDS/PAGE was carried out at 12% acrylamide concentration according to the method of Laemmli (33). The fiber samples were lysed in Laemmli's sample buffer by heating for 4 min at 95°C after freezing (−80°C) and thawing several times. The gel was stained with silver by using a staining kit (Amersham Pharmacia Biotech) or with Coomassie brilliant blue R-250. An optical densitometric scan was performed by using phoretix gel analysis software (1-D Gel Analysis, Phoretix International, Newcastle upon Tyne, England) calibrated by a photographic step tablet (21 steps; density range 0.05–3.03, Eastman Kodak). The linearity of the densitometric reading was confirmed by changing the amount of sample applied on the gel in steps.
Data Reduction and Error Analysis.
To determine the pCa (−log[Ca2+]) value at half-maximal force generation or ATPase activation (pCa50) and the Hill coefficient (nH), the force and ATPase activity were normalized to the maximum force in the same fiber and to the maximum ATPase activity in the same series of measurement, respectively, and the relative force or ATPase–pCa relationship in each fiber or in each series of ATPase measurement was fitted to the following form of the Hill equation by means of the Marquardt nonlinear least-squares method (graphpad prism Ver. 3.00 for Windows, GraphPad, San Diego):
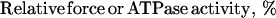 |
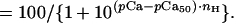 |
The mean values of pCa50 and nH and their SEM were then calculated. The value r2, a measure of goodness of fit, was in the range of 0.98–1.0 in all determinations.
Results
Fig. 1 shows SDS/PAGE of the bacterially expressed and purified human wild-type and DCM-causing deletion mutant ΔK210 cTnTs as well as the purified native rabbit cardiac Tn three subunits. Fig. 2 shows an SDS/PAGE analysis of the rabbit skinned cardiac muscle fibers before and after exchanging native cTnT with recombinant human wild-type or ΔK210 cTnT. Treatment of skinned fibers with an excess amount of recombinant human cTnT resulted in a decrease in the amount of endogenous cTnI and cTnC (lanes 3 and 4) because of displacement of the Tn complex with recombinant human cTnT, as demonstrated in our previous studies (10, 14, 30–32). Subsequent incubation with rabbit cTnI and cTnC accomplished the exchange of recombinant human cTnT into rabbit cardiac muscle fibers (lanes 5 and 6). Densitometric scans of the gels demonstrated that there was no difference in the extent of incorporation between wild-type and ΔK210 cTnTs determined from the decrease of cTnI and cTnC after cTnT treatment (P > 0.85, t test), and that exchange of wild-type and ΔK210 cTnTs into fibers did not change the stoichiometry of the three Tn components in muscle fibers (Fig. 3).
Figure 2.

SDS/PAGE of rabbit skinned cardiac muscle fibers before and after exchanging endogenous cTnT with recombinant human cTnT. Lane 1, purified rabbit cardiac Tn complex; lane 2, a control muscle fiber; lane 3, a muscle fiber treated with human wild-type cTnT; lane 4, a muscle fiber treated with human ΔK210 cTnT; lane 5, a muscle fiber treated with human wild-type cTnT and reconstituted with rabbit cTnI and cTnC, and lane 6, a muscle fiber treated with human ΔK210 cTnT and reconstituted with rabbit cTnI and cTnC. The gel was stained with silver. wt, wild-type; tr., treatment; MHC, myosin heavy chain; Tm, tropomyosin; LC1, myosin light chain 1; and LC2, myosin light chain 2.
Figure 3.

Stoichiometry of Tn components in rabbit skinned cardiac muscle fibers after treatment (tr.) with recombinant human cTnT and after subsequent reconstitution with rabbit cTnI and cTnC (+ TnI⋅TnC). SDS/PAGE gels shown in Fig. 2 were analyzed by an optical densitometric scan. The amounts of cTnI, cTnC, and cTnT were normalized to the amount of LC1 in each muscle fiber and are expressed as means ± SEM of three independent experiments.
Fig. 4 compares the force–pCa relationship in the fibers exchanged with the deletion mutant ΔK210 with that in the fibers exchanged with wild-type cTnT. We also determined the force–pCa relationship in the ΔE160 mutant cTnT-exchanged fibers as a control. The mutation ΔE160 is the only deletion mutation in cTnT associated with HCM, and we have demonstrated that this mutation increases the Ca2+ sensitivity of cardiac myofibrillar ATPase activity (13). As shown in Fig. 4A, the mutation ΔE160 shifted the force–pCa relationship to lower Ca2+ concentrations, as is the case of the other HCM-linked cTnT mutations showing the Ca2+-sensitizing effect (10–12, 14, 15, 34). In contrast, the force–pCa relationship in the ΔK210 mutant cTnT-exchanged fibers was shifted to higher Ca2+ concentrations compared to that in the wild-type cTnT-exchanged fibers, indicating that the deletion mutation ΔK210 decreases the Ca2+ sensitivity of force generation in cardiac muscle. The maximum force-generating capabilities of these three fiber groups were not statistically significantly different (Fig. 4B). Curve fittings of the data in Fig. 4A to the Hill equation revealed that the pCa value at half-maximal force generation (pCa50, an index of Ca2+ sensitivity) was statistically significantly lower in ΔK210 and higher in ΔE160 mutation than in wild type, whereas the Hill coefficient values (nH, an indicator of cooperativity or steepness of the curve) were not significantly different (Fig. 4 C and D). These results indicate that the deletion mutation ΔK210 in cTnT has a Ca2+-desensitizing effect on force generation in cardiac muscle without changing the maximum force-generating capability and cooperativity.
Figure 4.

Effects of mutations in cTnT on force generation of cardiac muscle. (A) Force–pCa relationships in the rabbit skinned cardiac muscle fibers into which human wild-type, ΔK210, or ΔE160 cTnT was incorporated. (B) Effects of mutations on maximum force-generating capability. Data are expressed as percentage of the mean maximum force generated by fibers with wild-type cTnT. Maximum force levels after cTnT exchange were 72.9 ± 3.3, 68.1 ± 4.9, and 67.7 ± 1.7% of the maximum force determined before exchange in the fibers with wild-type, ΔK210, and ΔE160 cTnTs, respectively. (C and D) Effects of mutations on Ca2+ sensitivity (pCa50) and steepness (nH) of the force–pCa relationships, respectively. The data represent the means ± SEM of measurements on five, six, and five fibers for wild-type, ΔK210, and ΔE160 cTnTs, respectively. Statistical significance was determined by ANOVA followed by the post-hoc Dunnett's multiple comparison test.
Next, we determined the ATPase–pCa relationship in the ΔK210 mutant cTnT-exchanged cardiac myofibrils (Fig. 5). The mutation ΔK210 also decreased the Ca2+ sensitivity of myofibrillar ATPase activity, as demonstrated by a rightward shift of the ATPase–pCa relationship with a statistically significant decrease in pCa50. The result clearly indicates that the deletion mutation ΔK210 has a depressant effect on the Ca2+ regulatory mechanism of cardiac muscle contraction at the level of actomyosin interaction.
Figure 5.

Effects of ΔK210 mutation in cTnT on ATPase activity of myofibrils. (A) ATPase–pCa relationships in the rabbit isolated cardiac myofibrils into which human wild-type or ΔK210 cTnT was incorporated. (B) Effects of ΔK210 mutation on Ca2+ sensitivity (pCa50) of ATPase–pCa relationships. The data represent the means ± SEM of five independent experiments. Statistical significance was determined by the t test. The Hill coefficients of the ATPase–pCa relationships are 1.76 ± 0.13 and 1.39 ± 0.16 for wild-type and Δ210 cTnTs, respectively, which are not significantly different (t test).
Discussion
DCM represents a heterogeneous group of inherited and acquired disorders characterized by cardiac dilation and systolic dysfunction. Idiopathic DCM is a relatively common disorder (≈50% of DCM) that accounts for >10,000 deaths annually by heart failure and sudden death in the United States (5-yr mortality = 40≈80%), being the primary indication for cardiac transplantation (35–38).
Recently, single gene defects that cause familial DCM (≈25–30% of idiopathic DCM) have been identified for cytoskeletal (dystrophin, desmin, tafazzin, and δ-sarcogycan) and sarcomeric (actin, tropomyosin, myosin, and troponin T) proteins (26, 39–45). Mutations in cytoskeletal proteins imply that deficits in transmission of force generated by sarcomere may be one mechanism for the pathogenesis of DCM. However, no direct evidence for this hypothesis has been provided experimentally, and no studies have been made on functional consequences of the DCM-causing mutations in these cytoskeletal and sarcomeric proteins.
In the present study, we used a technique for exchanging Tn components in sarcomere, which made it possible to explore the functional change of mutated cTnT in muscle fiber with its contractile apparatus being kept intact. The deletion mutation ΔK210 decreased the Ca2+ sensitivity of force generation without affecting the stoichiometry of three Tn components in sarcomere. This indicates that this mutation alters a relatively localized interaction within the Tn complex involving the Ca2+-sensitive disinhibition of the TnI action by TnC. The lysine residue 210 in cTnT is in a domain that is involved in the Ca2+-sensitive strong TnC binding and in the very weak tropomyosin and TnI binding (46). Thus, the deletion mutation ΔK210 might impair the Ca2+-induced interaction of TnC with this region on cTnT. However, more precise structural information on the Tn complex from x-ray crystallography or NMR is required to elucidate the exact molecular mechanism by which the mutation causes the depression of Ca2+ sensitivity of force generation.
The present study provides the first evidence, to our knowledge, that the decrease in Ca2+ sensitivity of force generation in sarcomere might be a primary mechanism for the pathogenesis of DCM in patients with the deletion mutation ΔK210 in cTnT. This mutation does not affect the maximum force-generating capability of sarcomere. However, even a small decrease in Ca2+ sensitivity is expected to cause a significant reduction in the force generation of the cardiac muscle in the heart, because the intact cardiac muscle is known to never be activated beyond the half-maximal level (47). The reduction in force generation by sarcomere with the deletion mutation ΔK210 in cTnT and the deficits of force transmission in cytoskeletal proteins might go through the same process of pathogenesis, leading to ventricular dilation as a compensatory mechanism for the decrease in stroke volume because of the reduction in contractile force of the heart. The decrease in Ca2+ sensitivity of force generation also is expected to facilitate the relaxation of the cardiac muscle at diastole and might directly explain the cardiac dilation itself to some extent. In contrast, we have previously shown that the mutations in cTnT associated with HCM have Ca2+-sensitizing effects on force generation in cardiac muscle (10–15). The increase in Ca2+ sensitivity of force generation is expected to impair the relaxation of cardiac muscle and thus cause a diastolic dysfunction, which might be directly involved in the high incidence of sudden death despite mild or no hypertrophy in HCM patients associated with cTnT mutations. Our studies indicate that the Ca2+ sensitivity of force generation in cardiac muscle plays an extremely important role in cardiac function, and even a sight change in the Ca2+ sensitivity in either direction caused by the mutations in the regulatory protein complex, troponin, may have a fatal effect on the cardiac system.
Acknowledgments
This study was supported by Special Coordination Funds from the Ministry of Education, Culture, Sports, Science and Technology, the Japanese Government. Q.-W.L. is an Uehara Memorial Foundation Research Fellow.
Abbreviations
- Tn
troponin
- cTnT
cardiac Tn T
- cTnI
cardiac Tn I
- HCM
hypertrophic cardiomyopathy
- DCM
dilated cardiomyopathy
- pCa
−log[Ca2+]
References
- 1.Ebashi S, Endo M. Prog Biophys Mol Biol. 1968;18:123–183. doi: 10.1016/0079-6107(68)90023-0. [DOI] [PubMed] [Google Scholar]
- 2.Ohtsuki I, Maruyama K, Ebashi S. Adv Protein Chem. 1986;38:1–67. doi: 10.1016/s0065-3233(08)60525-2. [DOI] [PubMed] [Google Scholar]
- 3.Potter J D, Gergely J. J Biol Chem. 1975;250:4628–4633. [PubMed] [Google Scholar]
- 4.Zot H G, Iida S, Potter J D. Chem Scr. 1983;21:133–136. [Google Scholar]
- 5.Morimoto S. J Biochem. 1991;109:120–126. doi: 10.1093/oxfordjournals.jbchem.a123331. [DOI] [PubMed] [Google Scholar]
- 6.Morimoto S, Ohtsuki I. Eur J Biochem. 1994;226:597–602. doi: 10.1111/j.1432-1033.1994.tb20085.x. [DOI] [PubMed] [Google Scholar]
- 7.Schachat F H, Diamond M S, Brandt P W. J Mol Biol. 1987;198:551–554. doi: 10.1016/0022-2836(87)90300-7. [DOI] [PubMed] [Google Scholar]
- 8.Pan B S, Potter J D. J Biol Chem. 1992;267:23052–23056. [PubMed] [Google Scholar]
- 9.Marian J, Roberts R. J Mol Cell Cardiol. 2001;33:655–670. doi: 10.1006/jmcc.2001.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morimoto S, Yanaga F, Minakami R, Ohtsuki I. Am J Physiol. 1998;275:C200–C207. doi: 10.1152/ajpcell.1998.275.1.C200. [DOI] [PubMed] [Google Scholar]
- 11.Yanaga F, Morimoto S, Ohtsuki I. J Biol Chem. 1999;274:8806–8812. doi: 10.1074/jbc.274.13.8806. [DOI] [PubMed] [Google Scholar]
- 12.Nakaura H, Yanaga F, Ohtsuki I, Morimoto S. J Biochem. 1999;126:457–460. doi: 10.1093/oxfordjournals.jbchem.a022473. [DOI] [PubMed] [Google Scholar]
- 13.Harada K, Takahashi-Yanaga F, Minakami R, Morimoto S, Ohtsuki I. J Biochem. 2000;127:263–268. doi: 10.1093/oxfordjournals.jbchem.a022603. [DOI] [PubMed] [Google Scholar]
- 14.Nakaura H, Morimoto S, Yanaga F, Nakata M, Nishi H, Imaizumi T, Ohtsuki I. Am J Physiol. 1999;46:C225–C232. doi: 10.1152/ajpcell.1999.277.2.C225. [DOI] [PubMed] [Google Scholar]
- 15.Morimoto S, Nakaura H, Yanaga F, Ohtsuki I. Biochem Biophys Res Commun. 1999;261:79–82. doi: 10.1006/bbrc.1999.1000. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi-Yanaga F, Morimoto S, Harada K, Minakami R, Shiraishi F, Ohta M, Lu Q-W, Sasaguri T, Ohtsuki I. J Mol Cell Cardiol. 2001;33:2095–2107. doi: 10.1006/jmcc.2001.1473. [DOI] [PubMed] [Google Scholar]
- 17.Tobacman L S, Lin D, Butters C, Landis C, Back N, Pavlov D, Homsher E. J Biol Chem. 1999;274:28363–28370. doi: 10.1074/jbc.274.40.28363. [DOI] [PubMed] [Google Scholar]
- 18.Szczesna D, Zhang R, Zhao J, Jones M, Guzman G, Potter J D. J Biol Chem. 2000;275:624–630. doi: 10.1074/jbc.275.1.624. [DOI] [PubMed] [Google Scholar]
- 19.Redwood C S, Lohman K, Bing W, Esposito G M, Elliott K, Abdulrazzak H, Knott A, Purcell I, Marston S, Watkins H. Circ Res. 2000;86:1146–1152. doi: 10.1161/01.res.86.11.1146. [DOI] [PubMed] [Google Scholar]
- 20.Miller T, Szczesna D, Housmans P R, Zhao J, de Freitas F, Gomes A V, Culbreath L, McCue J, Wang Y, Xu Y, et al. J Biol Chem. 2001;276:3743–3755. doi: 10.1074/jbc.M006746200. [DOI] [PubMed] [Google Scholar]
- 21.Chandra M, Rundell V L M, Tardiff J C, Leinwand L A, De Tombe P P, Solaro R J. Am J Physiol. 2001;280:H1653–H1659. doi: 10.1152/ajpheart.2001.280.2.H705. [DOI] [PubMed] [Google Scholar]
- 22.Elliott K, Watkins H, Redwood C S. J Biol Chem. 2000;275:22069–22074. doi: 10.1074/jbc.M002502200. [DOI] [PubMed] [Google Scholar]
- 23.James J, Zhang Y, Osinska H, Sanbe A, Klevtsky R, Hewett T E, Robbins J. Circ Res. 2000;87:805–811. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 24.Maas A, Leinwand L A. Curr Opin Cardiol. 2000;15:189–196. doi: 10.1097/00001573-200005000-00012. [DOI] [PubMed] [Google Scholar]
- 25.Hernandez O M, Housmans P R, Potter J D. J Appl Physiol. 2001;90:1125–1136. doi: 10.1152/jappl.2001.90.3.1125. [DOI] [PubMed] [Google Scholar]
- 26.Kamisago M, Sharma S D, DePalma S R, Solomon S, Sharma P, McDonough B, Smoot L, Mullen M P, Woolf P K, Wigle E D, et al. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 27.Horton R M. Methods Mol Biol. 1993;15:251–261. doi: 10.1385/0-89603-244-2:251. [DOI] [PubMed] [Google Scholar]
- 28.Morimoto S, Ohtsuki I. J Biochem. 1987;101:291–301. doi: 10.1093/oxfordjournals.jbchem.a121913. [DOI] [PubMed] [Google Scholar]
- 29.Tsukui R, Ebashi S. J Biochem. 1973;73:1119–1121. doi: 10.1093/oxfordjournals.jbchem.a130168. [DOI] [PubMed] [Google Scholar]
- 30.Hatakenaka M, Ohtsuki I. Biochem Biophys Res Commun. 1991;181:1022–1027. doi: 10.1016/0006-291x(91)92039-m. [DOI] [PubMed] [Google Scholar]
- 31.Shiraishi F, Kambara M, Ohtsuki I. J Biochem. 1992;111:61–65. doi: 10.1093/oxfordjournals.jbchem.a123719. [DOI] [PubMed] [Google Scholar]
- 32.Hatakenaka M, Ohtsuki I. Eur J Biochem. 1992;205:985–999. doi: 10.1111/j.1432-1033.1992.tb16865.x. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi-Yanaga F, Ohtsuki I, Morimoto S. J Biochem. 2001;130:127–131. doi: 10.1093/oxfordjournals.jbchem.a002950. [DOI] [PubMed] [Google Scholar]
- 35.Kasper E K, Agema W R, Hutchins G M, Deckers J W, Hare J M, Baughman K L. J Am Coll Cardiol. 1994;23:586–590. doi: 10.1016/0735-1097(94)90740-4. [DOI] [PubMed] [Google Scholar]
- 36.Manolio T A, Baughman K L, Rodeheffer R, Pearson T A, Bristow J D, Michels V V, Abelmann W H, Harlan W R. Am J Cardiol. 1992;69:1458–1466. doi: 10.1016/0002-9149(92)90901-a. [DOI] [PubMed] [Google Scholar]
- 37.Dec G W, Fuster V. N Engl J Med. 1994;331:1564–1575. doi: 10.1056/NEJM199412083312307. [DOI] [PubMed] [Google Scholar]
- 38.Gilbert E M, Bristow M R. In: The Heart. Hurst J W, editor. New York: MacGraw–Hill; 1994. pp. 1609–1619. [Google Scholar]
- 39.Towbin J A, Hejtmancik J F, Brink P, Gelb B, Zhu X M, Chamberlain J S, McCabe E R, Swift M. Circulation. 1993;87:1854–1865. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- 40.Li D, Tapscoft T, Gonzalez O, Burch P E, Quinones M A, Zoghbi W A, Hill R, Bachinski L L, Mann D L, Roberts R. Circulation. 1999;100:461–464. doi: 10.1161/01.cir.100.5.461. [DOI] [PubMed] [Google Scholar]
- 41.D'Adamo P, Fassone L, Gedeon A, Janssen E A, Bione S, Bolhuis P A, Barth P G, Wilson M, Haan E, Orstavik K H, et al. Am J Hum Genet. 1997;61:862–786. doi: 10.1086/514886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakamoto A, Ono K, Abe M, Jasmin G, Eki T, Murakami Y, Masaki T, Toyo-oka T, Hanaoka F. Proc Natl Acad Sci USA. 1997;94:13873–13878. doi: 10.1073/pnas.94.25.13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsubata S, Bowles K R, Vatta M, Zintz C, Titus J, Muhonen L, Bowles N E, Towbin J A. J Clin Invest. 2000;106:655–662. doi: 10.1172/JCI9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olson T M, Michels V V, Thibodeau S N, Tai Y S, Keating M T. Science. 1998;280:750–752. doi: 10.1126/science.280.5364.750. [DOI] [PubMed] [Google Scholar]
- 45.Olson T M, Kishimoto N Y, Whitby F G, Michels V V. J Mol Cell Cardiol. 2001;33:723–732. doi: 10.1006/jmcc.2000.1339. [DOI] [PubMed] [Google Scholar]
- 46.Tanokura M, Tawada Y, Ono A, Ohtsuki I. J Biochem. 1983;93:331–337. doi: 10.1093/oxfordjournals.jbchem.a134185. [DOI] [PubMed] [Google Scholar]
- 47.Rüegg J C. Calcium in Muscle Activation. Berlin: Springer; 1986. [Google Scholar]