

Abstract
The innate immune response is crucial for defense against microbial pathogens. To investigate the molecular choreography of this response, we carried out a systematic examination of the gene expression program in human peripheral blood mononuclear cells responding to bacteria and bacterial products. We found a remarkably stereotyped program of gene expression induced by bacterial lipopolysaccharide and diverse killed bacteria. An intricately choreographed expression program devoted to communication between cells was a prominent feature of the response. Other features suggested a molecular program for commitment of antigen-presenting cells to antigens captured in the context of bacterial infection. Despite the striking similarities, there were qualitative and quantitative differences in the responses to different bacteria. Modulation of this host-response program by bacterial virulence mechanisms was an important source of variation in the response to different bacteria.
The host–microbe encounter has shaped the evolution of humans as hosts for both commensal and pathogenic microorganisms. Innate responses are a crucial element in the body's defense against the daily threats posed by potential pathogens breaching epithelial barriers; many and perhaps most human cells respond to molecular signs of microbial invasion by initiating local defense mechanisms, and recruiting and activating the specialized cells of the immune system. A central component of the host's surveillance system for invading pathogens is an evolutionarily conserved family of Toll-like cell surface receptors, which recognize patterned microbial ligands ranging from cell wall components to bacterial DNA (1, 2). Through the differential and cooperative specificities of these receptors for distinct pathogen-associated molecular patterns, innate immune cells are also capable of rudimentary receptor-level discrimination of different classes of pathogens (3).
As a step toward developing a more complete molecular portrait of the host–microbe interaction and host capabilities for microbial discrimination, we used human cDNA microarrays to examine in detail the host transcriptional program in a simple laboratory model of the initial encounter between human immune cells and a variety of bacterial stimuli. Human peripheral blood mononuclear cells (PBMCs), encompassing a diverse repertoire of both innate and adaptive immune functions, have well-established roles in surveillance for infectious threats, both directly, through contact with infectious agents, and indirectly, through interactions with infected cells and tissues by means of secreted signaling molecules (4). By analyzing host gene expression responses, we addressed the following: (i) whether detection and discrimination of patterned bacterial determinants by immune cells is accompanied by distinct gene expression programs; (ii) which features of the host response to pathogens are common to diverse pathogens and which are variable; and (iii) how virulence mechanisms, which may be unique to pathogens, influence and modify these innate response programs.
Materials and Methods
Bacterial Strains and Culture Conditions.
Bordetella pertussis strains Bp338 (5), Minnesota 1, and isogenic mutant strains (6) derived from Bp338: Bp537 (7), BpTox6 (8), and BpA2–6 (9), were grown at 37°C on Bordet–Gengou agar (Difco), supplemented with sheep blood (13%, vol/vol), and then transferred to Stainer–Scholte liquid media and grown to late logarithmic phase. For experiments with heat-killed bacteria, the liquid culture was heated to 56°C for 30 min. The Escherichia coli and Staphylococcus aureus clinical strains were isolated from patient blood samples (Veterans Affairs Palo Alto Health Care System clinical lab) and grown and heat-killed in the same manner as the B. pertussis strains. B. pertussis lipopolysaccharide (LPS; List Biologicals), phorbol 12-myristate 13-acetate (PMA; Sigma), and ionomycin (Sigma) were diluted in fresh Stainer–Scholte media. After heat killing, bacteria were diluted in their original growth media and added to 1 × 108 PBMCs at 0.002–4 bacteria per human cell (see supporting information on the PNAS web site, www.pnas.org). LPS was added to the same number of PBMCs at a concentration ranging from 0.01 to 1 μg/ml. For experiments described in Figs. 1, 2, and 4, replicate samples were harvested just before treatment (7, 4, and 2 replicates, respectively), and subsequent samples were harvested after incubation at 37°C for 0.5, 1, 2, 4, 6, 12, and 24 hr. At harvesting, cells were scraped from the flask, diluted in cold PBS, pelleted at 400 × g for 5 min, and snap-frozen in liquid nitrogen. For the comparison of live and heat-killed B. pertussis, the bacterial culture was split, half of the bacteria were heat killed and added to PBMCs, and the other half were added live to PBMCs; samples were harvested at the same time points as above. U937 cells (American Type Culture Collection) were infected at a concentration of 50 live bacteria per human cell for all time courses with Bordetella mutant and wild-type strains.
Figure 1.

Stereotyped host responses to diverse heat-killed bacteria. PBMCs obtained by apheresis from a single healthy donor were treated with B. pertussis LPS (BpLPS; 1 μg/ml), and heat-killed B. pertussis virulent laboratory strain 338 (Bp338), B. pertussis Minnesota 1 clinical isolate (BpMinn1), E. coli clinical isolate, and S. aureus clinical isolates 1 and 2, each at a ratio of ≈1 microbial cell to 1 human cell, and ionomycin (1 μM) plus PMA (25 ng/ml). For the mock time course, PBMCs were treated with sterile liquid media. (a) Genes displayed are those exhibiting at least 2.5-fold change in level of expression from baseline (t = 0) in at least 4 (of 64) experiments. The expression pattern of the corresponding 920 cDNAs is displayed in hierarchical cluster format. Each row represents a single array element and each column, a separate experimental mRNA sample. Experiments are organized by increasing time within each time course as indicated by the key at the bottom. Each expression measurement represents the ratio of fluorescence from the hybridized experimental sample to the reference sample, and is displayed as relative to the averaged zero time point. Missing or excluded data are represented by gray squares. The gray vertical bar identifies a cluster of genes whose expression pattern varies between individuals, as discussed in the text. (b and c) The common induction (b) and common repression (c) clusters, which are expanded portions of the larger cluster (a). A blue/yellow version of this figure is published as Fig. 10 in the supporting information on the PNAS web site.
Figure 2.

Host responses to dose escalation. PBMCs isolated from an apheresis sample from a second healthy donor were treated with a dose titration of B. pertussis LPS at 0.01 μg/ml, 0.1 μg/ml, and 1 μg/ml, a dose titration of heat-killed Bp338 [0.004 bacteria per human cell (1×) to 4 bacteria per human cell (1000×)], and a dose titration of heat-killed S. aureus isolate 1 [0.002 bacteria per human cell (1×) to 2 bacteria per human cell (1000×)]. Cells were harvested at 0, 0.5, 2, 4, 6, and 12 hr (100× time courses also have 1-hr and 24-hr time points). Data were filtered as in Fig. 1, and the resulting 463 genes are displayed as a six-node self-organizing map (38, 39). The common induction, common repression, and donor variant clusters are again marked. A blue/yellow version of this figure is published as Fig. 11 in the supporting information on the PNAS web site.
Figure 4.

B. pertussis toxins modify the host response. (a) PBMCs isolated from whole blood from a third healthy donor were treated with live (≈20 bacteria per human cell) and heat-killed (≈20 bacteria per human cell) Bp338 from the same bacterial culture. Data were collected at 0, 0.5, 2, 4, 6, and 12 hr. Data were selected as in previous figures and for each gene, and the Euclidean distance between responses to the two treatment conditions was calculated. Genes with Euclidean distances greater than 2 (≈400 cDNAs), are displayed in hierarchical cluster format. The 0.5-hr time point is not displayed in the cluster because of lower data quality. The names of selected genes are displayed next to the cluster. (b) The average of eight independent measurements of TNFα expression during treatment with both live and killed Bp338 are displayed with error bars representing the SEM. (c) The average induction ratio of the TNFα expression measurements are displayed for PBMCs treated with live and killed B. pertussis (corresponding to b), and for U937 cells, treated with isogenic mutants of B. pertussis that lack toxins that influence levels of intracellular cAMP. Error bars represent the SEM of measurements made by independent TNFα cDNAs (n = 8 for PBMC, n = 3 for U937). A blue/yellow version of this figure is published as Fig. 12 in the supporting information on the PNAS web site.
Microarray Procedures.
mRNA was extracted from cell pellets with the FastTrack 2.0 kit (Invitrogen). mRNA from samples for the data set comparing live versus heat-killed bacteria was amplified as described (10). cDNA was fluorescently labeled and microarrays were hybridized as described (ref. 11 and supporting information at www.pnas.org). More detailed information can be found at http://genome-www.stanford.edu/hostresponse/.
Expression Profile Modeling.
To model simultaneously separate effects of the three treatment parameters on gene expression responses, let variables b, d, and t denote, respectively, bacterial type (b = 0 for B. pertussis and b = 1 for S. aureus), log10 treatment dose (d = 0, 1, 2, 3), and exposure time (t = 0.5, 2, 4, 6, 12 hr). For a particular gene, let the relationship
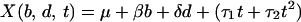 |
1 |
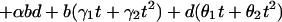 |
represent the expected gene expression response for given levels of the three factors. This model specifies a linear dose response in which dose is measured on logarithmic scale and a quadratic time response, and also allows for two-way interactions between each of the factors. When the model in Eq. 1 is used, the difference in the expected response for the two types of bacteria is
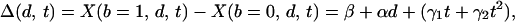 |
2 |
for a given treatment dose d and exposure time t. We used the method of least squares to fit the function in Eq. 1 to the zero-transformed expression data of a subset of genes with multiple representations on the array. For each gene, this method identified parameter values α, β, γ1, γ2, δ, τ1, τ2, θ1, and θ2, which minimize the sum of squared deviations between the observed responses and the fitted responses.
An expanded version of Materials and Methods, including methods for data analysis, is published as supporting information on the PNAS web site, www.pnas.org.
Results and Discussion
Stereotyped Host Responses to Diverse Bacteria.
In the context of complex host–pathogen interactions, the host genomic expression program reflects responses to both static pathogen-associated molecular patterns and active virulence-associated processes. To explore common themes and variations in the innate response to static molecular features of bacterial pathogens, we first compared expression profiles of PBMCs treated with selected heat-killed Gram-negative (B. pertussis and E. coli) or Gram-positive (S. aureus) bacteria. By using killed organisms, we eliminated potentially confounding effects of differential bacterial growth rate and in vitro cytotoxicity.
In a set of parallel experiments, we examined the responses of PBMCs to eight culture conditions, including six bacterial stimuli, each measured at eight time points over a 24-hr time course, using 64 cDNA microarrays. We refer to the resulting ≈1.2 million gene expression measurements as the “bacterial diversity” data set. Fig. 1 provides an overview of the gene expression responses of PBMCs to these diverse bacterial stimuli, focusing on the subset of genes whose expression level changed most dramatically during treatment (see supporting information at www.pnas.org). This group of ≈515 unique genes, as represented by 920 arrayed cDNAs, has been organized by agglomerative hierarchical clustering (12) (Fig. 1a). Most of the induced and repressed genes responded in a strikingly stereotyped manner to all of the bacterial treatments, and many of these changes were also seen in the response of these cells to ionomycin plus PMA, pharmacological stimulants that together mimic antigenic stimulation of leukocytes through pathways shared by adaptive and innate immune receptors (13).
Features of the Host Response: Common Induction.
A group of ≈206 unique genes, represented by 282 independent cDNA clones, was induced with stereotyped kinetics in response to each of the bacterial challenges, with a generally more pronounced induction in response to the Gram-negative bacteria (Fig. 1b). Products of the genes that contribute to this response have both systemic and local effects in vivo, reflecting the complexity and multifaceted nature of the physiological response to immune activation. One critical role of cells encountering invading bacteria is to recruit other leukocytes to the site of infection, to activate these cells, and to choreograph the multicellular physiologic response. Accordingly, within this cluster we found a preponderance of immune activation genes involved in cell–cell signaling, genes whose products participate in intercellular immunoregulatory signaling pathways, and other proinflammatory mediators of the immune response. The most prevalent classes among the commonly induced genes include cytokines [interleukin (IL)1α, IL1β, IL3, IL6, IL10, tumor necrosis factor α (TNFα), lymphotoxin, granulocyte colony-stimulating factor (CSF), macrophage CSF, and granulocyte–macrophage CSF] and chemokines [IL8, macrophage inflammatory protein (MIP)1α, MIP1β, MIP2α, MIP3α], as well as cell-surface receptors and ligands (CD40 and CD40 ligand, IL2 receptor α, tissue factor). The inflammatory response of immune cells to insults such as infection has been extensively studied, including several genome-level studies of host responses to bacterial stimuli (14–16), and the rich cohort of genes in this cluster represents the majority of the genes known to be induced in such a response.
A unifying feature of the characterized genes in the common induction response is that many are known to be regulated by the transcription factor NFκB (14, 17). NFκB factors have critical roles in orchestrating both acquired and innate immune responses; signals transduced both by antigen receptors on lymphocytes and by Toll-like receptors on innate immune cells activate this system (18, 19). Interestingly, the genes encoding most NFκB/Rel proteins (i.e., NFκB1, NFκB2, relB, cRel, IκBα) were also found within this cluster, reflecting the known autoregulation of this signaling pathway.
Treatment with a combination of ionomycin and PMA can activate lymphocytes, as well as other leukocytes, by mimicking the intracellular signals that occur during natural responses to antigen. Stimulation of these physiological signaling systems ultimately results in nuclear translocation of NFκB (13). While NFκB signaling has been implicated in the regulation of almost all genes that were induced by either heat-killed bacteria or ionomycin/PMA, there were distinct differences between the responses of PBMCs to these stimuli. A cohort of genes, including those encoding IL2, c-Myc, ornithine decarboxylase 1, and HSP70, were responsive to treatment with ionomycin/PMA, but not to any of the bacterial treatments. Conversely, a set of genes including adenosine deaminase and carboxypeptidase M were uniquely induced by exposure to bacteria. Some of these differences could reflect the activity of other signaling pathways, used either by Toll-like receptors or by alternative microbe-recognition receptors, which regulate transcripts through non-NFκB factors and contribute to the genomic response during a response to infection (see Fig. 5, which is published as supporting information on the PNAS web site).
Some of the differences between the gene expression responses of PBMCs to the various stimuli could be attributable to differential responses of specific cell types. While the utility of characterizing the responses of purified cell subpopulations is clear, a survey of the composite response profiles of a mixture of peripheral blood cell types provides an overview of this physiological system with some distinct advantages. For example, interactions between cell types significantly enhance the complexity of the expression programs in this model system, as in natural encounters of immune cells with bacteria.
Features of the Host Response: Common Repression.
The relative abundance of the transcripts from ≈96 genes, represented by 168 independent cDNA clones, decreased after pathogen exposure, in a remarkably consistent temporal program. A similar repression pattern was observed for many of these genes in independent experiments employing in vitro differentiated macrophages derived from human U937 cells (20) in response to live pathogenic bacteria (Fig. 6, which is published as supporting information on the PNAS web site). In addition, in a whole blood ex vivo human bacteremia model (21), diverse live and heat-killed pathogenic organisms similarly repressed most of the genes in this cluster (data not shown). Unlike the common induction response, the repression of this group of genes has not previously been identified as a canonical component of the transcriptional response of immune cells to bacterial or other stressful stimuli. The behavior of this cohort of genes has several interesting features, including a characteristic delay before changes are detected, a more prominent response to Gram-negative bacteria than to Gram-positive bacteria (at equivalent bacterial cell to host cell ratios), and an intriguing biological theme involving antigen processing and presentation (Fig. 1c).
Whereas many chemokines were consistently induced by the bacterial stimuli, a distinct subset of chemokines and chemokine receptors were selectively repressed, including the monocyte-attracting chemokines MCP1 and MCP3, the CC-chemokine receptor CCR1, and FPR, the high-affinity receptor for bacterial N-formylmethionyl peptides. In addition, several genes whose products play known roles in cell–cell adhesion, diapedesis, and extravasation of leukocytes were consistently repressed (CD156, CD11c, CD31), as were genes encoding surface molecules known to be important for both the recognition of bacterial pathogens (CD14), and their phagocytosis (CD64, the high-affinity receptor for the Fc fragment of IgG). Gelsolin and WASP, both central components of IgG-mediated phagocytosis (22, 23), were also among the repressed genes within this cluster.
Another distinctive group of repressed genes has well-known roles in the degradation and presentation of antigen. The genes encoding the heavy chain of cytochrome b-245 (CYBB) and neutrophil cytosolic factor 1 (NCF1), pivotal components of the respiratory burst machinery, were repressed, as were the genes encoding the lysosomal protease cathepsin B, and IP30, a lysosomal thiol reductase. Remarkably, this repression program also included many genes encoding both classical and nonclassical MHC class II molecules, as well as other components involved in the display of foreign peptides on the surface of the antigen-presenting cells (APCs). The repression of genes encoding the nonclassical MHC molecules HLA DMα and -β, which stabilize nascent and recycled classical MHC class II molecules and participate in loading peptide into the antigen cleft, was first observed at 4 hr. Subsequently, the repression of HLA DRα and -β and HLA DPα, which directly bind and present captured antigens, was observed at 12 hr.
In the setting of a host–pathogen encounter, the repression of genes whose products are crucial to host defense seems paradoxical. However, we believe that this repression program, like the common induction response, may serve an important functional role in regulating the activity of APCs during an encounter with a potential pathogen. We propose that, by attenuating the capacity for engulfment and processing of new particles, the common repression response serves to commit the APC to retaining and presenting the antigens captured in the context of pathogen contact. Several others have proposed analogous models for similar phenomena observed in specific immune cell subpopulations after contact with stimulatory microbial molecules (24–26).
A gene expression program leading to such antigen commitment might allow the APC to present the captured determinants to T cells not only at the site of infection, but also at secondary lymphoid organs. Indeed, the observed repression of genes involved in migration to, and retention at, the site of infection may serve to release APCs for migration to areas rich in adaptive immune cells. Accordingly, this gene expression program may play a role in vivo in the context of vaccine immunity, in which the inclusion of bacterial cell wall components as adjuvant strengthens adaptive immune responses to antigenic determinants with low intrinsic immunogenicity. Stimulation of this program at the systemic level may also play a role in the immune paralysis of APC function referred to as endotoxin tolerance in the context of bacteremia and sepsis (27).
The broad biological themes revealed by the clusters of induced and repressed genes (Fig. 7, which is published as supporting information on the PNAS web site) were independently supported by a statistical analysis measuring the enrichment of specific functional annotations associated with genes in these clusters (see Table 1, which is published as supporting information on the PNAS web site). The transcriptional programs revealed by genome-wide expression analysis not only paint a more complete portrait, but also reveal previously unknown and intriguing elements of the host response.
Response to a Range of Bacterial Doses.
The expression responses of PBMCs measured in the bacterial diversity data set were generally more pronounced during treatment with killed Gram-negative than with killed Gram-positive bacteria, although the number of bacterial cells in each treatment was comparable. To address the possibility that the differences in the magnitude of the response were simply due to a disparity in the potencies of the microbial stimuli, we examined expression responses across a wide range of doses of B. pertussis and S. aureus in a second set of time course experiments. The set of genes whose transcript levels changed in response to bacterial stimuli in this data set matched almost completely the genes identified in the bacterial diversity data set, although the PBMCs used in the two sets of experiments were collected from different donors.
The amplitude of the response for almost all of the induced and repressed genes varied with the stimulus dose in a graded manner (Fig. 2). The graded nature of the response implies that the number of microbial cells used to stimulate immune cells determines the level and/or probability of response in the latter. We observed a large disparity in the potency of Gram-positive and Gram-negative bacteria as inducers of this transcriptional response. The lowest dose of heat-killed Gram-negative bacteria (4 per 1,000 host cells) elicited a prominent expression response, whereas a comparable nominal dose of killed Gram-positive bacteria had little or no discernable effect on gene expression. However, at the highest dose, the two stimuli had a nearly or completely equivalent effect on the expression of many genes. This convergence raises the possibility that there exists a common PBMC response to microbial stimuli, and that any observed difference in the responses to different bacteria might simply be attributable to differences in effective dose. In this model, at a properly matched effective dose, host responses to the two different bacterial stimuli would be indistinguishable.
To explore this possibility, we analyzed gene expression patterns, over time and in response to increasing dose, using a linear modeling approach. With this method, we fitted smoothed surfaces to the experimentally measured responses of individual genes to the two bacterial stimuli. By subtracting the two “expression surfaces,” we obtained, for each gene, a surface representing the difference between the Gram-negative and Gram-positive response profiles. If an equivalent effective dose existed, the expected difference between the two surfaces would be constant under the model.
Fig. 3 depicts this comparison for CD64, one of the genes whose expression response was most divergent between the two bacterial stimuli. The repression of CD64 during treatment with heat-killed Bp338 increased in amplitude as dose increased and exposure time progressed. In contrast, induction was observed in response to treatment with heat-killed S. aureus, but the level of this induction was attenuated with increasing dose. Displays of the absolute difference between the two response profiles (Fig. 3 c and d) emphasize that the greatest divergence occurred at late time points and was relatively independent of stimulus dose. It is clear that for the observed CD64 expression profiles, a single matched effective dose for all measured time–dose points could not be defined. A broader analysis confirmed that while the response profiles of some genes suggested a matched effective dose (e.g., defensin α1, CXCR4), most genes had incongruent difference profiles such that no matched effective dose could be defined that would apply to all genes (Fig. 8, which is published as supporting information on the PNAS web site). Despite the overall similarity of the gene expression programs of PBMCs responding to different microbial stimuli, the distinct sensitivity of different genes to dose escalation revealed perceptual discrimination of the two types of heat-killed bacteria by PBMCs.
Figure 3.

Dose–response patterns reveal discrimination. The modeled expression profile of CD64 is shown in response to B. pertussis (a) and S. aureus (b) treatments as an example (surfaces fitted by using four independent measurements). The x axis represents time; the y axis, exposure dose; and the z axis, the expression level (log2) of the zero-transformed data. The surface represents the least-squares fit of the expression data to the model. The correlation of fitted to observed responses was 0.83. (c) The subtraction of the two expression surfaces, Δ(d, t). (d) The same fitted response difference as in c, but in the form of a two-dimensional color image and contour plot, in which a red to blue color scale represents high to low absolute difference, respectively. Further examples are shown in Fig. 8 in the supporting information.
Such discordance between the dose responses of different genes suggests that the ostensibly similar host responses to killed S. aureus and B. pertussis arise from distinct sensory mechanisms converging on a similar set of genes, or that different cell populations respond in a distinct manner to each of the two stimuli. Convergence of signals from distinct sensory mechanisms has been observed in the response of S. cerevisiae to diverse environmental stresses (28), as well as the response of human cell lines to distinct signaling domains of growth factor receptors (29). While subtle when viewed in this experimental model, such discrimination at the cellular level could potentially lead to quite different consequences at the level of the whole organism.
Donor Expression Differences.
The expression responses of PBMCs from different donors were remarkably consistent, although we identified a set of genes whose responses differed among donors. The significance of these differences is unclear. This “donor variant” response involved several genes related to IFN signaling, a subset of which have known functions in the antiviral response, including 2′,5′-oligoadenylate synthetase 1 (OAS1) and myxovirus resistance 1 (MX1). The unique induction of this group of genes in the response of the first donor to E. coli (Fig. 1a) was recapitulated in a second set of experiments with cells from this same donor. The response of this donor's PBMCs was consistently different from that of the other two donors, each of which showed induction of these genes in response to all treatments (Fig. 9, which is published as supporting information on the PNAS web site). These subtle differences in the responses of individual hosts could be attributable to variation in the basal expression of certain genes, or to differences in the relative populations of the diverse specialized cells in peripheral blood from normal individuals. Basal levels of various cytokines can vary significantly among healthy individuals (30) and may be related to the variations observed in physiological gene expression programs.
Active Modulation of Host Gene Expression by Bacteria.
Stimulation of cells with heat-killed bacteria allowed us to compare host responses over prolonged time periods to diverse organisms with different growth rates, but also eliminated contributions by active, virulence-associated processes, and potentially reduced the complexity in the observed responses. We therefore explored the effect of specific bacterial virulence attributes on innate host responses, beginning with a comparison of live and killed B. pertussis. We chose this organism because its slow growth in tissue culture media and low cytotoxicity allowed us to cocultivate live bacteria with PBMCs over a long time course. PBMCs isolated from a third healthy donor were exposed in parallel to nominally identical multiplicities of live and heat-killed B. pertussis.
While the observed responses to killed B. pertussis were largely similar to those seen in the PBMCs from the previous donors, distinct differences were seen between the host responses to live and killed B. pertussis (Fig. 4). In contrast to the remarkably similar gene expression programs induced by distinct heat-killed organisms, the host expression programs in response to live and dead organisms of the same species differed greatly. For example, genes encoding TNFα, MIP1β, IL1α, and IL1β were quickly induced after exposure to either live or killed bacteria. However, while the induction of these genes was sustained in PBMCs treated with killed bacteria, their transcripts rapidly diminished in the cells exposed to live B. pertussis (Fig. 4b). The ability of live B. pertussis to suppress expression of these important antimicrobial genes suggests active mechanisms used by the bacteria to influence the host response.
To address whether these differences were attributable to specific active virulence mechanisms of live organisms, we compared host responses to several isogenic B. pertussis mutant strains defective in toxin production. Avirulent-phase (Bvg−) B. pertussis lack expression of many virulence factors, including two major toxins, adenylate cyclase toxin and pertussis toxin, both of which are produced during infection (5). An increase in host intracellular cAMP levels through the actions of pertussis toxin and adenylate cyclase toxin is a crucial feature of B. pertussis virulence (31).
The disparity observed in the TNFα response of PBMCs to live and killed Bp338 was mimicked during infection of U937-derived macrophages with live Bp338 (virulent-phase) and live Bp537 (avirulent-phase) bacteria (Fig. 4c). Infection with live Bp537 is analogous to treatment with killed Bp338 in the sense that neither bacteria can produce cAMP-modulating toxins. Notably, mutants lacking only pertussis toxin (BpTox6) or adenylate cyclase toxin (BpA2-6) had an intermediate effect, further implicating these toxins in the active suppression of the programmed induction of TNFα that naturally occurs in response to bacterial stimulation. Previous studies have shown that pharmacological increases in intracellular cAMP levels can abrogate the LPS-stimulated induction of TNFα expression in several cell types, including primary human monocytes (32).
In contrast to the attenuation of TNFα induction in the response of PBMCs to live B. pertussis, the expression of genes encoding prostaglandin-endoperoxide synthase 2 (COX-2), phosphodiesterase 4D, and CXC chemokine receptor 4 was enhanced in response to treatment with live toxin-producing bacteria. The expression of all three genes has previously been shown to increase when intracellular cAMP levels rise (33–35), suggesting that the toxins of B. pertussis may account for their induction after infection with live organisms. We found that many additional genes displayed differential expression patterns that suggested modification by the toxins. For example, M-CSF and IP10, both known to be expressed during an inflammatory response and to stimulate immune cells, had expression patterns similar to TNFα. While the expression of these transcripts has not been shown to be influenced by B. pertussis toxins specifically, the expression of both genes has been shown to decrease as the level of intracellular cAMP increases (36, 37). Again, the suppressive influence of toxins on the expression of these and other genes could contribute to the virulence of B. pertussis. This simple comparison illustrates how dynamic interactions with live pathogens modulate stereotyped responses of the host, revealing previously uncharacterized specific gene expression responses.
Many human cells recognize and respond to common conserved molecular signatures of microorganisms through sensory systems such as Toll-like receptors. Despite recent evidence of specialization by Toll-like receptor subtypes for different patterned molecular ligands, our data argue for convergence of signaling through these receptors and a highly stereotyped host response in PBMCs. Pathogenic microbes have evolved diverse mechanisms to subvert, exploit, or evade such host defenses. As a result, different pathogens may have measurable and distinct “signatures” of two types: a specific set of microbial virulence factors, and a specific set of induced and manipulated host responses. Our data suggest that active virulence-associated processes might provide a basis for the recognition of, and discrimination between, human responses to distinct pathogens based on host gene expression patterns. Detailed characterization of the specific ways in which pathogens alter the gene expression programs involved in host defenses should provide important insights into mechanisms of pathogenesis.
The stereotyped gene expression responses of human PBMCs to live and killed bacteria in vitro reveal important features of innate immunity that may be relevant to the initial stages of contact between pathogens and local immune surveillance cells at skin and mucosal portals of entry. These responses suggest efforts on the part of host cells to recruit and activate multiple arms of the immune system, as well as to orchestrate effective antigen processing and presentation. The host seeks to contain local infection; pathogens seek niches in which they can continue to multiply or persist. Further analyses of the molecular interaction between microbes and host will help clarify the basis upon which the host classifies pathogens and other noxious stimuli, and provide a framework for the development of new strategies for detecting and thwarting invading pathogens.
Supplementary Material
Acknowledgments
We thank the Minnesota State Dept. of Health for providing B. pertussis strain Minnesota 1, and we thank members of the Brown, Relman, and Botstein laboratories for helpful discussions. We thank Drs. S. Morse and A. Rudolph and the U.S. Defense Advanced Research Projects Agency (DARPA) for their support of this work. This work was supported by DARPA Grant N65236-99-1-5428 to P.O.B and D.A.R., by National Institute of Allergy and Infectious Diseases Grant AI39587 to D.A.R., and by the Howard Hughes Medical Institute. P.O.B. is an Associate Investigator of the Howard Hughes Medical Institute. J.C.B. was supported by the Medical Scholars Program of Stanford University School of Medicine.
Abbreviations
- PBMCs
peripheral blood mononuclear cells
- LPS
lipopolysaccharide
- PMA
phorbol 12-myristate 13-acetate
- TNFα
tumor necrosis factor α
- APCs
antigen-presenting cells
References
- 1.Aderem A, Hume D A. Cell. 2000;103:993–996. doi: 10.1016/s0092-8674(00)00201-4. [DOI] [PubMed] [Google Scholar]
- 2.Janeway C A, Jr, Medzhitov R. Curr Biol. 1999;9:R879–R882. doi: 10.1016/s0960-9822(00)80073-1. [DOI] [PubMed] [Google Scholar]
- 3.Ozinsky A, Underhill D M, Fontenot J D, Hajjar A M, Smith K D, Wilson C B, Schroeder L, Aderem A. Proc Natl Acad Sci USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. . (First Published November 28, 2000; 10.1073/pnas.250476497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Medzhitov R, Janeway C., Jr N Engl J Med. 2000;343:338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 5.Stibitz S, Aaronson W, Monack D, Falkow S. Nature (London) 1989;338:266–269. doi: 10.1038/338266a0. [DOI] [PubMed] [Google Scholar]
- 6.Boschwitz J S, Batanghari J W, Kedem H, Relman D A. J Infect Dis. 1997;176:678–686. doi: 10.1086/514090. [DOI] [PubMed] [Google Scholar]
- 7.Relman D A, Domenighini M, Tuomanen E, Rappuoli R, Falkow S. Proc Natl Acad Sci USA. 1989;86:2637–2641. doi: 10.1073/pnas.86.8.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Relman D, Tuomanen E, Falkow S, Golenbock D T, Saukkonen K, Wright S D. Cell. 1990;61:1375–1382. doi: 10.1016/0092-8674(90)90701-f. [DOI] [PubMed] [Google Scholar]
- 9.Gross M K, Au D C, Smith A L, Storm D R. Proc Natl Acad Sci USA. 1992;89:4898–4902. doi: 10.1073/pnas.89.11.4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang E, Miller L D, Ohnmacht G A, Liu E T, Marincola F M. Nat Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- 11.Alizadeh A A, Eisen M B, Davis R E, Ma C, Lossos I S, Rosenwald A, Boldrick J C, Sabet H, Tran T, Yu X, et al. Nature (London) 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 12.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acuto O, Cantrell D. Annu Rev Immunol. 2000;18:165–184. doi: 10.1146/annurev.immunol.18.1.165. [DOI] [PubMed] [Google Scholar]
- 14.Pahl H L. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 15.Detweiler C S, Cunanan D B, Falkow S. Proc Natl Acad Sci USA. 2001;98:5850–5855. doi: 10.1073/pnas.091110098. . (First Published April 24, 2001; 10.1073/pnas.091110098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diehn M, Relman D A. Curr Opin Microbiol. 2001;4:95–101. doi: 10.1016/s1369-5274(00)00171-5. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh S, May M J, Kopp E B. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 18.Medzhitov R, Preston-Hurlburt P, Janeway C A. Nature (London) 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 19.Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning C J. J Biol Chem. 1999;274:17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 20.Koren H S, Anderson S J, Larrick J W. Nature (London) 1979;279:328–331. doi: 10.1038/279328a0. [DOI] [PubMed] [Google Scholar]
- 21.Ison C A, Heyderman R S, Klein N J, Peakman M, Levin M. Microb Pathog. 1995;18:97–107. doi: 10.1016/s0882-4010(95)90093-4. [DOI] [PubMed] [Google Scholar]
- 22.Serrander L, Skarman P, Rasmussen B, Witke W, Lew D P, Krause K H, Stendahl O, Nusse O. J Immunol. 2000;165:2451–2457. doi: 10.4049/jimmunol.165.5.2451. [DOI] [PubMed] [Google Scholar]
- 23.Lorenzi R, Brickell P M, Katz D R, Kinnon C, Thrasher A J. Blood. 2000;95:2943–2946. [PubMed] [Google Scholar]
- 24.Chu R S, Askew D, Noss E H, Tobian A, Krieg A M, Harding C V. J Immunol. 1999;163:1188–1194. [PubMed] [Google Scholar]
- 25.Askew D, Chu R S, Krieg A M, Harding C V. J Immunol. 2000;165:6889–6895. doi: 10.4049/jimmunol.165.12.6889. [DOI] [PubMed] [Google Scholar]
- 26.Wyant T L, Tanner M K, Sztein M B. Infect Immun. 1999;67:3619–3624. doi: 10.1128/iai.67.7.3619-3624.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolk K, Docke W D, von Baehr V, Volk H D, Sabat R. Blood. 2000;96:218–223. [PubMed] [Google Scholar]
- 28.Gasch A P, Spellman P T, Kao C M, Carmel-Harel O, Eisen M B, Storz G, Botstein D, Brown P O. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fambrough D, McClure K, Kazlauskas A, Lander E S. Cell. 1999;97:727–741. doi: 10.1016/s0092-8674(00)80785-0. [DOI] [PubMed] [Google Scholar]
- 30.Yaqoob P, Newsholme E A, Calder P C. Cytokine. 1999;11:600–605. doi: 10.1006/cyto.1998.0471. [DOI] [PubMed] [Google Scholar]
- 31.Kerr J R, Matthews R C. Eur J Clin Microbiol Infect Dis. 2000;19:77–88. doi: 10.1007/s100960050435. [DOI] [PubMed] [Google Scholar]
- 32.Zidek Z. Eur Cytokine Netw. 1999;10:319–328. [PubMed] [Google Scholar]
- 33.Hinz B, Brune K, Pahl A. Biochem Biophys Res Commun. 2000;278:790–796. doi: 10.1006/bbrc.2000.3885. [DOI] [PubMed] [Google Scholar]
- 34.Sette C, Conti M. J Biol Chem. 1996;271:16526–16534. doi: 10.1074/jbc.271.28.16526. [DOI] [PubMed] [Google Scholar]
- 35.Cole S W, Jamieson B D, Zack J A. J Immunol. 1999;162:1392–1400. [PubMed] [Google Scholar]
- 36.Boorsma D M, Flier J, van den Brink E N, Sampat S, Walg H L, Willemze R, Tensen C P, Stoof T J. Cytokine. 1999;11:469–475. doi: 10.1006/cyto.1998.0463. [DOI] [PubMed] [Google Scholar]
- 37.Kamthong P J, Wu F M, Wu M C. Biochem J. 2000;350:115–122. [PMC free article] [PubMed] [Google Scholar]
- 38.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 39.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T R. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.