

An 8-year-old boy who had immigrated from China a year earlier with his family was referred for assessment of possible developmental and learning difficulties. His grade 2 teacher had expressed concerns regarding the boy's distractibility and difficulties with attention span, task completion and organizational skills. He was also showing significant delays in reading, writing, spelling and arithmetic and exhibiting fine motor difficulties (pencil control, letter formation) as well as gross motor incoordination (in physical education classes and sports) and clumsiness.
The child's mother reported an unremarkable pregnancy and delivery. Weighing 3.1 kg at birth, the boy had no significant neonatal problems. There was no apparent delay in his early developmental milestones. His mother reported that he had always been a very active and demanding child. His medical history had been unremarkable apart from myringotomy tube placement for recurrent otitis media. There was no history of seizures or developmental regression; however, his parents described with few details the fact that a physician in China had diagnosed a disorder that might affect the boy's brain and development. The family's medical history was unremarkable, including an absence of developmental delay or neurologic disease.
The boy's height was 114.5 cm (3rd centile), weight 21.7 kg (10th centile) and skull circumference 52.3 cm (50th centile). Findings on physical examination were unremarkable except for multiple (> 25) café au lait spots larger than 5 mm in diameter on the skin of his trunk and limbs as well as axillary freckling (Fig. 1). The combination of multiple café au lait spots with axillary freckling, the short stature, the relative macrocephaly and the developmental difficulties led to the suspicion of neurofibromatosis type 1 (NF-1). Other features of NF-1, such as Lisch nodules (iris hamartomas) or neurofibromata (benign peripheral nerve sheath tumours), were not evident. Examination of the parents did not reveal any features of NF-1.
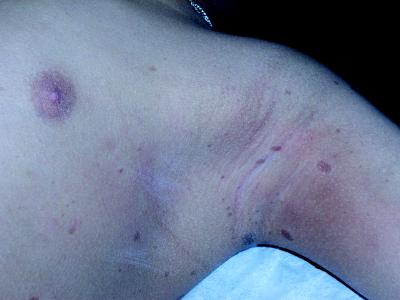
Figure 1. Photo: Courtesy: Stephen Wainer
NF-1 is a common autosomal dominant disorder affecting about 1 in 3500 people and involves abnormalities of the skin, bones, eyes, and central and peripheral nervous systems. Although the incidence of mental retardation in NF-1 is only slightly higher than that in the general population (4.8%–8% v. 3%), the average intelligence quotient of children with NF-1 is reduced, ranging from 89 to 94.1,2,3 Learning disabilities are seen in 30%–61% of children with the disorder,4,5 and children with NF-1 have been shown to have reading disabilities, visuospatial and neuromotor deficits as well as problems with attention span, self-organization, speech (articulation) and motor coordination.6,7 There is no reliable predictor of learning difficulties or cognitive deficits in NF-1, although there is a possible association with the number and location of areas of increased signal intensity on T2-weighted images on cranial MRI scans.6
The diagnosis is clinical and based on defined National Institutes of Health criteria.8 Although most of the clinical features are not present in infancy, affected individuals tend to accumulate features of the disease over time, with 90% meeting the diagnostic criteria by 7 years of age.8 The majority of affected children will show axillary or inguinal freckling by the age of 2, and by 11 years most children will have 2 or more neurofibromata of any type or 1 plexiform neurofibroma (slow-growing tumours that involve multiple nerves and fascicles).8 Neurofibromata may be widely distributed and give rise to a variety of complications related to their size, location and extent of local tissue encroachment (including pain, cosmetic defects and functional deficits).9 NF-1 is also associated with an increased risk of cancer (fibrosarcoma, leukemia), short stature (in 33% of cases), blindness (due to optic gliomas), kyphosis and scoliosis (leading to diminished pulmonary function), pseudoarthroses, hypertension, macrocephaly (in 50% of cases) and seizures.10,11 The multisystem, and life-long, nature of these complications mandates careful and regular health surveillance.10,12 There is extreme variability in the clinical expressivity of NF-1, even within the same family, and prediction of the impact of the disease on an individual is not possible. In general, about half of the patients with NF-1 are mildly affected, and about one-third have serious complications.10
Although café au lait spots are associated with NF-1, they are by no means diagnostic of it: solitary spots are common in the general population, with 13%–27% of children under 10 years of age having at least one spot.11 NF-1 is the predominant cause of multiple café au lait spots; however, they can also occur with neurofibromatosis type 2, segmental neurofibromatosis, tuberous sclerosis, McCune Albright syndrome, Fanconi anemia, Bloom syndrome and ataxia telangiectasia.11
The NF-1 gene is located on chromosome 17 at band q11.2. There is a high rate of spontaneous mutation (in 50% of cases), and most NF-1 mutations are unique to a particular family, with no clear phenotype–genotype correlation.11 Although genetic testing for NF-1 is available (through linkage analysis, direct mutation analysis or protein truncation assay), this is not usually required or indicated.
Stephen Wainer Assistant Clinical Professor Department of Pediatrics University of Calgary Alberta Children's Hospital Calgary, Alta.
References
- 1.North K, Joy P, Yuille D, Cocks N, Hutchins P. Cognitive function and academic performance in children with neurofibromatosis type 1. Dev Med Child Neurol 1995;37:427-36. [DOI] [PubMed]
- 2.Ferner RE, Hughes RAC, Wenman J. Intellectual impairment in neurofibromatosis 1. J Neurol Sci 1996;138:125-33. [DOI] [PubMed]
- 3.Eldridge R, Denckla MB, Bien E, Myers S, Kaiser-Kupfer MI, Pikus A, et al. Neurofibromatosis type 1 (Recklinghausen's disease): neurologic and cognitive assessment with sibling controls. Am J Dis Child 1989;143:833-7. [PubMed]
- 4.Moore BD, Ater JL, Needle MN, Slopis J, Copeland DR. Neuropsychological profile of children with neurofibromatosis, brain tumor or both. J Child Neurol 1994; 9:368-77. [DOI] [PubMed]
- 5.Legius EM, Desceemaeker MJ, Spaepen A, Casaer P, Fryns JP. Neurofibromatosis type 1 in childhood: a study of the neuropschological profile in 45 children. Genet Couns 1994;5:51-60. [PubMed]
- 6.NF1 Cognitive Disorders Task Force. Cognitive function and academic performance in neurofibromatosis 1. Neurology 1997;48:1121-7. [DOI] [PubMed]
- 7.Hofman KJ, Harris EL, Bryan N, Denckla MB. Neurofibromatosis type 1: the cognitive phenotype. J Pediatr 1994;124:S1-8. [DOI] [PubMed]
- 8.DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000;105:608-14. [DOI] [PubMed]
- 9.Packer RJ, Gutmann DH, Rubinstein A, et al. Plexiform neurofibromas in NF1: Toward biologic-based therapy. Neurology 2002;58:1461-70. [DOI] [PubMed]
- 10.Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics 1995; 96: 368-72. [PubMed]
- 11.Tekin M, Bodurtha JN, Riccardi VM. Café au lait spots: the pediatrician's perspective. Pediatr Rev 2001;22:82-9. [DOI] [PubMed]
- 12.Guidelines/Outcomes Committee. Guidelines of care for neurofibromatosis type 1. J Am Acad Dermatol 1997;37:625-30. [DOI] [PubMed]