

Abstract
The total genetic diversity of a species is a key factor in its persistence and conservation. Because realistic sample sizes are far smaller than the total population, it is impractical to exhaustively characterize diversity of most populations. Here, we demonstrate the possibility of calculating the genetic diversity of a spatial population from a sample using genealogical models. We trace the history of a population by simulating the locations of the ancestors of a particular sample of the population backwards in time. We use this method to estimate the genetic diversity of the global population of Pseudomonas bacteria. The same results are obtained whether using a global sample or a subsample restricted to a particular geographic region (California). The results are also validated by comparing additional predictions of the model to the data. Furthermore, we use these results to show that the level of genetic diversity in a population depends strongly on the size of its habitat, much more strongly than does biodiversity as measured by the number of species. The strong dependence of diversity on habitat area has significant implications for conservation strategies.
Keywords: habitat loss, spatial populations, biodiversity, diversity estimation, coalescent
In this paper, we study the total genetic diversity of populations using simulations and analytic studies of genealogical trees, a method known as coalescent theory (1–6). We (i) present a method for estimating the total diversity from a spatial sample and (ii) study the dependence of diversity on habitat area. Genetic diversity is an important factor in the persistence of a species (7, 8); thus, estimating the total genetic diversity of a species and the reduction of its diversity due to habitat loss is important to its conservation. There are indications that reduced diversity increases susceptibility to disease (9) and reduces individual fitness through inbreeding (8, 10). Diversity is also believed to confer adaptability in the face of environmental changes (8, 11). Although demographic stochasticity is a greater immediate threat to the survival of endangered species, diversity loss puts species at increased risk of extinction (10) and is thus important to their long-term survival. Hence, the results we obtain may inform conservation strategies. In this article, we adopt a simple genealogical model and derive from it properties of the diversity. We show by analysis that our results are robust to the introduction of additional biological realism, and we compare the results with data from field populations, demonstrating that they capture key aspects of the behavior of genetic diversity. There are many factors that affect genetic diversity that are not included in the model, including environmental changes and interactions with other species. Still, our results suggest that the genealogical model is a useful foundation for studies that incorporate the impact of other factors on diversity.
Our studies are based on a spatially explicit model for two reasons. First, limited dispersal is known, theoretically (12) and from field data (13), to increase genetic diversity even without explicit barriers to gene flow (14). Second, area is a primary determinant of biodiversity above the species level, as measured by the number of species (15), suggesting that it is also likely to be important within species.
Estimating Diversity from a Sample
We first present a method of estimating the diversity of a spatial population from a sample, using analytic results (see Box 1: Analytic Results) and simulations, and compare predictions of our method with genetic data from field populations. The estimate of diversity is rough; however, it may not be possible to be more precise because diversity naturally undergoes large fluctuations (6). Methods exist to estimate quantities relevant to diversity, such as the effective population size of well mixed (16, 17) and various kinds of subdivided (18) populations. The method introduced here, however, gives a more direct characterization of diversity, is applicable to continuous spatial populations, and estimates diversity at any level of genetic resolution. In addition to the diversity, the approach also yields other predictions that can be verified against the genetic data itself.
Methods. Genealogical model. We consider the genealogical tree of a population looking backward in time (6). Each individual's parent is at a location given by a random step in space with a distribution of distances determined by dispersal. Therefore, a line of descent is a random walk with a distribution of step sizes given by dispersal characteristics. What is different from the usual random walk is that two organisms can have the same parent. This occurs when two of the random walks step onto the same location. Subsequent steps are made together—the random walks are coalescing. The common ancestor is represented by the single walker remaining after all walkers have coalesced (Fig. 1).
Fig. 1.

Illustration of the simulation of a spatial genealogy. Only 3 of the 248 lineages are shown for clarity, corresponding to three of the Pseudomonas samples. At the present (at right), their locations correspond to locations where samples were taken. They are simulated in two spatial dimensions backward in time (shown as a third dimension) until all have coalesced.
The genealogical tree we described could result from a wide range of models of how organisms reproduce and disperse, and, therefore, properties that apply to all such genealogical trees can be used to infer widely applicable properties of the diversity, particularly scaling properties. For concreteness, we can describe a particular simple model for how organisms reproduce and disperse that gives rise to such genealogies. We consider the population of organisms to be located on a lattice in space with each site containing a single individual. At each time step, each individual reproduces into all neighboring sites and its own site and then expires, but only one randomly chosen offspring in each site survives. Thus, each individual is the offspring of a parent in a small neighborhood. When viewed backward in time, the genealogical tree of this simple model population corresponds to the backward-looking description we gave of coalescing random walks.
However, the backward time picture of genealogies as coalescing random walks does not depend on this simple version of population reproduction. Instead, it is robust to the addition of a variety of aspects of particular biological populations. For example, consider a population that has a highly variable fecundity, with most organisms having no offspring and some having many, as is often found in nature. We can ask whether it is necessary for the random walk model to have the same variability to have similar genealogical properties. For a highly variable fecundity, when we trace lineages back in time, we initially sometimes see many lineages merging into one, corresponding to an organism that had many offspring with descendants in the present. However, this only occurs in the most recent few generations. Beyond these generations, the ancestors of the present population are only a small fraction of the entire population at that time. Under these conditions, in any generation the likelihood that two of the ancestors have the same parent is small, and the probability that three have the same parent is negligible. Thus, the degree of variability in fecundity does not affect the description of the genealogical tree before the most recent generations. The absolute rate of pairwise coalescence may differ between different populations; however, this does not affect the results that we give below. Similarly, many of the specific properties of organism dispersal and organism life history do not affect the results, because the properties of random walks are highly robust. Thus, lineages show the same diffusion-like behavior for a wide range of dispersal properties and generation times. As we show below, there exist some properties of dispersal that can affect the results. In particular, if the dispersal occurs predominantly over a length that is close to the size of the habitat, the population will behave as well mixed rather than spatial, with different scaling behavior; dispersal that is limited to one dimension also leads to distinct scaling behavior. We have used simulations to test the results using a variety of assumptions about the specific details of the model. A formal derivation of the robustness of the results can be given by using scaling arguments and other methods as described in part in Box 1: Analytic Results and in the supporting information, which is published on the PNAS web site. The scaling behavior of well mixed models has been shown to be similarly robust (1).
A key property of the diversity is the number of organisms L living at a time t1 in the past that have descendants in the present. In the genealogical model, this is the number of remaining random walkers. We write T = t – t1 to represent the number of generations before the present time t. Given a constant mutation rate, L(T) is the number of genotypes in the present that are distinct at a resolution corresponding to the amount of genetic divergence that accumulates over time T. We will use L(T) to estimate the number of distinct genotypes in present population.
For a well mixed population, Fisher (19) showed that the number of lineages decreases inversely with T: L(T) ∼ 1/T. The mathematical study of coalescing random walks has provided the scaling of the number of remaining walkers with time for spatial systems (20). The results are highly robust and depend only on the spatial dimension of the dispersal. They do not depend on the distribution of dispersal distances or times between steps as long as the dispersal is small compared with the size of the habitat. For spatial populations in two dimensions, the result implies L(T) ∼ log(T)/T (see Box 1: Analytic Results), which differs from the well mixed population result by a logarithmic factor. This implies that at any genetic resolution, there are more distinct genotypes than in a well mixed case, and that the tree is deeper. In general, a logarithmic factor is a weak correction, but in this case, the number of generations to the common ancestor can be large (e.g., 10,000 for a small lattice of 50 × 50 dispersal distances), and, therefore, it can be significant. In one dimension the effect is even greater because the number of lineages decreases inversely as the square root of 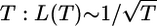 . These scaling relationships are for the entire population, but we can also obtain L(T) for a sample by directly simulating the ancestral tree of the sample. The coalescence of the ancestral lineages of the samples is independent of their coalescence with the rest of the population because each coalescence occurs independently in this simple genealogical model.
. These scaling relationships are for the entire population, but we can also obtain L(T) for a sample by directly simulating the ancestral tree of the sample. The coalescence of the ancestral lineages of the samples is independent of their coalescence with the rest of the population because each coalescence occurs independently in this simple genealogical model.
Comparison with genetic data. The analytic results for L(T) can be directly compared with genetic data from field populations, and the results of the comparison can then be used to estimate the diversity of the population. We used data of Cho and Tiedje (21) consisting of 248 samples of Pseudomonas soil bacteria from multiple locations on five continents. From their dendrogram, we obtained counts of the number of ancestors at a particular effective genomic similarity (r value) as measured by this fingerprinting technique to obtain L(r), corresponding to the number of ancestors that existed at a time such that their living descendants have diverged to a similarity value of r. We then obtained L(T) from L(r) as described in the supporting information. We normalized T by dividing by T0, the time to the smallest genetic difference considered (r = 0.95). The final results are shown as circles in Fig. 2. To compare this data with the theoretical results, we simulated the genealogical tree of the sample (Fig. 1), with the current generation represented at points corresponding to the locations where the samples were obtained and the locations of ancestors modeled as a coalescing random walk (6). [The idea of considering the location of ancestors has previously been used to investigate genetic distances (2) and the geographic origin of a lineage (22).]
Fig. 2.

Number of lineages as a function of time in the past, L(T). (a) Genetic data (circles) and a spatial simulation of the sampled population (solid line). The dashed line corresponds to theory for the whole population. (b) A comparison of the spatial and well mixed cases. L(T) for a subset of the samples from one geographic region (California, squares) is compared with a spatial (dashed line) and well mixed (dotted line) simulation of the subset by using the same parameters as in a. The number of simulation time steps per unit, T0, of biological time is 160, and the coalescence probability, pc, is 0.15.
Two properties of the genealogical model were adjustable: the number of simulation time steps, NT, corresponding to one unit, T0, of biological time, and the probability, pc, of two lineages in the same site coalescing. The parameters were set by a simple fitting procedure that adjusts the intercepts of L(T) at the L and T axes but does not affect the shape of the curve. The parameters were adjusted to simultaneously fit both L(T) and a different property of the same genealogical tree: the distribution of genetic uniqueness, that is, the number U(T) of samples whose most closely related sample diverged from it T generations ago (6). The fit of the model to both of these properties using the same parameters provides an additional verification of the model. The simulated tree is shown in Fig. 1.
Results and Discussion. Our simulation of a key property of the genealogical tree, the number of lineages as a function of time L(T), agrees with the genetic data over the full range of time. The simulation result is shown as a solid line in Fig. 2, and the genetic data are shown as circles. This curve consists of two parts, corresponding to the recent and deep parts of the tree. A sample should be a complete representation of the deepest part of the tree (4), and indeed the deep (T/T0 > 50) part of the genetic data matches the scaling expected for the full population (dashed line in Fig. 2a). Because this scaling behavior is robust and not sensitive to the details of the model, we can extrapolate this part of the curve to a particular resolution to estimate the total diversity at that resolution. At the level of resolution of the genetic data (T/T0 = 1), on the order of 1,000 genotypes could be distinguished. Although the short-term part of the L(T) curve may vary depending on details of reproduction, simulations show that the amount of variability is similar to the size of natural fluctuations in diversity (6).
At recent times, a sample should underrepresent the tree of the whole population. Indeed, L(T) is lower than the scaling result at short times. The degree of underrepresentation depends on the spatial structure of the population. Although a simulation of a well mixed population matches the data from global samples, it does not match a subsample of isolates from California (squares in Fig. 2b). The diversity of the California samples is lower than it would be if the population were well mixed. A spatial simulation of the ancestry of the California samples alone, using the same parameters as for the full simulation, does match the data for the California samples, confirming the importance of spatial structure and validating the choice of a spatial (as opposed to a well mixed) model. Because the long-time tail of both the global and California simulations match, both sample sets give the same estimate of total diversity, supporting the validity of using a sample to determine the total diversity.
This method can be used to estimate the diversity of a population when there are enough samples to determine that the power-law scaling of L(T) holds. The example of Pseudomonas suggests that this is possible for remarkably few samples. We note that this method is not applicable to populations that have been exponentially growing for much of their histories, because this growth effectively “cuts off” the deep part of the tree that would follow the scaling behavior.
Dependence of Diversity on Habitat Area
To determine the implications of our results for conservation, we studied the dependence of genetic diversity on habitat area. Biological diversity has often been quantified by using the number of species, and field studies show that species diversity, S, increases with area, A (the species–area relationship), as S ∼ Az, with z typically 0.25 and ranging from 0.15 to 0.4 on intermediate scales but possibly closer to 1 on large scales (15). This scaling has been modeled theoretically (15, 23). A key difference between species and genetic diversity is that the former treats all species as equally distinct, not considering the degree to which species are different from each other. This is also the case for measures of genetic diversity that count types, such as allelic diversity (24). The measure used here [branch length or segregating sites (17)] counts mutations along a lineage that make a descendant progressively more different from its ancestor and relatives.
Methods. We use the genealogical model described above, and analytically derive and simulate the scaling dependence of the total branch length of the genealogical tree for spatial populations from L(T) (see Box 1: Analytic Results).
Results and Discussion. The diversity of a population in a two-dimensional habitat scales as
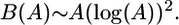 |
Thus, B grows somewhat faster than area, by a slowly increasing factor of log2(A). Still, this implies genetic diversity increases faster with area than does species diversity. Fig. 3 shows that this function well approximates the average diversity of a simulated population.
Fig. 3.

The dependence of diversity on habitat area. Shown is the average branch length, B, of the genealogical tree of a two-dimensional population simulated for 500,000 generations as a function of habitat area, A (squares), compared with the analytic result, B = A(log(A))2 (solid line). For comparison, the scaling S ∼ A0.25 typically observed for species counts, S, as a function of sample area is plotted (dashed line).
A number of habitats are topologically one-dimensional, such as river banks and coastal or tidal zones (25), as long as the width of the habitat is not much more than the dispersal distance. In one dimension, the branch length scales as
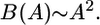 |
Thus, B grows much faster than length or population size; it quadruples when length or population size is doubled. This is significantly different from well mixed populations, whose diversity scales as N log N, where N is the population size (17). River banks and coastlines are often fractals; for species that live in such restricted habitats but disperse by distance in two dimensions (such as airborne dispersal of seeds of riverbank-dwelling plants), we expect an intermediate scaling between that for one and two dimensions (26).
The diversity–area relationship is essentially the same in sexual and asexual populations. For the same per-genome mutation rate, the number of mutations is unaffected by recombination. Selection can affect the results in two ways. A high frequency of advantageous mutations can reduce diversity by wiping out the existing diversity before an equivalent amount of diversity has time to develop in the mutant's descendants (see the supporting information). [This is known as periodic selection or genetic hitchhiking (27, 28).] Deleterious mutations (known as background mutations) only occur in a bounded subset of the population. With a fixed rate of occurrence, they contribute to diversity in proportion to the population size and thus the habitat area, a weaker scaling than those obtained above. They may reduce the effective population size for neutral mutations (29); this does not affect the scaling dependence.
Our analysis, confirmed by simulations, shows that these scaling results are robust to many possible changes in the model as long as the dispersal distance is significantly smaller than the habitat size. Incorporating frequent long-range dispersal causes a transition to the well mixed behavior. Still, even in this case, diversity grows somewhat faster than area. Our results support the growing recognition that loss of habitat causes a dramatic loss of diversity. Indeed, while the observed number of species scales as a weak power of the area, so that a reduction in area by a factor of 16 causes a reduction in number of species by a factor of 2 (15), the effect on genetic diversity is much more dramatic. A reduction in area by a factor of 2 causes a reduction by more than a factor of 2, and the effect is even more dramatic in one-dimensional habitats where three-quarters of the diversity is lost.
Box 1: Analytic Results
Number of Lineages. L(T), the expected number of ancestors of the current generation living T generations in the past, or their density p(T), can be calculated based on the characteristics of the coalescent process of random walkers (20) representing lineages. A conceptual argument, which gives the leading scaling result in one and two dimensions, is that a random walk travels a characteristic length l(T) ∼ T1/2 in a time T. If the random walk were to coalesce with all of the others in an area of this radius, we would be left with one lineage for each part of the habitat of area l(T)d, where d is the dimensionality. This gives a density p(T) ∼ 1/Td/2. Logarithmic corrections are important in two dimensions (see the supporting information).
Diversity–Area Relationship. We obtain the total branch length B of the genealogy of the population by summing up the number of lineages L(T) over all generations to the expected time TA of the most recent common ancestor of the population. Thus,
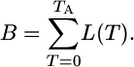 |
If the rate of mutation is sufficiently high, such that multiple mutations can occur at a single locus within the tree of the currently living individuals, a correction is necessary: D(B) ≅ μ/μL(1 – e–μ0B), where D(B) is the total diversity, B is the total branch length of the tree, and μ0 is the probability of a particular mutation arising at a particular locus. [This is akin to the Jukes–Cantor correction for estimating the divergence time between two sequences, but in reverse (30).] TA is the expected time at which there is only a single lineage and is obtained by setting L(TA) = 1. In two dimensions, L(T) scales as A log(T)/T, where area A is the number of lattice sites available to the population. This implies that TA scales as A log(A) [this can be shown by insertion into the expression A log(TA)/TA ∼ 1 and neglecting log(log(A)) compared with log(A)], giving
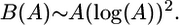 |
In one dimension, 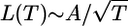 . The expected time of the most recent common ancestor, TA, scales as A2; therefore, the branch length scales as
. The expected time of the most recent common ancestor, TA, scales as A2; therefore, the branch length scales as
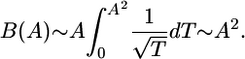 |
Supplementary Material
Acknowledgments
We thank Stephen Hubbell, Stuart Pimm, John Wakeley, Mehran Kardar, and Charles Goodnight for valuable comments and Jae-Chang Cho and James Tiedje for valuable comments and for providing the original figure with data from ref. 21. This work was supported, in part, by National Science Foundation Grant 0083885.
Author contributions: E.M.R. and Y.B. designed research, performed research, contributed new reagents/analytic tools, analyzed data, and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hudson, R. R. (1990) Oxford Surv. Evol. Biol. 7, 1–44. [Google Scholar]
- 2.Barton, N. H. & Wilson, I. (1995) Philos. Trans. R. Soc. London B 349, 49–59. [DOI] [PubMed] [Google Scholar]
- 3.Notohara, M. (2001) J. Appl. Prob. 38, 1–17. [Google Scholar]
- 4.Wakeley, J. & Aliacar, N. (2001) Genetics 159, 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wakeley, J. & Takahashi, T. (2003) Mol. Biol. Evol. 20, 208–213. [DOI] [PubMed] [Google Scholar]
- 6.Rauch, E. M. & Bar-Yam, Y. (2004) Nature 431, 449–452. [DOI] [PubMed] [Google Scholar]
- 7.Faith, D. P. (1994) Biol. Conserv. 68, 69–74. [Google Scholar]
- 8.Amos, W. & Balmford, A. (2001) Heredity 87, 257–265. [DOI] [PubMed] [Google Scholar]
- 9.Altizer, S., Harvell, D. & Friedle, E. (2003) Trends Ecol. Evol. 18, 589–596. [Google Scholar]
- 10.Keller, L. F. & Waller, D. M. (2002) Trends Ecol. Evol. 17, 230–241. [Google Scholar]
- 11.Frankham, R., Lees, K., Montgomery, M. E., England, P. R., Lowe, E. H. & Briscoe, D. A. (1999) Anim. Conserv. 2, 255–260. [Google Scholar]
- 12.Wright, S. (1968) Evolution and the Genetics of Populations (Univ. of Chicago Press, Chicago), Vol. 1.
- 13.Avise, J. C. (2000) Phylogeography: The History and Formation of Species (Harvard Univ. Press, Cambridge, MA).
- 14.Epperson, B. K. (1993) Evol. Biol. 27, 95–155. [Google Scholar]
- 15.Rosenzweig, M. L. (1995) Species Diversity in Space and Time (Cambridge Univ. Press, Cambridge, U.K.).
- 16.Kuhner, M. K., Yamato, J. & Felsenstein, J. (1995) Genetics 140, 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watterson, G. A. (1975) Theor. Popul. Biol. 7, 256–276. [DOI] [PubMed] [Google Scholar]
- 18.Wang, J. L. & Caballero, A. (1999) Heredity 82, 212–226. [Google Scholar]
- 19.Fisher, R. A. (1930) Proc. R. Soc. Edinburgh 50, 205–220. [Google Scholar]
- 20.Bramson, M. & Griffeath, D. (1980) Z. Wahrscheinlichkeitstheorie 53, 183–196. [Google Scholar]
- 21.Cho, J. C. & Tiedje, J. M. (2000) Appl. Environ. Microbiol. 66, 5448–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neigel, J. C. & Avise, J. C. (1993) Genetics 135, 1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hubbell, S. P. (2001) The Unified Neutral Theory of Biodiversity and Biogeography (Princeton Univ. Press, Princeton).
- 24.Maruyama, T. (1970) Ann. Hum. Genet. 34, 201–219. [DOI] [PubMed] [Google Scholar]
- 25.Clausing, G., Vickers, K. & Kadereit, J. W. (2000) Mol. Ecol. 9, 1823–1833. [DOI] [PubMed] [Google Scholar]
- 26.O'Shaughnessy, B. & Procaccia, I. (1985) Phys. Rev. Lett. 54, 455–458. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan, N. L., Hudson, R. R. & Langley, C. H. (1989) Genetics 123, 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majewski, J. & Cohan, F. M. (1999) Genetics 152, 1459–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charlesworth, B., Morgan, M. T. & Charlesworth, D. (1993) Genetics 134, 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jukes, T. H. & Cantor, C. R. (1969) in Mammalian Protein Metabolism, ed. Munro, H. N. (Academic, New York), pp. 21–132.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}