

Abstract
Models of rheumatoid arthritis (RA) in laboratory animals are important tools for research into pathogenic mechanisms and the development of effective, safe therapies. Rodent models (rats and mice) have provided important information about the pathogenic mechanisms. However, the evolutionary distance between rodents and humans hampers the translation of scientific principles into effective therapies. The impact of the genetic distance between the species is especially seen with treatments based on biological molecules, which are usually species-specific. The outbred nature and the closer anatomical, genetic, microbiological, physiological, and immunological similarity of nonhuman primates to humans may help to bridge the wide gap between inbred rodent strain models and the heterogeneous RA patient population. Here we review clinical, immunological and pathological aspects of the rhesus monkey model of collagen-induced arthritis, which has emerged as a reproducible model of human RA in nonhuman primates.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease of unknown etiology [1,2]. Once established, immune reactions against joint components contribute significantly to the pathological hallmarks of the disease, being synovial hyperplasia (pannus formation) and a variable degree of destruction and remodeling of joint cartilage and bone. RA affects approximately 1% of people in Western countries, with a 2:1 prevalence in females over males. The ageing societies in the developed countries create a growing need for safer and more effective therapies to treat chronic diseases such as RA. The advent of biotechnology has fuelled the search for drugs that act more specifically to overcome the considerable side effects of nonspecific anti-inflammatory and immunosuppressive drugs. Especially for immune-mediated diseases, biotechnology-based therapies have a great therapeutic potential. The preclinical development of immunomodulatory compounds often begins with an observation in vitro, after which proof of therapeutic principle is obtained in animal models, usually in inbred strains of rats or mice.
Unfortunately, the promising effects of new therapeutics observed in rodents are often not reproduced on testing in patients. There is a growing awareness that the evolutionary gap between inbred rodent strains and the human population is too wide for direct translation of data from rodents to humans [3]. Because of the closer evolutionary and immunological proximity to humans, nonhuman primates may help to bridge this gap [4-6]. Trans-species antigen presentation of human antigen-presenting cells to rhesus T cells and vice versa [7,8] nicely illustrates the immunological proximity of rhesus monkeys and humans [9-11].
It is of critical importance for preclinical safety testing that the selected animal model is sensitive to the pharmacological action of the tested drug and that the tissue distribution and pharmacological properties of the molecules targeted by the treatment are comparable to those observed in patients [12]. Parallel to the advent of biotechnology in recent decades, the interest in nonhuman primate models of human disease, in which highly specific new treatments can be tested, has increased. It is remarkable that whereas in transplantation research nonhuman primates are considered an essential preclinical model in the development of new therapies, the selection of therapies for a chronic disease such as RA relies mainly on inbred rodent models [6]. Many new therapeutic reagents, such as antibodies, cytokines, and cytokine antagonists but also more specifically acting small molecules, are active only in humans and some closely related nonhuman primate species.
Nonhuman primates spontaneously develop several of the arthritic diseases that affect the human population [9,13]. However, spontaneous manifestations of arthritis in a large outbred population of rhesus monkeys (>1,000 individuals) kept at the Biomedical Primate Research Centre in Rijswijk (the Netherlands) are rare. The low incidence and unpredictable nature of spontaneous arthritis prompted us to develop a model that can be induced at will and that is suitable for testing new therapies for safety and efficacy.
Arthritis models in nonhuman primates
Initial attempts were aimed at the reproduction of well-established arthritis models in rats and mice, to test whether these were experimentally feasible and would be compatible with ethical and practical standards. Widely used models, such as streptococcal-cell-wall-induced or mycobacterium-induced reactive arthritis in Lewis rats, could not be reproduced in rhesus monkeys [14].
A frequently used model of joint inflammation in rodents is antigen-induced arthritis (AIA). In a preliminary experiment, intra-articular injection of methylated ovalbumin (OVA) into OVA-sensitized rhesus monkeys induced macroscopic arthritis in one of two monkeys (MPM Vierboom, personal observation). The AIA model may provide a useful model, causing less discomfort to the animals than systemic polyarthritis, for the assessment of the immunogenic properties of new products to assist in the repair of the joint under local inflammatory conditions or therapeutics that are administered locally to suppress inflammation.
The clinical expression of arthritis induced by collagen type II (CII) in rodent strains is strongly influenced by their genetic background [15-17]. Immunization with heterologous CII induces reproducible autoimmune-mediated arthritis in a variety of genetically susceptible strains of mice and rats and in macaques [18,19]. Interestingly, immunization with bovine CII induced spondylitis without joint involvement in Buffalo rats (RT1b), while Wistar rats (RT1u) developed chronic joint inflammation without marked involvement of the spinal column. The F1 offspring of both strains developed inflammation at both locations (B 't Hart, personal observation).
While inbred rodent strains are genetically uniform and essentially represent a single individual in an outbred population, an outbred colony of rhesus macaques more closely resembles the human population in its heterogeneity. Predictably, the incidence and clinical presentation of collagen-induced arthritis (CIA) in a random sample (more than 50) of the large, outbred rhesus monkey colony at our institute (more than 1,200 animals) appeared heterogeneous, as is observed for human RA. In about half of randomly selected animals from the outbred colony of genetically typed rhesus monkeys at our institute, CIA could be induced. On the basis of these data, the CIA model in rhesus monkeys was further developed as a preclinical model of human RA.
The rhesus monkey model of collagen-induced arthritis
CIA is induced in rhesus monkeys by immunization with 3 to 5 mg bovine collagen type II (b-CII) dissolved in 0.5 ml 0.1 M acetic acid and emulsified in an equal volume of complete Freund's adjuvant (CFA). This emulsion is injected into the dorsal skin, distributed over 10 spots to reduce the formation of ulcerative skin lesions.
The time of onset and severity of clinical signs varies, most likely reflecting the outbred nature of the colony. Whereas RA susceptibility, more precisely the disease severity, in the human population maps to the major histocompatibility complex (MHC) class II alleles HLA-DR1 to HLA-DR4, no apparent association with the MHC class II region has been observed in rhesus monkeys thus far. This was rather unexpected, because 'shared epitopes' that confer a high risk to RA (QKRAA and QRRAA) are present at the correct location in several Mamu-DRB1 alleles (see the IPD-MHC sequence database [20]). In contrast, a strong influence of the MHC class I region on the susceptibility to CIA was found. Young animals (of Indian origin) from our colony that were positive for the Mamu-A26 serotype appeared completely resistant to the disease even after several booster immunizations. This resistance may be age dependent, since Mamu-A26+ monkeys more than 20 years old developed CIA, though the disease was less severe than in animals lacking this marker. Furthermore, the resistance is specific for the immunizing antigen, since Mamu-A26+ and Mamu-A26- monkeys are equally susceptible to experimental autoimmune encephalomyelitis (EAE) induced with human myelin basic protein (Table 1). Selection of A26- monkeys thus allows reproducible induction of CIA in >95% of animals. Furthermore, the Mamu-A26-associated CIA resistance was mainly observed in rhesus monkeys of Indian origin (B 't Hart, RE Bontrop, personal observation). We recently found that the A26 serotype defines a region configuration encoding multiple Mamu-B molecules, which has been renamed Mamu-B26 [21]. Hence it is possible that this resistance is defined by a particular combination of MHC class I molecules or by a closely linked gene. A gender bias as observed in humans for the risk of developing RA was not found for CIA in rhesus monkeys, although a prevalence in females has been found in the closely related cynomolgus macaque.
Table 1.
Young Mamu-A26+ rhesus monkeys are sensitive to collagen-induced arthritis
| Collagen-induced arthritis | Experimental autoimmune encephalomyelitis | |||
| Serotype | Positive | Negative | Positive | Negative |
| A26+ | 2 | 9 | 6 | 5 |
| A26- | 25 | 3 | 9 | 3 |
| P < 0.0001 | P < 0.31 | |||
Biomarkers for inflammation and joint destruction
Several surrogate markers for CIA have been developed, which reflect different pathological aspects of the model, that is, markers for inflammation, bone degradation, and clinical wellbeing. These markers help to determine the therapeutic efficacy of a new therapy. Consistent improvement of a biomarker in the experimental group versus a control group without a direct clinical effect can nevertheless indicate a therapeutic effect. The relation between various biomarkers, the clinical manifestation of arthritis in the model, and the response to treatment is illustrated by data collected over the past decade.
Serum CRP as a biomarker of CIA severity
The serum level of C-reactive protein (CRP), an acute-phase protein produced in the liver under conditions of systemic inflammation, is increased in patients with clinically active RA [22]. A low but significant increase of serum CRP concentration can be observed years before the onset of RA symptoms [23]. This protein is a very useful marker of inflammation and a potential biomarker for the anti-inflammatory effect of a new therapy, because the half-life remains unchanged under conditions of health and disease. Moreover, the serum CRP concentration directly reflects the intensity of the pathological process. After a small increase of CRP in the first week after CIA induction without clinical signs, a second increase is observed in CIA-sensitive animals, which is more pronounced and precedes the onset of clinical arthritis. In the CIA model, CRP is elevated before macroscopic clinical signs are observed and can be used as an early marker for disease onset. On the basis of the CRP pattern, three types of CIA responders are discerned in an outbred animal group: group I animals (n = 6) are early responders, showing an increase in CRP above an arbitrary threshold of 50 mg/l before day 14. Group II animals (n = 12) are moderate responders, showing an increase in CRP above 50 mg/l between days 14 and 21. The third group (group III; n = 13) are late responders, showing an increase in CRP of more than 50 mg/l between days 21 and 35. The highest peak CRP levels were observed in the early responders (Fig. 1, top panel, squares); the lowest, in the late responders (circles).
Figure 1.
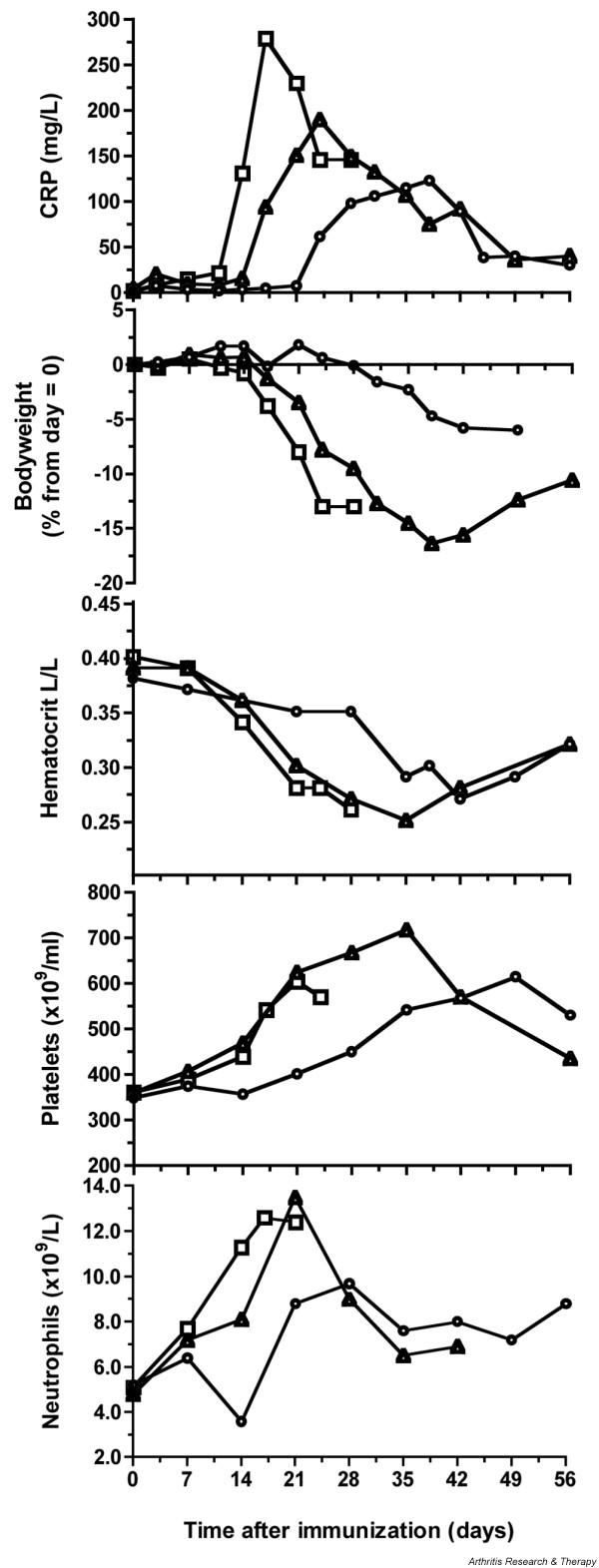
Clinical and hematological markers of collagen-induced arthritis. (Top panel) Early responders to challenge (group I, square points) demonstrated a sharp and larger increase in C-reactive protein (CRP) than was observed for intermediate and late responders (respectively, group II, triangles; and group III, circles). (Panel 2) Animals with an early increase in CRP showed an early weight loss. Group III showed only a minor weight loss after day 28. The hematocrit values (panel 3) were decreased during periods of active inflammation, while platelets (panel 4) and neutrophils (panel 5) were increased Each data point in the graphs represents a mean value for at least 3 animals for group I (n = 6) and 5 animals for groups II and III (n = 12 and n = 13, respectively). Squares, early responders (group I); triangles, intermediate responders (group II); circles, late responders (group III).
Body weight as a general disease marker
Early CIA responders display a rapid weight loss between days 14 and 28 (Fig. 1, panel 2). In early experiments, those monkeys that were not humanely killed at the height of the disease showed, after a disease episode of variable length, a body weight increase that was associated with remission of clinical signs of arthritis, such as pain or apathy. Hence, body weight is a useful objective biomarker of the general disease status.
Hematological and chemical markers of disease
Neutrophils, platelets, hematocrit
Once a week, a complete hematological and serological analysis is performed, which provides additional information on the disease status and the general physical condition. An increase of platelets and neutrophils marks episodes of active inflammation (Fig. 1, panels 4,5). Furthermore, active periods of the disease are associated with decreased hematocrit values (Fig. 1, panel 3).
Albumin and alkaline phosphatase alkaline phosphatase
The production of the serum protein albumin is affected by the induction of acute-phase proteins such as CRP (Fig. 2, top panel). Predictably, serum levels of albumin are found to be decreased during active joint inflammation (Fig. 2, panel 2). Alkaline phosphatase (AP) is a marker for the evaluation of bone metabolism. AP is mainly produced in the liver and by osteoblasts. When liver enzymes are unaltered, changes in AP can be indicative of increased bone metabolism as a consequence of ongoing destructive joint erosion (Fig. 2, panel 3).
Figure 2.
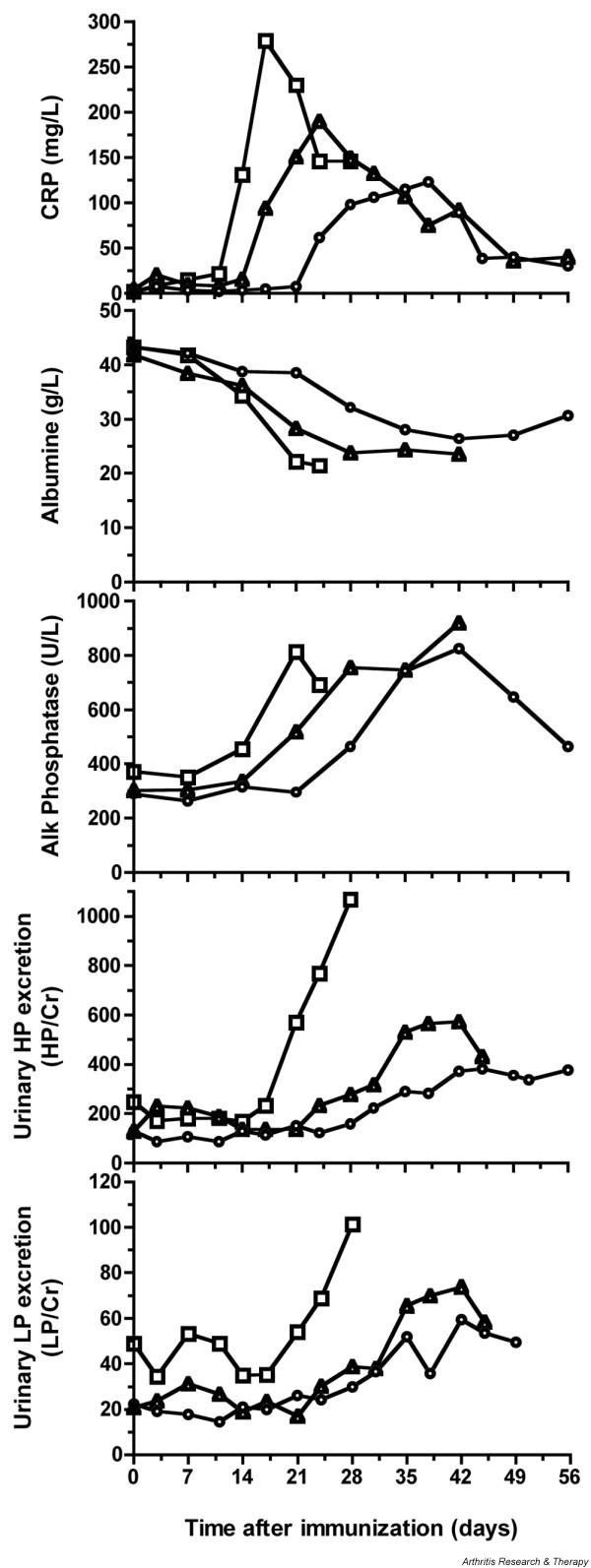
Serological and urinary markers of collagen-induced arthritis. (Panel 2) The serum albumin concentration was negatively correlated with the production of acute-phase proteins such as C-reactive protein (CRP) (top panel). (Panel 3) Alkaline phosphatase (AP) is mainly produced by liver and osteoblasts. When liver function is normal, changes in AP can be indicative of bone remodeling processes as a result of bone degradation. (Panels 4,5) Increased urinary excretion rates of hydroxylysylpyridinoline (HP) and lysylpyridinoline (LP) cross-link products, expressed relative to creatinine (Cr), were observed during the active phase of the disease. Each data point in the graphs is a mean of at least 3 animals for group I (n = 6) and 5 animals for groups II and III (n = 12 and n = 13, respectively). Squares, early responders (group I); triangles, intermediate responders (group II); circles, late responders (group III).
Urinary excretion rates of collagen cross-links as a biomarker of joint erosion
Joint tissues contain different quantities of the major cross-links hydroxylysylpyridinoline (HP) and lysylpyridinoline (LP), which are degradation products of collagen contained in cartilage and bone and are excreted into the urine. Urinary excretion rates of these metabolites can therefore serve as biomarkers of joint destruction. About 95% of the cross-links in the joint cartilage of the rhesus monkey consists of HP (HP/LP ratio = 55), while the HP/LP ratio in bone is 3.8 [24]. As the excretion rate of the cross-link product varies during the day, urine samples for analysis were collected overnight and stored frozen at -20°C. Unhydrolyzed urine samples were used for the measurement of collagen cross-links with reverse-phase high-performance liquid chromatography (HPLC) essentially according to the method of Black and colleagues [25]. Increased excretion rates of HP and LP, expressed relative to creatinine, were observed during the active phase of CIA (Fig. 2, two lower panels). In particular, the excretion rates of HP were associated with CIA severity. A fivefold increase in the HP/Cr ratio relative to baseline values (from about 200 to 1,000) was observed in early responders (group I). The LP excretion rate followed the same course but increased only twofold (from about 45 to 100), suggesting a prominence of cartilage degradation. The HP excretion rate correlated with the number of affected joints per animal in each group. In the early responders, the mean number of affected joints was approximately 26. It was lower, (approximately 16) in group II, and lowest (10) in group III.
Immunological evaluation: collagen specific IgM and IgG
A clear contribution of autoantibodies to the immunopathogenesis of CIA was found in arthritic animals. The resistance to CIA observed in Mamu-B26 (formerly A26)-positive animals is most likely associated with the failure to produce adequate levels of immunoglobulin (Ig)M antibodies against the immunizing antigen [26,27]. Interestingly, CIA-resistant animals mount a normal collagen-specific IgG response, both quantitatively and qualitatively, that is, reactivity profile with epitopes in the CB11 fragment of collagen [28]. Unpublished data indicate that the subclass of anticollagen IgG antibodies in resistant monkeys may resemble human IgG4, which does not efficiently fix complement. As the IgG4-like antibodies bind to the same epitopes as the complement-fixing IgG1/3-like antibodies in CIA-susceptible animals, these IgG4-like antibodies may protect the joint cartilage against opsonization.
Histology of CIA-affected joints
For routine histology, the patellae of both knee joints and the proximal interphalangal and distal interphalangeal joints of the third digit of the hand and foot were processed and analyzed. Figure 3 shows different phases of the destructive erosion of cartilage and subchondral bone in an arthritic finger joint. We use the pathology grading system published by Pettit and colleagues [29]. This system quantifies the degree of inflammation, bone destruction, and cartilage degradation on an arbitrary scale from 0 to 5.
Figure 3.
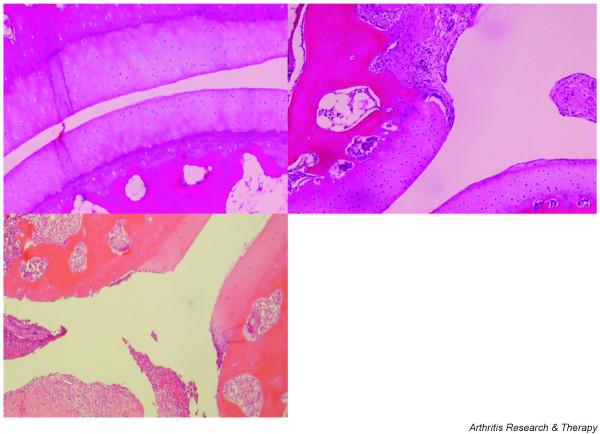
Histology of a proximal interphalangeal joint, showing phases of the degenerative process in CIA. (Top left) A healthy, unaffected joint in a rhesus monkey with collagen-induced arthritis (CIA) shows intact cartilage and no marked activity of the subchondral bone. (Top right) Joint destruction starts where the synovium overgrows the cartilage. A hyperplastic synovium resulting in pannus formation produces factors such as cytokines and matrix metalloproteases mediating the destruction of the cartilage. (Bottom left) In the late phase of the disease, cartilage can be completely eroded and also the underlying bone can be seriously damaged.
The arthritic joints of CIA-affected rhesus monkeys display essentially the same histopathological hallmarks as RA joints. In the early phase of active CIA, hyperplasia of the synovium and pannus formation were already observed [30]. These preceded the dramatic destruction of cartilage and bone in advanced CIA (Fig. 3, bottom left).
Ethical management
The ethical management of the rhesus monkey CIA model relies on a semiquantitative clinical scoring system that represents the overall disease status of the animals (Table 2). Clinical signs that were monitored daily are body weight, body temperature, and the amount of pain relief used. Macroscopic signs of inflammation, that is, the number of joints showing soft-tissue swelling, warmth, and redness, were recorded twice weekly. Medication to minimize the discomfort during the experiment was given at the indication of the institute's veterinarians. Pain relief medication consisted of buprenorphine (Temgesic; an opiate). Ulcerative skin lesions developing at the immunization sites were sprayed daily with disinfectant wound spray (Acederm) to prevent further contamination.
Table 2.
Scheme for clinical and ethical management of rhesus monkeys with collagen-induced arthritis
| Disease score | Characteristics | Monitoring | Maximal durationa |
| 0 | No disease symptoms | Daily | Length of experiment |
| 0.5 | Fever (>0.5°C) | 2 × per week | 12 weeks |
| 1 | Apathy; lessened mobility; loss of appetite | Daily | 10 weeks |
| 2 | Weight loss; warm extremitiesb; treatable pain without STS | 2 × per weekc, or daily | 6 weeks |
| 3 | Redness of joints (with STS)b; normal flexibility of extremities | 2 × per week | 4 weeks |
| 4 | Severe STS of joints (plus redness); joint stiffnessb | 2 × per week | 2 weeks |
| 5 | Untreatable pain; immobility of jointsb; weight loss >25% | 2 × per weekc, or daily | 18 hours |
aThe duration of discomfort is calculated cumulatively. bCan be assessed only in the sedated monkey and therefore cannot be done more than twice a week for ethical reasons. cFor characteristics requiring sedation. STS, soft-tissue swelling.
Prophylactic treatment with a promising compound can result in a marked reduction of the clinical score, signifying improved clinical wellbeing, as was recently described for a low-molecular-weight CCR5 antagonist [31].
All these markers can be used to evaluate various aspects of the disease, allowing us to differentiate between disease-modifying drugs affecting bone degradation or therapies affecting inflammation.
Pathogenic mechanisms in CIA
The variety of intervention studies performed in the past decade have provided insight into the pathogenic mechanisms operating in the rhesus monkey CIA model. As proposed for RA by Choy and Panayi [1], we like to distinguish three phases in the etiopathogenesis of the CIA model (Fig. 4).
Figure 4.
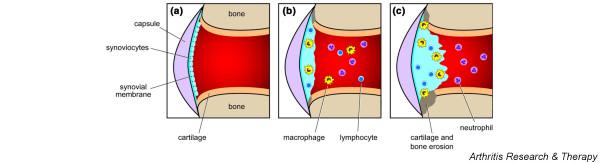
Schematic presentation of immune factors in the arthritic process. (a) A healthy joint. The histology of a healthy rhesus monkey diarthrodial joint resembles that of humans, namely, a thin synovium lining the synovial cavity and a rather thick layer of hyaline cartilage covering the articular bone. No marked activity of the bone marrow is observed. (b) The early phase of collagen-induced arthritis (CIA). Synovitis with marked infiltration of T lymphocytes and macrophages is already present in clinically unaffected joints. (c) The late phase of CIA. We have little information about this stage of the disease, because monkeys are usually killed earlier, for welfare reasons.
Synovitis
As in RA, a hyperplastic synovium staining positively for CD3+ and CD68+ infiltrated cells can be found in joints lacking macroscopic signs of arthritis [30]. When the hyperplastic synovium is removed – for example, by intra-articular injection of thymidine-kinase-expressing adenovirus followed by gancyclovir infusion – joint inflammation is abolished [32]. This finding illustrates that similar to RA, CIA probably starts with synovitis.
Leukocyte infiltration
Histological analysis of arthritic joints has revealed the presence of several leukocyte subsets, such as T cells, B cells, macrophages, and neutrophils. Lymphocyte migration to the site of inflammation is directed by chemokines. Effector T helper type 1 (Th1) cells expressing chemokine receptors CCR5 and CXCR3 have been found enriched in synovial joints of RA patients. Ligands for both chemokine receptors are elevated in inflamed synovial tissue and synovial fluid [33-35]. A low-molecular-weight CCR5 antagonist that prevents the binding of its ligand, and hence the migration of these destructive T cells, was tested in the CIA model of rhesus monkeys. Prophylactic treatment with this compound resulted in diminished severity of disease compared with that in nontreated controls [31] and in better control of several disease markers, such as serum levels of CRP and body weight (Fig. 5a,b), serum albumin and alkaline phosphatase (Fig. 5c,d), and the urinary excretion rates of the collagen cross-link products HP and LP (Fig. 5e,f).
Figure 5.
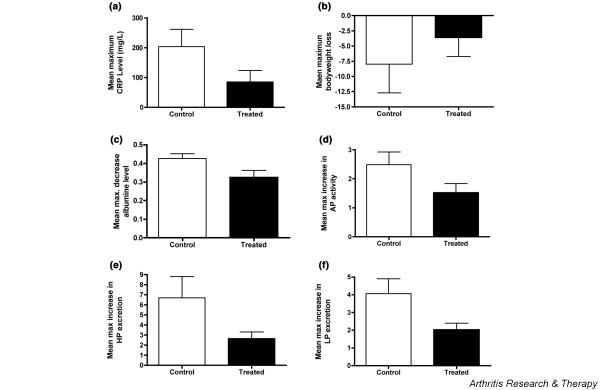
Effect of a CCR5 antagonist on clinical, inflammatory, and bone remodeling processes of collagen-induced arthritis (CIA). CIA was induced in 10 susceptible rhesus monkeys. One group of five animals received prophylactic treatment with the low-molecular-weight CCR5 antagonist SCH-X twice daily for 45 days by intramuscular injection, while a second group of five animals received saline solution for the same period. Treatment was started on the day of CIA induction. Results are expressed as the mean maximum change (± standard deviation), which was deduced from the highest measured increase or decrease of the depicted parameter relative to the start of treatment. The figure shows a significant effect (Student's t-test; P < 0.05) of SCH-X treatment on serum levels of C-reactive protein (CRP) (a), albumin (c), and alkaline phosphatase (AP) (d), as well as the excretion rates of hydroxylysylpyridinoline (HP) (e) and lysylpyridinoline (LP) (f). The effect on body weight (b) was not statistically significant. Histology confirmed the lower-joint destruction in the group treated with SCH-X (see ref [31]).
T cells
The role of T cells in the onset of CIA in rhesus monkeys was shown in two separate studies. Early treatment with ciclosporin A, a strong inhibitor of T-cell immunity, prevented the development of CIA [36]. However, treatment of animals during clinically active CIA had no effect on the disease. In a separate study we showed a beneficial effect of daclizumab, a humanized antibody directed against the Tac antigen on the IL-2 receptor α chain [37]. Both studies underline that T cells present in the early inflammatory synovium play an important role in the onset of arthritis. The poor proliferative response of rhesus monkey blood mononuclear cells to CII has hampered the generation of stable cell lines. Hence, the precise specificity analysis and MHC restriction of cellular autoimmune mechanisms could not be systematically evaluated.
Neutrophils
Activated neutrophils produce highly toxic reactive oxygen species that destroy tissue inhibitors of metalloproteases (TIMPs) and thus make the joint more vulnerable to metalloproteases [38]. Interestingly, early treatment of CIA-affected rats via the drinking water with the oxidative-burst antagonist apocynin protects against the arthritis but leaves T-cell (delayed-type hypersensitivity; DTH) or B-cell functions (serum antibodies) intact [39].
Autoantibodies
A newly emerging target of therapy is the B cell, for example using rituximab, a depleting antibody directed against CD20. Initially used for the treatment of B-cell lymphomas, this antibody has now proven effective in the treatment of RA [40]. That collagen-specific antibodies, in particular those of the IgM isotype, have a pivotal role in the rhesus monkey model of CIA appears from two findings. The absence of anti-CII IgM production in CIA-resistant monkeys is highly suggestive of a causal link [27]. This is supported by the observation that monkeys and rats presensitized with CII, in which conformational B-cell epitopes had been destroyed by heating, are protected against CIA [41]. Control animals, which had been presensitized with albumin and subsequently immunized with native CII in CFA, developed CIA and produced normal anti-CII IgM and IgG antibody levels. However, the protected animals failed to produce IgM antibodies, but produced normal levels of anti-CII IgG antibodies.
The fine specificity of anti-CII IgG was determined by analyzing the reactivity of immune sera from CIA-susceptible and resistant monkeys with synthetic peptides based on the CB11 fragment of bovine CII [28]. Sera from both groups reacted with the same peptides, including peptide 260–273, which contains a dominant T-cell epitope in murine CIA.
How can the role of IgM antibodies be explained? Most binding sites of anti-CII antibodies on the surface of intact human articular cartilage are protected by proteinaceous material from the synovial fluid. This layer can be removed by neutrophil elastase digestion. We believe that the CII epitope density on the intact cartilage surface is too low for classical-route complement fixation by bound anti-CII IgG antibodies. However, surface binding with one of the five available antigen-binding sites of an anti-CII IgM molecule is sufficient for complement fixation, and neutrophil binding via Fc receptor and/or C3 receptor can take place. Erosion of the cartilage surface under the influence of neutrophil elastase enhances the exposure of antibody-binding sites on collagen and other cartilage antigens. It can thus be envisaged that IgG antibodies can enhance inflammation and degradation of an already affected joint [42,43].
Interferon β
The current state-of-the-art biological treatment in RA is with inhibitors of proinflammatory cytokines such as TNF-α and IL-1. We have not directly tested antagonists of these cytokines in the model, as treatments that have already been approved for use in patients are usually not tested in nonhuman primates. However, we have tested mammalian-cell-derived IFN-β. This interferon inhibits the production or the effects of proinflammatory cytokines such as IL-12. It also inhibits the secretion of TNF-α and exerts a variety of immunomodulatory effects, which underlie the therapeutic benefit in multiple sclerosis [44].
We have tested recombinant human IFN-β (REBIF®, Ares-Serono, Geneva, Switzerland) as a treatment for CIA in four rhesus monkeys. At the tested dose of 107 units per day administered via subcutaneous injection during one week, the cytokine showed a clear beneficial effect on clinically manifest arthritis in two monkeys and abolished arthritis in one monkey [45]. A clinical trial in RA patients of fibroblast-derived IFN-β (Frone®, Ares-Serono) combined with methotrexate failed to reproduce the promising effects of IFN-β observed in the monkeys [46], but negative interaction of the two medications cannot be excluded.
Towards a treatment for chronic inflammation
In the later stages of the disease, the cartilage is severely damaged, requiring repair of the damage for restoration of function. A possible strategy to treat this condition is the introduction of a matrix that provides a scaffold for chondrocytes and that stimulates the regeneration of the cartilage. The main obstacle will be the introduction of such a matrix under inflammatory and destructive conditions. Conceptually, the regenerating substrate not only provides a scaffold for rebuilding the cartilage but also provides immunomodulatory signals that help to restore homeostatic mechanisms maintaining tolerance to joint antigens. Implantation of a tolerogenic collagen matrix, for example, would obviate the need for further immunosuppressive regimens.
A still-unresolved question is why the inflammation in RA is chronic. We have postulated a model in which the dendritic cell (DC) plays a pivotal role in governing the response to released self-antigens. The reasoning is that DC maturation is regulated by the interaction of C-type lectin receptors (CLRs), binding glycan epitopes on self-antigens and pathogens, and Toll-like receptors (TLRs), which recognize conserved molecular patterns on pathogens [47]. This notion has led to the Yin-Yang hypothesis for the regulation of autoimmunity and tolerance by DCs [48]. In the concept, testable hypotheses were formulated for the maintenance of tolerance to self-antigens in a resting immune system and the induction of reactive or chronic inflammation by infection. The three depicted paradigms mentioned below describe extreme situations. In clinical reality, subtle variants may occur.
One paradigm, the tolerance paradigm, postulates that in a resting immune system, immature DCs present in lymhoid organs, and their equivalents in the joint, such as the type A synovial lining cells, continuously sample glycoproteins released from the normal tissue turnover via their CLRs. When presented in the absence of DC maturation signals, T cells recognizing the presented glycoproteins attain a regulatory function. By this mechanism, the nonresponsiveness of the immune system is maintained. This mechanism may explain why a pig collagen matrix implanted into the knee joint of a healthy rhesus monkey does not evoke anticollagen autoimmunity (our own unpublished observations). In addition, the robust tolerance that is induced when rhesus monkeys are pretreated with attenuated collagen in incomplete adjuvant – that is, in the absence of a TLR ligand – may be explained by this model. Interestingly, the observation that rats injected with T cells from rats immunized with attenuated CII produced lower anti-CII antibody levels suggests a regulatory function of the transferred cells [41].
The infection paradigm postulates that disturbance of the CLR/TLR balance by a viral or bacterial infection induces DC maturation. Consequently, self-antigens sampled by the DCs will be presented in the context of costimulatory signals expressed by mature DCs and induce Th1 cell activation. Immunohistochemical analysis of the arthritic joints of RA patients reveals that high quantities of the TLR2/Nod1,2 ligand peptidoglycan are present in the arthritic joint [49]. Peptidoglycan is arthritogenic by itself in susceptible rodent strains and can enhance a specific autoimmune reaction to central nervous system myelin, giving rise to monophasic autoimmune encephalomyelitis [50]. Importantly, when the TLR stimulus is cleared, the CLR/TLR balance is restored, and tolerance can be restored by the induction of regulatory T cells (Treg) specific for joint components. This paradigm could explain why in monkeys that have recovered from CIA it is almost impossible to induce exacerbation of the arthritis by a second immunization with bovine collagen type II [26,51].
Another paradigm is that of altered glycosylation. Glycosylation is the most important post-translational modification of secreted proteins, which are expressed on the cell membrane or part of the extracellular matrix. Glycosylation of self-glycoproteins is not constant, but varies with time (ageing) and place (organ-specific glycosylation). Moreover, the normal glycosylation patterns can change under pathological conditions that cause stress to cells, such as infection, or on exposure to certain hormones or cytokines [52,53]. The disturbance of the normal glycosylation under these conditions may affect the restoration of the delicate CLR/TLR balance after clearance of the TLR ligand and thus impair remission of the inflammation. RA is one of the clinical disorders in which abnormal glycosylation of self-antigen (agalactosyl IgG) has been suggested as a cause of autoimmunity [54]. It was shown in a mouse CIA model that the arthritogenic and tolerogenic capacity of CII depends on the glycosylation of the immunizing autoantigen. We hypothesize that the disturbed glycosylation may not be confined to IgG but may also affect other self-antigens in the joint, such as CII.
Future perspectives
CIA in rhesus monkeys is a very useful preclinical model of human arthritis, but it is suboptimal in a number of respects. First, the arthritis can be very severe. In addition, the large size of the monkeys, which weigh 6 to 10 kg at adulthood, is an advantage for blood collection or invasive techniques (arthroscopy). However, this implies that large amounts of test compound are required to observe a clinical effect. Another disadvantage of using CIA in rhesus monkeys is the aggressive nature of these monkeys, which usually have to be sedated for each handling, limiting the frequency of experimental interventions. Moreover, the model is usually monophasic, although in some cases exacerbation could be induced by booster immunization [26]. Finally, because of the high susceptibility to mycobacterial components of adjuvant, rhesus monkeys develop severe ulcerating skin lesions where the CII/CFA inoculum is injected.
We have encountered similar phenomena with the rhesus monkey EAE model, which was developed as a model of multiple sclerosis [37]. To overcome this problem, we have developed an EAE model in a New World nonhuman primate species, the common marmoset [55]. This is a small animal (weighing approximately 400 g), which is ideal for efficacy testing in an early development phase of a new drug, when only limited amounts of the compound are available. The new EAE model appeard to lack all the negative aspects of the rhesus monkey EAE models and to represent the human disease multiple sclerosis much better. A unique aspect of marmosets is that they normally give birth to nonidentical twins or triplets, which, because they share the placental bloodstream, are complete and stable bone marrow chimeras. The resulting allo-tolerance between twins allows the exchange of cells and tissues without rejection. We expect that the same differences observed for the EAE models also hold true in CIA and have started to explore the susceptibility of marmosets to arthritis induction with bovine collagen type II. Whether the success of the marmoset model for multiple sclerosis can be reproduced in RA will become clear in the coming years.
Abbreviations
AIA = antigen-induced arthritis; AP = alkaline phosphatase; CFA = complete Freund's adjuvant; CIA = collagen-induced arthritis; CII = collagen type II; CLR = C-type lectin receptor; CRP = C-reactive protein; DC = dendritic cell; EAE = experimental autoimmune encephalomyelitis; HP = hydroxylysylpyridinoline; HPLC = high-performance liquid chromatography; IFN = interferon; Ig = immunoglobulin; IL = interleukin; LP = lysylpyridinoline; MHC = major histocompatibility complex; OVA = ovalbumine; RA = rheumatoid arthritis; Th1 = T helper type 1; TIMP = tissue inhibitor of metalloproteases; TLR = Toll-like receptor.
Competing interests
The author(s) declare that they have no competing interests.
Acknowledgments
Acknowledgements
We would like to thank the long list of collaborators and funding bodies that have contributed to the development of the rhesus monkey CIA model and Mr Henk Westbroek for the artwork. We appreciate the helpful criticism by Dr Sandra Amor during the preparation of this manuscript.
References
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358:903–911. doi: 10.1016/S0140-6736(01)06075-5. [DOI] [PubMed] [Google Scholar]
- Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 't Hart B, Amor S, Jonker M. Evaluating the validity of animal models for research into therapies for immune-based disorders. Drug Discov Today. 2004;9:517–524. doi: 10.1016/S1359-6446(04)03112-5. [DOI] [PubMed] [Google Scholar]
- Bontrop RE. Non-human primates: essential partners in biomedical research. Immunol Rev. 2001;183:5–9. doi: 10.1034/j.1600-065x.2001.1830101.x. [DOI] [PubMed] [Google Scholar]
- Sachs DH. Tolerance: Of mice and men. J Clin Invest. 2003;111:1819–1821. doi: 10.1172/JCI200318926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 't Hart BA, Elferink DG, Drijfhout JW, Storm G, van Blooijs L, Bontrop RE, de Vries RR. Liposome-mediated peptide loading of MHC-DR molecules in vivo. FEBS Lett. 1997;409:91–95. doi: 10.1016/S0014-5793(97)00493-6. [DOI] [PubMed] [Google Scholar]
- Geluk A, Elferink DG, Slierendregt BL, van Meijgaarden KE, de Vries RR, Ottenhoff TH, Bontrop RE. Evolutionary conservation of major histocompatibility complex-DR/peptide/T cell interactions in primates. J Exp Med. 1993;177:979–987. doi: 10.1084/jem.177.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 't Hart BA, Losen M, Brok HPM, de Baets MH. Chronic diseases. In: Wolfe-Coote SP, editor. The Laboratory Primate. Amsterdam: Elsevier Science; [Google Scholar]
- Bontrop RE, Elferink DG, Otting N, Jonker M, de Vries RR. Major histocompatibility complex class II-restricted antigen presentation across a species barrier: conservation of restriction determinants in evolution. J Exp Med. 1990;172:53–59. doi: 10.1084/jem.172.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontrop RE, Otting N, de Groot NG, Doxiadis GG. Major histocompatibility complex class II polymorphisms in primates. Immunol Rev. 1999;167:339–350. doi: 10.1111/j.1600-065x.1999.tb01403.x. [DOI] [PubMed] [Google Scholar]
- Roth GS, Mattison JA, Ottinger MA, Chachich ME, Lane MA, Ingram DK. Aging in rhesus monkeys: relevance to human health interventions. Science. 2004;305:1423–1426. doi: 10.1126/science.1102541. [DOI] [PubMed] [Google Scholar]
- 't Hart BA, Vervoordeldonk M, Heeney JL, Tak PP. Gene therapy in nonhuman primate models of human autoimmune disease. Gene Therapy. 2003;10:890–901. doi: 10.1038/sj.gt.3302017. [DOI] [PubMed] [Google Scholar]
- Bakker NP, van Erck MG, Zurcher C, Faaber P, Lemmens A, Hazenberg M, Bontrop RE, Jonker M. Experimental immune mediated arthritis in rhesus monkeys. A model for human rheumatoid arthritis? Rheumatol Int. 1990;10:21–29. doi: 10.1007/BF02274777. [DOI] [PubMed] [Google Scholar]
- Holmdahl R. Genetics of susceptibility to chronic experimental encephalomyelitis and arthritis. Curr Opin Immunol. 1998;10:710–717. doi: 10.1016/S0952-7915(98)80093-9. [DOI] [PubMed] [Google Scholar]
- Holmdahl R. Dissection of the genetic complexity of arthritis using animal models. J Autoimmun. 2003;21:99–103. doi: 10.1016/S0896-8411(03)00096-9. [DOI] [PubMed] [Google Scholar]
- Holmdahl R, Andersson ME, Goldschmidt TJ, Jansson L, Karlsson M, Malmstrom V, Mo J. Collagen induced arthritis as an experimental model for rheumatoid arthritis. Immunogenetics, pathogenesis and autoimmunity. APMIS. 1989;97:575–584. doi: 10.1111/j.1699-0463.1989.tb00446.x. [DOI] [PubMed] [Google Scholar]
- Cremer MA, Townes AS, Kang AH. Collagen-induced arthritis in rodents. A review of clinical, histological and immunological features. Ryumachi. 1984;24:45–56. [PubMed] [Google Scholar]
- Yoo TJ, Kim SY, Stuart JM, Floyd RA, Olson GA, Cremer MA, Kang AH. Induction of arthritis in monkeys by immunization with type II collagen. J Exp Med. 1988;168:777–782. doi: 10.1084/jem.168.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immuno Polymorphism Database, Non-Human Major Histocompatibility Complex http://www.ebi.ac.uk/ipd/mhc/nhp/align.html
- Otting N, Heijmans CM, Noort RC, de Groot NG, Doxiadis GG, van Rood JJ, Watkins DI, Bontrop RE. Unparalleled complexity of the MHC class I region in rhesus macaques. Proc Natl Acad Sci USA. 2005;102:1626–1631. doi: 10.1073/pnas.0409084102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollheim FA. Markers of disease in rheumatoid arthritis. Curr Opin Rheumatol. 2000;12:200–204. doi: 10.1097/00002281-200005000-00007. [DOI] [PubMed] [Google Scholar]
- Nielen MM, van Schaardenburg D, Reesink HW, Twisk JW, van de Stadt RJ, van der Horst-Bruinsma IE, de Gast T, Habibuw MR, Vandenbroucke JP, Dijkmans BA. Increased levels of C-reactive protein in serum from blood donors before the onset of rheumatoid arthritis. Arthritis Rheum. 2004;50:2423–2427. doi: 10.1002/art.20431. [DOI] [PubMed] [Google Scholar]
- 't Hart BA, Bank RA, De Roos JA, Brok H, Jonker M, Theuns HM, Hakimi J, Te Koppele JM. Collagen-induced arthritis in rhesus monkeys: evaluation of markers for inflammation and joint degradation. Br J Rheumatol. 1998;37:314–323. doi: 10.1093/rheumatology/37.3.314. [DOI] [PubMed] [Google Scholar]
- Black D, Duncan A, Robins SP. Quantitative analysis of the pyridinium crosslinks of collagen in urine using ion-paired reversed-phase high-performance liquid chromatography. Anal Biochem. 1988;169:197–203. doi: 10.1016/0003-2697(88)90274-6. [DOI] [PubMed] [Google Scholar]
- Bakker NP, van Erck MG, Botman CA, Jonker M, 't Hart BA. Collagen-induced arthritis in an outbred group of rhesus monkeys comprising responder and nonresponder animals. Relationship between the course of arthritis and collagen-specific immunity. Arthritis Rheum. 1991;34:616–624. doi: 10.1002/art.1780340514. [DOI] [PubMed] [Google Scholar]
- Bakker NP, van Erck MG, Otting N, Lardy NM, Noort RC, 't Hart BA, Jonker M, Bontrop RE. Resistance to collagen-induced arthritis in a nonhuman primate species maps to the major histocompatibility complex class I region. J Exp Med. 1992;175:933–937. doi: 10.1084/jem.175.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner S, Bakker NP, 't Hart BA, Holt PJ, Morgan K. Identification of antibody epitopes in the CB-11 peptide of bovine type II collagen recognized by sera from arthritis-susceptible and -resistant rhesus monkeys. Clin Exp Immunol. 1994;96:275–280. doi: 10.1111/j.1365-2249.1994.tb06553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159:1689–1699. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraan MC, Versendaal H, Jonker M, Bresnihan B, Post WJ, 't Hart BA, Breedveld FC, Tak PP. Asymptomatic synovitis precedes clinically manifest arthritis. Arthritis Rheum. 1998;41:1481–1488. doi: 10.1002/1529-0131(199808)41:8<1481::AID-ART19>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Vierboom MP, Zavodny PJ, Chou CC, Tagat JR, Pugliese-Sivo C, Strizki J, Steensma RW, McCombie SW, Celebi-Paul L, Remarque E, et al. Inhibition of the development of collagen-induced arthritis in rhesus monkeys by a small molecular weight antagonist of CCR5. Arthritis Rheum. 2005;52:627–636. doi: 10.1002/art.20850. [DOI] [PubMed] [Google Scholar]
- Goossens PH, Schouten GJ, 't Hart BA, Bout A, Brok HP, Kluin PM, Breedveld FC, Valerio D, Huizinga TW. Feasibility of adenovirus-mediated nonsurgical synovectomy in collagen-induced arthritis-affected rhesus monkeys. Hum Gene Ther. 1999;10:1139–1149. doi: 10.1089/10430349950018139. [DOI] [PubMed] [Google Scholar]
- Patel DD, Zachariah JP, Whichard LP. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin Immunol. 2001;98:39–45. doi: 10.1006/clim.2000.4957. [DOI] [PubMed] [Google Scholar]
- Shadidi KR, Aarvak T, Henriksen JE, Natvig JB, Thompson KM. The chemokines CCL5, CCL2 and CXCL12 play significant roles in the migration of Th1 cells into rheumatoid synovial tissue. Scand J Immunol. 2003;57:192–198. doi: 10.1046/j.1365-3083.2003.01214.x. [DOI] [PubMed] [Google Scholar]
- Wedderburn LR, Robinson N, Patel A, Varsani H, Woo P. Selective recruitment of polarized T cells expressing CCR5 and CXCR3 to the inflamed joints of children with juvenile idiopathic arthritis. Arthritis Rheum. 2000;43:765–774. doi: 10.1002/1529-0131(200004)43:4<765::AID-ANR7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Bakker NP, Van Besouw N, Groenestein R, Jonker M, 't Hart LA. The anti-arthritic and immunosuppressive effects of cyclosporin A on collagen-induced arthritis in the rhesus monkey. Clin Exp Immunol. 1993;93:318–322. doi: 10.1111/j.1365-2249.1993.tb08179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brok HP, Bauer J, Jonker M, Blezer E, Amor S, Bontrop RE, Laman JD, 't Hart BA. Non-human primate models of multiple sclerosis. Immunol Rev. 2001;183:173–185. doi: 10.1034/j.1600-065x.2001.1830114.x. [DOI] [PubMed] [Google Scholar]
- Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 't Hart BA, Simons JM, Knaan-Shanzer S, Bakker NP, Labadie RP. Antiarthritic activity of the newly developed neutrophil oxidative burst antagonist apocynin. Free Radic Biol Med. 1990;9:127–131. doi: 10.1016/0891-5849(90)90115-Y. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 't Hart BA, Bakker NP, Jonker M, Bontrop RE. Resistance to collagen-induced arthritis in rats and rhesus monkeys after immunization with attenuated type II collagen. Eur J Immunol. 1993;23:1588–1594. doi: 10.1002/eji.1830230729. [DOI] [PubMed] [Google Scholar]
- Jasin HE, Noyori K, Takagi T, Taurog JD. Characteristics of antitype II collagen antibody binding to articular cartilage. Arthritis Rheum. 1993;36:651–659. doi: 10.1002/art.1780360512. [DOI] [PubMed] [Google Scholar]
- Noyori K, Koshino T, Takagi T, Okamoto R, Jasin HE. Binding characteristics of antitype II collagen antibody to the surface of diseased human cartilage as a probe for tissue damage. J Rheumatol. 1994;21:293–296. [PubMed] [Google Scholar]
- Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352:1498–1504. doi: 10.1016/S0140-6736(98)03334-0. [DOI] [PubMed] [Google Scholar]
- Tak PP, Hart BA, Kraan MC, Jonker M, Smeets TJ, Breedveld FC. The effects of interferon beta treatment on arthritis. Rheumatology (Oxford) 1999;38:362–369. doi: 10.1093/rheumatology/38.4.362. [DOI] [PubMed] [Google Scholar]
- van Holten J, Pavelka K, Vencovsky J, Stahl H, Rozman B, Genovese M, Kivitz AJ, Alvaro J, Nuki G, Furst DE, et al. A multicentre, randomised, double blind, placebo controlled phase II study of subcutaneous interferon beta-1a in the treatment of patients with active rheumatoid arthritis. Ann Rheum Dis. 2005;64:64–69. doi: 10.1136/ard.2003.020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, Van Vliet SJ, Engering A, 't Hart BA, Van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- 't Hart BA, van Kooyk Y. Yin-yang regulation of autoimmunity by DCs. Trends Immunol. 2004;25:353–359. doi: 10.1016/j.it.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Schrijver IA, Melief MJ, Tak PP, Hazenberg MP, Laman JD. Antigen-presenting cells containing bacterial peptidoglycan in synovial tissues of rheumatoid arthritis patients coexpress costimulatory molecules and cytokines. Arthritis Rheum. 2000;43:2160–2168. doi: 10.1002/1529-0131(200010)43:10<2160::AID-ANR3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Visser L, Jan de Heer H, Boven LA, van Riel D, van Meurs M, Melief MJ, Zahringer U, van Strijp J, Lambrecht BN, Nieuwenhuis EE, et al. Proinflammatory bacterial peptidoglycan as a cofactor for the development of central nervous system autoimmune disease. J Immunol. 2005;174:808–816. doi: 10.4049/jimmunol.174.2.808. [DOI] [PubMed] [Google Scholar]
- Bakker NP, van Erck MG, 't Hart LA, Jonker M. Acquired resistance to type II collagen-induced arthritis in rhesus monkeys is reflected by a T cell low-responsiveness to the antigen. Clin Exp Immunol. 1991;86:219–223. doi: 10.1111/j.1365-2249.1991.tb05799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels MA, Hogquist KA, Jameson SC. Sweet 'n' sour: the impact of differential glycosylation on T cell responses. Nat Immunol. 2002;3:903–910. doi: 10.1038/ni1002-903. [DOI] [PubMed] [Google Scholar]
- Lowe JB, Marth JD. A genetic approach to mammalian glycan function. Annu Rev Biochem. 2003;72:643–691. doi: 10.1146/annurev.biochem.72.121801.161809. [DOI] [PubMed] [Google Scholar]
- Rademacher TW, Williams P, Dwek RA. Agalactosyl glycoforms of IgG autoantibodies are pathogenic. Proc Natl Acad Sci USA. 1994;91:6123–6127. doi: 10.1073/pnas.91.13.6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 't Hart BA, Laman JD, Bauer J, Blezer ED, van Kooyk Y, Hintzen RQ. Modelling of multiple sclerosis: lessons learned in a non-human primate. Lancet Neurol. 2004;3:588–597. doi: 10.1016/S1474-4422(04)00879-8. [DOI] [PubMed] [Google Scholar]