

Recently, Akiyama et al . [1] described defects in thymic negative selection and in CD4+CD25+ regulatory T cell production in mice deficient for tumor necrosis factor (TNF) receptor associated factor (TRAF)6. Signaling through cell surface receptors to activate nuclear factor (NF)κB and mitogen-activated protein (MAP) kinases through adaptor molecules, including TRAF6, is of critical importance to survival and activation of all cells in the body, from those regulating the immune response to epithelial cells, with which immunocytes interact (Fig. 1). Because the same cell signaling pathways regulate survival and activation in the periphery and in the thymus, however, mutations or polymorphisms in the pathway can have outcomes for the immune system that might have been difficult to predict. This is because survival and activation of key antigen presenting cells (APCs), medullary thymic epithelial cells (mTECs) and dendritic cells (DCs), involved in thymic negative selection and peripheral immunity are regulated by a similar network of genes, which map a pathway from TRAF6 to the NFκB family member RelB.
Figure 1.
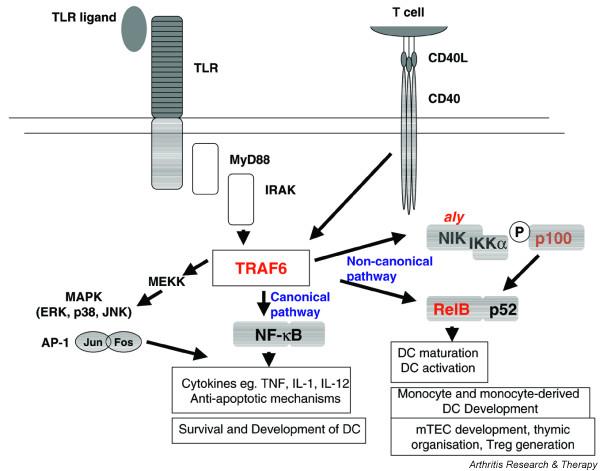
The NFκB pathway regulates inflammation, dendritic cell (DC) development and function, and thymic selection and regulatory T cell production. The pathway is described in the text. Deficient strains marked in red display defects in thymic organization and negative selection with increased numbers of peripheral autoreactive T cells. The two main NFκB activation pathways are marked in blue. IL, interleukin; IRAK, IL-1 receptor-associated kinase; MAPK, mitogen-activated protein kinase; mTEC, medullary thymic epithelial cell; NFκB, nuclear factor κB; NIK, NFκB inhibitory kinase; TLR, toll-like receptor; TNF, tumor necrosis factor; TRAF6, TNF receptor associated factor 6.
Thymic selection and autoimmunity
The vast majority of T cells arise in the thymus. In the fetal and neonatal period, 'central' tolerance is actively maintained in the thymus [2]. During this process, a repertoire of T cells restricted to self-MHC displayed by the thymic cortical epithelial cells (cTECs) is selected in each individual. In addition, those T cells reactive to self-antigen presented by medullary APCs, which include mTECs and medullary DCs, are deleted by negative selection above a threshold of affinity for self antigens presented by those APCs [3]. Because an affinity threshold applies for central deletion of self-reactive T cells, circulation of low-affinity self-reactive T cells in the periphery is inevitable. Ectopic low-level expression of self-antigens normally expressed by peripheral somatic cells in mTECs is very common, transcriptionally activated by the aire gene [4]. Thymic selection defects feature in the pathogenesis of many if not all mouse models of spontaneous autoimmune arthritis and lupus. In these models, including the non-obese diabetic (NOD) × K/B TCR transgenic strain [5], the New Zealand Black (NZB) lupus-prone strain [6], and the SKG ZAP70 mutant model of spontaneous arthritis [7], a variety of defects in the interaction of APCs and thymocytes interfere with the normal process of negative selection, thus permitting the release of dangerously autoreactive T cells into the periphery, where subsequent environmental events, such as infection, more readily trigger autoimmune disease [8]. For example, NOD mouse thymocytes fail to induce the pro-apoptotic gene bim after encountering high-affinity autoantigen, thus raising the threshold for deletion [9]. ZAP70 mutant thymocytes are signaled by APCs bearing self with lower affinity, also raising the threshold for deletion, but are subsequently activated in the periphery by fungal beta-glucans [7]. In contrast, NZB mice demonstrate a defective NFκB/RelB pathway, leading to disorganization of the thymus with associated selection defects [6].
Signaling through NFκB
NFκB is a transcription factor family whose members exist as homodimers or heterodimers of p50/p105, p52/p100, p65 (RelA), RelB and c-Rel. Key events leading to NFκB activation after stimulation of APCs are shown in Fig. 1. In resting APCs, NFκB dimers are sequestered in cytoplasm in complex with the inhibitory (I)κB family, which include the IκBα, IκBβ, IκBε inhibitors of the canonical (or 'standard') activation pathway, and the p100 inhibitory precursor of p52, which participates with RelB in the non-canonical activation pathway (Fig. 1) [10]. Infectious toll-like receptor (TLR) ligands, proinflammatory cytokines such as TNF and IL-1, or T-cell derived CD40-ligand, activate NFκB through phos-phorylation and eventual degradation of IκB, or processing of p100 to p52 as a result of phosphorylation by NFκB inhibitory kinase (NIK), in both cases allowing translocation of released NFκB dimers to the nucleus [10-12]. After binding DNA, active NFκB transcription factors promote the expression of many genes, of which the majority participate through the canonical pathway as 'central mediators of the immune response' [13]. These include cytokines, adhesion and costimulatory molecules, and genes regulating the oxidative burst [14].
Although other NFκB molecules contribute, the RelB subunit has been most directly associated with functional DC differentiation and activation, and with monocyte and monocyte-derived DC development, with no effect on other myeloid differentiation pathways [15-18]. However, while maturation of DCs is defective in RelB deficient mice and DCs from these mice induce T cell tolerance in wild-type mice, RelB deficient mice also display defective thymic organogenesis, reduced numbers of mTECs, reduced negative selection, increased numbers of autoreactive T cells in the periphery, and multi-organ inflammation [19-21].
TRAF6 and NFκB deficient mice define elements of the central tolerance pathway
TRAF6 acts like a junction, transducing signals from the TNF receptor superfamily, TLR/IL-1R family and CD40 to activate the transcription factors NFκB and AP1 (Fig. 1). TRAF6-deficient mice also demonstrate autoantibody production, and inflammation of multiple organs including liver, lung and pancreas. Akiyama et al . [1] show that TRAF6 is required for the development of mTECs but not thymic medullary DCs. Using thymic grafts depleted of hemopoietic cells, they show that the grafted TRAF6 thymic stroma is sufficient to recapitulate the negative selection defect, and the development of autoimmunity, in nude mice with intact TRAF6. They go on to show that RelB expression is undetectable in TRAF6-deficient thymic stroma, and that RelB expression is restored when TRAF6 is introduced into knockout mTEC lines. In addition, the number of CD4+CD25+regulatory T cells in TRAF6 thymus is markedly reduced. Their data suggest that reduced regulatory T cell development, and reduced negative selection as a result of an absence of the selecting mTEC, are two potential mechanisms of autoimmunity in these mice.
Implications for autoimmune disease pathogenesis
The analysis by Akiyama et al . [1] of TRAF6-deficient mice adds to a body of literature implicating the non-canonical pathway of NFκB activation, not only in DC development and function, but also in the processes of thymic organization, mTEC development, negative selection and regulatory T cell production. When DCs were compared in TRAF6 and RelB deficient mice by Kobayashi et al . [22], both were shown to have a specific defect in development of the CD4+CD11c+ subset of splenic DCs as a direct effect of TRAF6/RelB in hemopoietic cell differentiation. In contrast, thymic medullary DCs and other subsets of splenic DCs developed normally. As expected from the central role of TRAF6 in the NFκB activation pathway, DCs from TRAF6 deficient mice are unable to produce pro-inflammatory cytokines or to upregulate expression of costimulatory molecules in response to TLR ligands or CD40 ligand [22]. In addition, the TRAF6-NIK-RelB pathway has marked effects on stromal cells. TRAF6 and RelB are required for the development of mTECs and for organization of the thymic medulla, as well as for development of lymph nodes [19,22]. Similar disorganization is seen in the aly/aly mutant, which carries a functional mutation in NIK, which makes it unable to bind IKKα and thereby to phosphorylate p100 (Fig. 1). Aly thymic grafts also induce autoimmunity in wild-type nude recipient mice [23]. These different mice provide a powerful demonstration of the development of spontaneous autoimmunity due, at least in part, to central T cell dysregulation, even when TLR ligands and CD40 ligand are unable to activate DCs.
Could pathways related to TRAF-RelB impact on mTEC?
Vanin-1 is a thymic epithelial cell ectoenzyme that generates the amino-thiol cysteamine, an important mediator of oxidative stress. It was recently shown that vanin-1 deficient mice, which lack cysteamine in tissues, exhibit resistance to oxidative injury due to elevated stores of glutathione, the most potent cellular antioxidant [24]. Of interest here, vanin-1 is normally expressed at low levels by mTECs and is upregulated by oxidative stress [24]. These data raise the possibility that mTEC function may be regulated by oxidative stress. The implication is that genes or environmental factors that alter the oxidative/reductive state of mTECs in utero and in neonates may influence the outcome of thymic negative selection. Potential genetic factors have been described. A functional polymorphism in the ncf1 gene reduces the responsiveness of cells in rats and mice susceptible to arthritis to oxidative stress [25]. Environmental factors are less well characterized, although exposure to certain infections or possibly cigarette smoke in utero or in neonates may be sufficient to alter mTEC function.
Implications for human autoimmune diseases
Given that the same cell signaling pathways regulate survival and activation in the periphery and in the thymus, the immune system must balance antigen presentation and pro-inflammatory outcomes in the periphery in response to pathogens and other environmental inflammatory events, along with correct signaling of TRAF6-NIK-RelB in neonates to prevent excessive autoreactivity of the T cell repertoire and the appropriate development and function of peripheral APCs. The TRAF6-RelB deficient mice provide striking examples of how an apparent immune deficiency – in NFκB function – can also cause autoimmunity. Thus, emerging data suggest strongly that deficiencies of the TRAF6-NFκB pathway, and regulation of oxidation/reduction, will continue to be found in human autoimmune diseases, and that the thymus is implicated in its pathogenesis.
Abbreviations
APC = antigen presenting cell; cTEC = thymic cortical epithelial cell; DC = dendritic cell; IκB = inhibitory κB; IL = interleukin; mTEC = medullary thymic epithelial cell; NFκB = nuclear factor κB; NIK = NFκB inhibitory kinase; NOD = non-obese diabetic; NZB = New Zealand black; TLR = toll-like receptor; TNF = tumor necrosis factor; TRAF6 = TNF receptor associated factor 6.
Competing interests
The author(s) declare that they have no competing interests.
References
- Akiyama T, Maeda S, Yamane S, Ogino K, Kasai M, Kajiura F, Mat-sumoto M, Inoue J. Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science. 2005;308:248–251. doi: 10.1126/science.1105677. [DOI] [PubMed] [Google Scholar]
- Ardavin C. Thymic dendritic cells. Immunol Today. 1997;18:350–361. doi: 10.1016/S0167-5699(97)01090-6. [DOI] [PubMed] [Google Scholar]
- Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273–280. doi: 10.1016/0092-8674(87)90568-X. [DOI] [PubMed] [Google Scholar]
- Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/S0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- Valero R, Baron ML, Guerin S, Beliard S, Lelouard H, Kahn-Perles B, Vialettes B, Nguyen C, Imbert J, Naquet P. A defective NF-kappa B/RelB pathway in autoimmune-prone New Zealand black mice is associated with inefficient expansion of thymocyte and dendritic cells. J Immunol. 2002;169:185–192. doi: 10.4049/jimmunol.169.1.185. [DOI] [PubMed] [Google Scholar]
- Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, Sakaguchi S. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426:454–460. doi: 10.1038/nature02119. [DOI] [PubMed] [Google Scholar]
- Yoshitomi H, Sakaguchi N, Kobayashi K, Brown GD, Tagami T, Sakihama T, Hirota K, Tanaka S, Nomura T, Miki I, et al. A role for fungal {beta}-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J Exp Med. 2005;201:949–960. doi: 10.1084/jem.20041758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A, Lesage S, Gray DH, O'Reilly LA, Strasser A, Fahrer AM, Boyd RL, Wilson J, Baxter AG, Gallo EM, et al. Generalized resistance to thymic deletion in the NOD mouse; a polygenic trait characterized by defective induction of Bim. Immunity. 2004;21:817–830. doi: 10.1016/j.immuni.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G. Modulation of NF-kappaB activity by exchange of dimers. Mol Cell. 2003;11:1563–1574. doi: 10.1016/S1097-2765(03)00227-2. [DOI] [PubMed] [Google Scholar]
- O'Sullivan BJ, Thomas R. CD40 ligation conditions dendritic cell antigen-presenting function through sustained activation of NF-kappaB. J Immunol. 2002;168:5491–5498. doi: 10.4049/jimmunol.168.11.5491. [DOI] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Iles KE, Forman HJ. Macrophage signaling and respiratory burst. Immunol Res. 2002;26:95–105. doi: 10.1385/IR:26:1-3:095. [DOI] [PubMed] [Google Scholar]
- Platzer B, Jorgl A, Taschner S, Hocher B, Strobl H. RelB regulates human dendritic cell subset development by promoting monocyte intermediates. Blood. 2004;104:3655–3663. doi: 10.1182/blood-2004-02-0412. [DOI] [PubMed] [Google Scholar]
- Pettit AR, Quinn C, MacDonald KP, Cavanagh LL, Thomas G, Townsend W, Handel M, Thomas R. Nuclear localization of RelB is associated with effective antigen-presenting cell function. J Immunol. 1997;159:3681–3691. [PubMed] [Google Scholar]
- Carrasco D, Ryseck RP, Bravo R. Expression of relB transcripts during lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development. 1993;118:1221–1231. doi: 10.1242/dev.118.4.1221. [DOI] [PubMed] [Google Scholar]
- Martin E, O'Sullivan BJ, Low P, Thomas R. Antigen-specific suppression of a primed immune response by dendritic cells mediated by regulatory T cells secreting interleukin-10. Immunity. 2003;18:155–167. doi: 10.1016/S1074-7613(02)00503-4. [DOI] [PubMed] [Google Scholar]
- Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- Laufer TM, DeKoning J, Markowitz JS, Lo D, Glimcher LH. Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex. Nature. 1996;383:81–85. doi: 10.1038/383081a0. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Walsh PT, Walsh MC, Speirs KM, Chiffoleau E, King CG, Hancock WW, Caamano JH, Hunter CA, Scott P, et al. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity. 2003;19:353–363. doi: 10.1016/S1074-7613(03)00230-9. [DOI] [PubMed] [Google Scholar]
- Kajiura F, Sun S, Nomura T, Izumi K, Ueno T, Bando Y, Kuroda N, Han H, Li Y, Matsushima A, et al. NF-kappa B-inducing kinase establishes self-tolerance in a thymic stroma-dependent manner. J Immunol. 2004;172:2067–2075. doi: 10.4049/jimmunol.172.4.2067. [DOI] [PubMed] [Google Scholar]
- Berruyer C, Martin FM, Castellano R, Macone A, Malergue F, Garrido-Urbani S, Millet V, Imbert J, Dupre S, Pitari G, et al. Vanin-1-/- mice exhibit a glutathione-mediated tissue resistance to oxidative stress. Mol Cell Biol. 2004;24:7214–7224. doi: 10.1128/MCB.24.16.7214-7224.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson P, Holmberg J, Tordsson J, Lu S, Akerstrom B, Holmdahl R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet. 2003;33:25–32. doi: 10.1038/ng1058. [DOI] [PubMed] [Google Scholar]