

Abstract
Chemical modification of biological molecules is a general mechanism for cellular regulation. A quantitative approach has been developed to measure the extent of modification on HS (heparan sulphates). Sulphation on HS by sulphotransferases leads to variable sulphation levels, which allows cells to tune their affinities to various extracellular proteins, including growth factors. With stable isotope labelling and HPLC-coupled MS, modification degrees at various O-sulphation sites could be determined. A bovine kidney HS sample was first saturated in vitro with 34S by an OST (O-sulphotransferase), then digested with nitrous acid and analysed with HPLC-coupled MS. The 34S-labelled oligosaccharides were identified based on their unique isotope clusters. The modification degrees at the sulphotransferase recognition sites were obtained by calculating the intensities of isotopic peaks in the isotope clusters. The modification degrees at 3-OST-1 and 6-OST-1 sites were examined in detail. This approach can also be used to study other types of chemical modifications on biological molecules.
Keywords: chemical modification, heparan sulphate, MS, modification degree, stable isotope, sulphotransferase
Abbreviations: aManR, 2,5-anhydromannitol; DBA, dibutylamine; GlcNS, N-sulphated glucosamine; HexA, hexuronic acid; HS, heparan sulphate(s); IdoA, L-iduronic acid; LC, liquid chromatography; 3-OST, 3-O-sulphotransferase; PAPS, 3′-phosphoadenosine 5′-phosphosulphate; TIC, total ion current; XIC, extracted ion current
INTRODUCTION
Chemical modification of biological molecules is a general mechanism for cellular regulation, but it is often difficult to measure quantitatively the extent of modification. An approach to measure the sulphation degrees at particular sites on HS (heparan sulphates) is described here. Sulphation of glycans by sulphotransferases is a widespread regulatory modification that can convert the underlying molecule from one functional state into another [1,2]. HS are linear polysaccharides covalently attached to the core proteins of proteoglycans [3–5]. HS have been found to play important roles in diverse biological systems, such as organ development, lipid metabolism, wound healing, inflammation and viral pathogenesis [6,7]. HS exert their biological functions through binding to various extracellular proteins, such as fibroblast growth factors, morphogens, lipoprotein lipase, cytokines, selectins and viral envelope proteins [6–10]. The affinities of HS for these proteins depend on the sulphation levels at specific sites on the HS chains.
HS are initially synthesized in the Golgi apparatus as non-sulphated polymers by the sequential addition of GlcA alternating with GlcNAc [5]. The chains then go through a series of modifications, including N-deacetylation and N-sulphation of the glucosamine, epimerization of the GlcA to IdoA (L-iduronic acid), 2-O-sulphation of IdoA, and 6-O- and 3-O-sulphation of the glucosamine [5,11]. These modifications are catalysed by different enzymes, which include a 2-OST (2-O-sulphotransferase), an epimerase, five 3-OSTs, three 6-OSTs and four NDSTs (N-deacetylases/N-sulphotransferases) [5,12]. The sulphate donor for sulphation is PAPS (3′-phosphoadenosine 5′-phosphosulphate). These modifications do not occur uniformly along the HS chains but focus on separate regions; therefore alternating intensively modified and rarely modified domains are formed. It is the intensively modified domains where various extracellular proteins usually bind.
To a certain bound protein, some sulphates on HS may be more important than others for binding [13]. Loss of these sulphates will abolish the affinity of HS for the protein; hence sulphation degrees at these sites are directly related to the activity of an HS sample. A classic example is the anti-thrombin III-binding region on HS, where a 3-O-sulphate is critical for binding to anti-thrombin III [11,14,15]. Recently, it has been found that the 6-O-sulphation levels on HS are dynamically regulated to control specific cell signalling [16] and specification of embryonic progenitor lineages [17–20]. This finding again emphasizes the importance of investigating the particular sulphation levels on HS chains.
The measurement of sulphation level at a particular site is achieved through the in vitro saturation of HS samples with the stable isotope 34S by a sulphotransferase of interest. To describe quantitatively a chemical modification, modification degree (d) at a specific site on a biological molecule is defined as the percentage of this molecule that is modified at this site in vivo to the total number of this molecule (Figure 1A). Obviously, on a nascent HS chain, before modification, all the d values should be 0%; in contrast, on heparin, where most of the modifications are nearly complete, the d values should be close to 100%.
Figure 1. A schematic view of using stable isotope labelling to determine the modification degree at a specific site on a biological molecule.

(A) A modification degree (d) at a specific site on a biological molecule. ●, modification introduced in vivo; ○, stable isotope containing modification introduced in vitro. (B) After a sulphation site is saturated with 34S in vitro, the m/z cluster of an oligosaccharide that carries this site can be considered to be a composite of two. One m/z cluster belongs to the portion of the oligosaccharide generated in vivo (hatched area) and the other belongs to the portion generated in vitro (dark grey area). Intensities of isotopic peaks M and M+2 are I1 and I3 respectively. The enhancement of peak M+2 after the 34S saturation is ΔI3.
MATERIALS AND METHODS
Materials
Bovine kidney HS, PEP (phosphoenolpyruvate), pyruvate kinase, inorganic pyrophosphatase and sulphurylase were purchased from Sigma (St. Louis, MO, U.S.A.). HS sulphotransferases 3-OST-1 and 6-OST-1 were cloned, expressed and purified with a baculovirus expression system as described previously [21]. The stable isotope 34S was obtained from Isonics Corp. (Columbia, MD, U.S.A.). PAPS was prepared as described previously [22]. APS kinase was a gift from Dr I. Segel (University of California, Davis, CA, U.S.A.).
Stable isotope labelling of HS to saturation
The labelling buffer (2×) contained 50 mM Mes (pH 7.0), 1% (w/v) Triton X-100, 5 mM MgCl2, 5 mM MnCl2, 2.5 mM CaCl2, 0.075 mg/ml protamine chloride and 1.5 mg/ml BSA. For a typical 20 μl saturation labelling reaction, 10 μg of HS, 10 μl of labelling buffer, 70 ng of the expressed sulphotransferase, 2 μl of PAPS (3 mM) and an appropriate amount of water were mixed. The mixture was then incubated for 2 h at 37 °C. The modified HS was purified on a DEAE column.
Digestion of HS with nitrous acid
Nitrous acid digestion was performed at pH 1.5 to cleave exclusively at GlcNS (N-sulphated glucosamine) residues. Nitrous acid solution was prepared by taking the supernatant of a mixture of equal volumes of 0.2 M Ba(NO2)2 and 0.2 M H2SO4. The nitrous acid solution was used within 10 min of preparation. To perform nitrous acid digestion, 10 μl of nitrous acid was added to 10 μg of HS sample and the mixture was held at room temperature (23 °C) for 10 min. The reaction was then neutralized with 1 M NaOH and reduced with 1 M NaBH4 in 0.01 M NaOH at room temperature for 15 min. Residual NaBH4 was eliminated by the addition of 1 μl of 1 M H2SO4, and the pH was adjusted to approx. 7.0.
Capillary LC (liquid chromatography)/MS
HPLC–MS (HPLC-coupled MS) was described previously [22,23]. Separations were performed on an Ultimate capillary HPLC workstation (Dionex, Sunnyvale, CA, U.S.A.), employing DBA (dibutylamine) as an ion-pairing agent. A gradient elution was performed, using a binary solvent system composed of water (eluent A) and 70% methanol (eluent B), both containing 8 mM acetic acid and 5 mM ion-pairing reagent. HPLC separations were performed on a 0.3 mm×250 mm C18 column (MS; 5 μm) at a rate of 5 μl/min and a sample volume of 6.3 μl. The elution profile was 0% B for 5 min, 6% B for 19 min, 18% B for 17 min, 34% B for 13 min and 55% B for 16 min. After each run, the column was washed with 90% B for 15 min and equilibrated with 100% A for 28 min.
Mass spectra were acquired on a Mariner BioSpectrometry Workstation ESI (electrospray ionization) time-of-flight mass spectrometer (PerSeptive Biosystems, Framingham, MA, U.S.A.). Nitrogen was used as a desolvation gas as well as a nebulizer. Conditions for electrospray ionization MS were as follows: nebulizer gas (N2) flow rate, 1 litre/min; nozzle temperature, 140 °C; drying gas (N2) flow rate, 0.6 litre/min; spray tip potential, 2.8 kV; and nozzle potential, 70 V. Negative ion spectra were acquired every 4 s by scanning m/z from 40 to 4000. Total ion chromatograms and mass spectra were processed with the Data Explorer software version 3.0.
Computer simulation of the natural isotope clusters of HS oligosaccharides
The natural isotope clusters of oligosaccharides were simulated with Isotope Calculator in the Data Explorer software. After the isotope cluster of a oligosaccharide was generated, the intensities of all isotopic peaks were normalized to that of the monoisotopic peak.
RESULTS
Theoretical calculation of a modification degree at a site of interest on a biological molecule
If a chemical modification on a biological molecule is not complete in vivo, it is possible to complete the modification in vitro with a stable isotope-containing a donor, which causes the enhancement of a specific peak in the isotope cluster of the molecule. For example, if a stable isotope such as 18O or 34S is used for labelling, the isotopic peak M+2 will be enhanced (Figure 1B). Because the in vivo- and in vitro-modified portions cannot be separated by HPLC, the m/z cluster of the molecule can be considered as a composite of the two. On labelling of an HS oligosaccharide with 34S by a sulphotransferase, the monoisotopic peak M in the cluster is not affected by the process and thus represents the in vivo-modified portion; in contrast, the enhancement of isotopic peak M+2 represents the in vitro-modified portion (Figure 1B). Assuming that the intensity of the monoisotopic peak M is I1 and the enhancement of peak M+2 is ΔI3, the modification degree (d) at the sulphotransferase recognition site will be
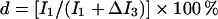 |
1 |
Because the natural abundance of each stable isotope is relatively constant, the initial intensity of the peak M+2 is a constant too. The relative initial I3 value of the oligosaccharides could be obtained through computer simulation on the basis of natural abundance of each stable isotope.
m/z values of nitrous acid-generated HS oligosaccharides
At pH 1.5, nitrous acid selectively attacks GlcNS on a HS chain, whereas N-acetylated and unmodified glucosamine residues are not affected [24]. Nitrous acid treatment converts a GlcNS residue into 2,5-anhydromannose and results in the cleavage of the corresponding glucosaminidic linkage. Further reduction by NaBH4 converts the unstable 2,5-anhydromannose into aManR (2,5-anhydromannitol). The net result is various oligosaccharides with HexA (hexuronic acid) at the non-reducing ends and aManR at the reducing ends (Figure 2); therefore a nitrous acid-generated oligosaccharide can be considered as a collection of one HexA-aManR, a number of internal HexA-GlcNAc, a number of internal HexA-GlcNH2 and a number of sulphates. Based on the major stable isotopes of 12C, 1H, 16O, 14N and 32S, HexA-aManR (C12H20O11), internal HexA-GlcNAc (C14H21O11N1) and internal HexA-GlcNH2 (C12H19O10N1) have molecular masses of 340.09, 379.11 and 337.10 Da respectively and sulphation increases the mass by 79.96 (addition of S1O3). For convenience, a nitrous acid- generated oligosaccharide can be designated as
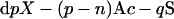 |
where X is the number of sugar residues, p is the number of internal disaccharides, X=2(p+1), n is the number of free amino groups, q is the number of sulphates, Ac stands for the acetyl group and S stands for sulphate. Accordingly, the m/z value of a negatively charged oligosaccharide will be
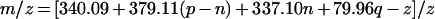 |
2 |
The calculated value should correspond to the monoisotopic peak M in the m/z cluster of an oligosaccharide (Figure 1B, and Supplemental Files 1 and 2 at http://www.BiochemJ.org/bj/389/bj3890383add.htm).
Figure 2. Structure of low-pH nitrous acid digestion-generated oligosaccharides.

A nitrous acid digestion-generated oligosaccharide can be considered as a combination of one HexA-aManR, numbers of internal GlcA-GlcNAc and GlcA-GlcNH2 and sulphates. p, number of internal disaccharides; q, number of sulphates; n, number of free amino groups; z, number of negative charges on the molecule. The linkage between the non-reducing end uronic acid and the next glucosamine residue is not specified since the uronic acid could be either GlcA or IdoA.
The modification degrees at the 3-OST-1 sites on bovine kidney HS
Bovine kidney HS was first saturated with a stable isotope 34S by 3-OST-1 (see Supplemental Files 5 and 7 at http://www.BiochemJ.org/bj/389/bj3890383add.htm) and then digested with nitrous acid at pH 1.5. The digest was analysed with HPLC–MS. Two 34S-labelled oligosaccharides were found with greatly enhanced isotopic peaks M+2 at m/z 439.59 and 479.57. The monoisotopic peak M of the two isotope clusters was at m/z 438.59 and 478.59 respectively. The distance between neighbouring peaks in the two clusters was 0.5 unit, indicating the double-charged ions (z=2). After these values were fitted into eqn (2), the parameters were found to be X=4, p=1, n=0 and q=2 or 3. The two oligosaccharides were then designated as dp4-1Ac-2S and dp4-1Ac-3S (Table 1).
Table 1. Identification of 3-OST-1- and 6-OST-1-labelled oligosaccharides on bovine kidney HS.
The relationship between the m/z values of the monoisotopic peaks, the number of internal disaccharides (p), the number of NH2-containing disaccharides (n) and the number of sulphates (q) was calculated based on eqn (2), m/z=[340.09+379.11(p−n)+337.10n+79.96q−z]/z.
| Enzyme | Monoisotopic peak (m/z) | Elution time (min) | z | n | p | q | Formula |
|---|---|---|---|---|---|---|---|
| 3-OST-1 | 438.59 | 53.3 | 2 | 0 | 1 | 2 | dp4-1Ac-2S |
| 478.59 | 60.3 | 2 | 0 | 1 | 3 | dp4-1Ac-3S | |
| 6-OST-1 | 499.05 | 41.7 | 1 | 0 | 0 | 2 | dp2-2S(a)* |
| 499.05 | 44.2 | 1 | 0 | 0 | 2 | dp2-2S(b)* | |
| 708.19 | 55.0 | 1 | 0 | 0 | 3 | dp2-3S:1DBA* |
* Only major molecular species were listed.
An XIC (extracted ion current) chromatogram was then obtained at m/z 439.59 and 479.57 from the TIC (total ion current) chromatogram (Figure 3A). Based on peak areas in XIC, the two oligosaccharides were estimated to be in the ratio 5:1 (semi-quantitative estimation, because the peak intensity in an XIC is not only related to the quantity but also to the charge state and ionization condition). Sodium adducts were observed for both oligosaccharides and a sulphate loss was observed for the second oligosaccharide (Figure 3B, and Supplemental File 3 at http://www.BiochemJ.org/bj/389/bj3890383add.htm). The intensities of the isotopic peaks in the m/z clusters were directly obtained from data acquisition. The relative intensities of isotopic peak M+2 (I3) of the two labelled oligosaccharides were found to be 6.12 and 27.86 respectively (Table 2).
Figure 3. The modification degrees at 3-OST-1 sites.

(A) TIC of the nitrous acid-digested, 34S/3-OST-1-saturated HS sample is followed by XIC at m/z 439.59±0.2 and 479.58±0.2. The XIC had peaks at T53.3 and T60.3. (B) The mass spectra and m/z clusters of the two 3-OST-1-labelled oligosaccharides. The two oligosaccharides were identified to be a dp4-1Ac-2S and a dp4-1Ac-3S. The peak at T60.3 in the XIC exhibited m/z 439.60, 479.57 and 490.58. The m/z 439.60 was caused by the loss of a sulphate from m/z 479.57. (C) The isotope clusters of dp4-1Ac-2S and dp4-1Ac-3S, generated by a computer program according to the natural abundances of stable isotopes.
Table 2. Calculation of the modification degrees (d) at 3-OST-1 and 6-OST-1 sites.
The intensities of isotopic peaks for 34S-saturated oligosaccharides by 3-OST-1 and 6-OST-1 were obtained through data acquisition. The initial intensities of the isotopic peaks were obtained by computer simulation. Peak intensities of monoisotopic peaks were arbitrarily set at 1 unit.
| m/z clusters | |||||||
|---|---|---|---|---|---|---|---|
| Enzymes | Oligosaccharide formula | Isotopic peak | m/z | Intensity | Initial intensity | ΔI3 | d (%) |
| 3-OST-1 | dp4-1Ac-2S | M | 438.59 | 1 | |||
| (C26H41O28N1S2) | M+2 | 439.59 | 6.12 | 0.20 | 5.92 | 14 | |
| dp4-1Ac-3S | M | 478.59 | 1 | ||||
| (C26H41O31N1S3) | M+2 | 479.58 | 27.86 | 0.25 | 27.61 | 4 | |
| 6-OST-1 | dp2-2S (a) | M | 499.05 | 1 | |||
| (C12H20O17S2) | M+2 | 501.05 | 0.80 | 0.13 | 0.67 | 60 | |
| dp2-2S (b) | M | 499.06 | 1 | ||||
| (C12H20O17S2) | M+2 | 501.06 | 0.71 | 0.13 | 0.58 | 63 | |
| dp2-3S:1DBA | M | 708.19 | 1 | ||||
| (C20H39O20N1S3) | M+2 | 710.20 | 1.17 | 0.21 | 0.96 | 51 | |
The isotope clusters of dp4-1Ac-2S (C26H41O28N1S2) and dp4-1Ac-3S (C26H41O31N1S3) were also generated with the Isotope Calculator according to the natural abundance of each stable isotope (Figure 3C), and the initial relative intensities of the peaks M+2 were determined to be 0.20 and 0.25 respectively. The modification degrees at these two 3-OST-1 sites were then calculated to be 14 and 4% respectively (Table 2) using eqn (1).
Modification degrees at 6-OST-1 sites on bovine kidney HS
The modification degrees at 6-OST-1 sites on bovine kidney HS were also determined (see Supplemental Files 6 and 8 at http://www.BiochemJ.org/bj/389/bj3890383add.htm). Three oligosaccharides were labelled by 6-OST-1 (Figure 4). The three M+2 peaks in the isotope clusters of the oligosaccharides were at m/z 501.05, 501.05 and 710.19. An XIC (Figure 4A) showed the proportion of the three oligosaccharides to be 20:1.5:1 according to the peak intensities (semi-quantitative estimation).
Figure 4. The modification degrees at 6-OST-1 sites.

(A) TIC of the nitrous acid-digested, 34S/6-OST-1-saturated HS sample is followed by the XIC at m/z 501.05±0.2 and 710.20±0.2. (B) The mass spectra and isotope clusters of the three 6-OST-1-labelled oligosaccharides. The three oligosaccharides were identified to be two dp2–2S and a dp2–3S, with the first dp2–2S being the dominant one. Each dp2–2S could form a complex with one DBA and the dp2–3S could form a complex with one or two DBAs. The mass spectrum of the second dp2–2S was similar to that of the first one and was omitted. Sodium adducts were also observed (not labelled). (C) The isotope clusters of dp2–2S and dp2–3S:1DBA, generated by a computer program according to the natural abundances of stable isotopes.
In the mass spectra, sodium and DBA adducts were observed for the three oligosaccharides (Figure 4B). The distance between the neighbouring peaks in all isotope clusters was 1 unit, indicating single-charged ions (z=1). The first oligosaccharide had two isotope clusters with the monoisotopic peak M at 499.05 and 628.22 respectively. Since the difference between the two values was 129.17, the second m/z cluster was probably from the complex between this oligosaccharide and the ion-pairing reagent DBA [NH(C4H9)2; molecular mass, 129.15 Da] (see Supplemental File 4 at http://www.BiochemJ.org/bj/389/bj3890383add.htm). By fitting the data into eqn (2), the parameters for this oligosaccharide were found to be X=2, p=0, n=0 and q=2; therefore the oligosaccharide was a dp2-2S (Table 1). The second oligosaccharide had a very similar mass spectrum and was also a dp2-2S. The first oligosaccharide was then designated dp2-2S(a) and the second one was designated dp2-2S(b). The third oligosaccharide had two isotope clusters with the monoisotopic peaks M at 708.19 (579.04+129.15) and 837.33 [579.03+(129.15×2)], indicating the formation of complexes between the oligosaccharide and one or two molecules of DBA. This oligosaccharide was identified as a dp2-3S (Table 1). The relative intensities of the isotopic peaks M+2 of the dp2-2S(a), dp2-2S(b) and dp2-3S:1DBA (complex between the dp2-3S and one DBA) were determined to be 0.80, 0.71 and 1.17 respectively according to their m/z clusters (Table 2).
When the isotope clusters of dp2-2S and dp2-3S:1DBA were simulated with Isotope Calculator (Figure 4C), the initial intensities of the peaks M+2 were found to be 0.13 and 0.21 respectively (Table 2). The modification degrees at the three 6-OST-1 sites were then determined to be 60, 63 and 51% respectively (Table 2).
DISCUSSION
There were two different sulphation sites for 3-OST-1 on bovine kidney HS. The modification degree at the site in dp4-1Ac-2S was 14%, whereas the modification degree at the site in dp4-1Ac-3S was only 4%. Considering that the amount of dp4-1Ac-2S was approx. 5-fold that of dp4-1Ac-3S (Figure 3A), it is clear that most of the oligosaccharides modified by 3-OST-1 in vivo were dp4-1Ac-2S. Recently, the 3-OST-1 knockout mice were reported to have normal anticoagulant activities [25], suggesting that 3-OST-1-modified oligosaccharides might have other biological functions. The fact that the modification levels at 3-OST-1 sites were significantly increased in retinoic acid-stimulated F9 embryonal carcinoma cells, indicated that 3-OST-1 might have a role in the differentiation of this cell line [26]. Overall, the modification degrees at the two 3-OST-1 sites on bovine kidney HS were very low, which leaves a relatively larger portion of the sites in the unmodified state and may allow a rapid increase in the 3-O-sulphation levels in response to stimuli such as retinoic acid [26].
On the other hand, 6-OST-1 had three different sulphation sites on bovine kidney HS. All modification degrees at these sites were more than 50%, which are significantly higher than those at the 3-OST-1 sites. The higher modification degrees at 6-OST-1 sites left less room for further addition of sulphates. However, it was found that the modification degrees at the 6-O sites can also be adjusted by HS 6-O-endosulphatase [17] that takes away sulphates from these 6-O sites [18,20]. This regulation of sulphation levels by sulphotransferases/sulphatases is very similar to the regulation of phosphorylation levels by kinases/phosphatases [2].
We found that stable isotope labelling and LC/MS have been applied recently for measuring the phosphorylation levels on proteins and peptides [27,28]. The novelty of our method lies in saturating the unmodified site with a stable isotope in vitro, which renders the site modified in vitro chemically identical with the site modified in vivo. Subsequently, both sites will appear in a same isotope cluster during LC/MS, and the ratio between the in vivo- and in vitro-modified sites can be quantitatively determined.
In summary, sulphation degrees at specific sites on HS are important parameters for cells to tune their affinities for various extracellular proteins; the measurement of sulphation degrees opens a new avenue to study the HS structure–function relationship; stable isotope labelling and LC/MS can also be used to measure other chemical modifications on biological molecules.
Online data
Acknowledgments
We thank Dr R. Rosenberg (Staten Island, NY, U.S.A.) for material support and D.L. Beeler (Division of Molecular and Vascular Medicine, Beth Israel Deaconess Medical Center, Boston, MA, U.S.A.) for technical assistance. Special thanks to Professor A. Rich (Massachusetts Institute of Technology, Cambridge) for his insightful comments and encouragement and Dr K. Lowenhaupt (Massachusetts Institute of Technology) for a critical reading of this paper.
References
- 1.de Graffenried C. L., Laughlin S. T., Kohler J. J., Bertozzi C. R. A small-molecule switch for Golgi sulfotransferases. Proc. Natl. Acad. Sci. U.S.A. 2004;101:16715–16720. doi: 10.1073/pnas.0403681101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honke K., Taniguchi N. Sulfotransferases and sulfated oligosaccharides. Med. Res. Rev. 2002;22:637–654. doi: 10.1002/med.10020. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl U., Kusche-Gullberg M., Kjellen L. Regulated diversity of heparan sulfate. J. Biol. Chem. 1998;273:24979–24982. doi: 10.1074/jbc.273.39.24979. [DOI] [PubMed] [Google Scholar]
- 4.Bernfield M., Gotte M., Park P. W., Reizes O., Fitzgerald M. L., Lincecum J., Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 5.Esko J. D., Selleck S. B. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 6.Perrimon N., Hacker U. Wingless, hedgehog and heparan sulfate proteoglycans. Development. 2004;131:2509–2511. doi: 10.1242/dev.01225. [DOI] [PubMed] [Google Scholar]
- 7.Gallagher J. T. Heparan sulfate: growth control with a restricted sequence menu. J. Clin. Invest. 2001;108:357–361. doi: 10.1172/JCI13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Z. L., Zhang L., Yabe T., Kuberan B., Beeler D. L., Love A., Rosenberg R. D. The involvement of heparan sulfate (HS) in FGF1/HS/FGFR1 signaling complex. J. Biol. Chem. 2003;278:17121–17129. doi: 10.1074/jbc.M212590200. [DOI] [PubMed] [Google Scholar]
- 9.Esko J. D., Lindahl U. Molecular diversity of heparan sulfate. J. Clin. Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnbull J., Powell A., Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/s0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg R. D., Shworak N. W., Liu J., Schwartz J. J., Zhang L. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J. Clin. Invest. 1997;100:S67–S75. [PubMed] [Google Scholar]
- 12.Kramer K. L., Yost H. J. Heparan sulfate core proteins in cell-cell signaling. Annu. Rev. Genet. 2003;37:461–484. doi: 10.1146/annurev.genet.37.061103.090226. [DOI] [PubMed] [Google Scholar]
- 13.Wu Z. L., Zhang L., Beeler D. L., Kuberan B., Rosenberg R. D. A new strategy for defining critical functional groups on heparan sulfate. FASEB J. 2002;16:539–545. doi: 10.1096/fj.01-0807com. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L., Schwartz J. J., Miller J., Liu J., Fritze L. M., Shworak N. W., Rosenberg R. D. The retinoic acid and cAMP-dependent up-regulation of 3-O-sulfotransferase-1 leads to a dramatic augmentation of anticoagulantly active heparan sulfate biosynthesis in F9 embryonal carcinoma cells. J. Biol. Chem. 1998;273:27998–28003. doi: 10.1074/jbc.273.43.27998. [DOI] [PubMed] [Google Scholar]
- 15.Atha D. H., Lormeau J. C., Petitou M., Rosenberg R. D., Choay J. Contribution of monosaccharide residues in heparin binding to antithrombin III. Biochemistry. 1985;24:6723–6729. doi: 10.1021/bi00344a063. [DOI] [PubMed] [Google Scholar]
- 16.Viviano B. L., Paine-Saunders S., Gasiunas N., Gallagher J., Saunders S. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J. Biol. Chem. 2004;279:5604–5611. doi: 10.1074/jbc.M310691200. [DOI] [PubMed] [Google Scholar]
- 17.Dhoot G. K., Gustafsson M. K., Ai X., Sun W., Standiford D. M., Emerson C. P., Jr Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science. 2001;293:1663–1666. doi: 10.1126/science.293.5535.1663. [DOI] [PubMed] [Google Scholar]
- 18.Ai X., Do A. T., Lozynska O., Kusche-Gullberg M., Lindahl U., Emerson C. P., Jr QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J. Cell Biol. 2003;162:341–351. doi: 10.1083/jcb.200212083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Izvolsky K. I., Zhong L., Wei L., Yu Q., Nugent M. A., Cardoso W. V. Heparan sulfates expressed in the distal lung are required for Fgf10 binding to the epithelium and for airway branching. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;285:L838–L846. doi: 10.1152/ajplung.00081.2003. [DOI] [PubMed] [Google Scholar]
- 20.Wang S., Ai X., Freeman S. D., Pownall M. E., Lu Q., Kessler D. S., Emerson C. P., Jr QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4833–4838. doi: 10.1073/pnas.0401028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L., Beeler D. L., Lawrence R., Lech M., Liu J., Davis J. C., Shriver Z., Sasisekharan R., Rosenberg R. D. 6-O-sulfotransferase-1 represents a critical enzyme in the anticoagulant heparan sulfate biosynthetic pathway. J. Biol. Chem. 2001;276:42311–42321. doi: 10.1074/jbc.M101441200. [DOI] [PubMed] [Google Scholar]
- 22.Wu Z. L., Lech M., Beeler D. L., Rosenberg R. D. Determining heparan sulfate structure in the vicinity of specific sulfotransferase recognition sites by mass spectrometry. J. Biol. Chem. 2004;279:1861–1866. doi: 10.1074/jbc.M311398200. [DOI] [PubMed] [Google Scholar]
- 23.Kuberan B., Lech M., Zhang L., Wu Z. L., Beeler D. L., Rosenberg R. D. Analysis of heparan sulfate oligosaccharides with ion pair-reverse phase capillary high performance liquid chromatography-microelectrospray ionization time-of-flight mass spectrometry. J. Am. Chem. Soc. 2002;124:8707–8718. doi: 10.1021/ja0178867. [DOI] [PubMed] [Google Scholar]
- 24.Westling C., Lindahl U. Location of N-unsubstituted glucosamine residues in heparan sulfate. J. Biol. Chem. 2002;277:49247–49255. doi: 10.1074/jbc.M209139200. [DOI] [PubMed] [Google Scholar]
- 25.HajMohammadi S., Enjyoji K., Princivalle M., Christi P., Lech M., Beeler D., Rayburn H., Schwartz J. J., Barzegar S., de Agostini A. I., et al. Normal levels of anticoagulant heparan sulfate are not essential for normal hemostasis. J. Clin. Invest. 2003;111:989–999. doi: 10.1172/JCI15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L., Yoshida K., Liu J., Rosenberg R. D. Anticoagulant heparan sulfate precursor structures in F9 embryonal carcinoma cells. J. Biol. Chem. 1999;274:5681–5691. doi: 10.1074/jbc.274.9.5681. [DOI] [PubMed] [Google Scholar]
- 27.Hegeman A. D., Harms A. C., Sussman M. R., Bunner A. E., Harper J. F. An isotope labeling strategy for quantifying the degree of phosphorylation at multiple sites in proteins. J. Am. Soc. Mass Spectrom. 2004;15:647–653. doi: 10.1016/j.jasms.2003.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Bonenfant D., Schmelzle T., Jacinto E., Crespo J. L., Mini T., Hall M. N., Jenoe P. Quantitation of changes in protein phosphorylation: a simple method based on stable isotope labeling and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2003;100:880–885. doi: 10.1073/pnas.232735599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.