

Abstract
Doxorubicin (DOX), a widely used antitumour drug, causes dose-dependent cardiotoxicity. Cardiac mitochondria represent a critical target organelle of toxicity during DOX chemotherapy. Proposed mechanisms include generation of ROS (reactive oxygen species) and disturbances in mitochondrial calcium homoeostasis. In the present study, we probed the mechanistic link between mitochondrial ROS and calcium in the embryonic rat heart-derived H9c2 cell line and in adult rat cardiomyocytes. The results show that DOX stimulates calcium/calcineurin-dependent activation of the transcription factor NFAT (nuclear factor of activated T-lymphocytes). Pre-treatment of cells with an intracellular calcium chelator abrogated DOX-induced nuclear NFAT translocation, Fas L (Fas ligand) expression and caspase activation, as did pre-treatment of cells with a mitochondria-targeted antioxidant, Mito-Q (a mitochondria-targeted antioxidant consisting of a mixture of mitoquinol and mitoquinone), or with adenoviral-over-expressed antioxidant enzymes. Treatment with GPx-1 (glutathione peroxidase 1), MnSOD (manganese superoxide dismutase) or a peptide inhibitor of NFAT also inhibited DOX-induced nuclear NFAT translocation. Pre-treatment of cells with a Fas L neutralizing antibody abrogated DOX-induced caspase-8- and -3-like activities during the initial stages of apoptosis. We conclude that mitochondria-derived ROS and calcium play a key role in stimulating DOX-induced ‘intrinsic and extrinsic forms’ of apoptosis in cardiac cells with Fas L expression via the NFAT signalling mechanism. Implications of ROS- and calcium-dependent NFAT signalling in DOX-induced apoptosis are discussed.
Keywords: doxorubicin, NFAT, calcium, apoptosis, oxidative stress, Fas ligand (Fas L)
Abbreviations: Ac-DEVD (or -IETD, or -LEHD)-pNA, N-acetyl-Asp-Glu-Val-Asp (or Ile-Glu-Thr-Asp, or Leu-Glu-Thr-Asp) p-nitroanilide; BAPTA-AM, bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid tetrakis(acetoxymethyl ester); (carboxy-)DCF, (carboxy-)2′,7′-dichlorofluorescein; carboxy-H2DCFDA, carboxy-2′,7′-dichlorodihydrofluorescein diacetate; c-FLIP, cellular FLICE-inhibitory protein; CoQ, coenzyme Q10; CsA, cyclosporin A; C3i, caspase 3 inhibitor; C8i, caspase 8 inhibitor; DOX, doxorubicin; DPBS, Dulbecco's phosphate-buffered saline; DPI, diphenyleneiodonium; ECL, enhanced chemiluminescence; Fas L, Fas ligand; FBS, fetal bovine serum; GFP, green fluorescent protein; Gpx-1, glutathione peroxidase 1; HRP, horseradish peroxidase; Mito-Q, a mitochondria-targeted antioxidant consisting of a mixture of mitoquinol and mitoquinone; MnSOD, manganese superoxide dismutase; NFAT, nuclear factor of activated T-lymphocytes; NFAT-I, NFAT inhibitory peptide; NF-κB, nuclear factor κB; ROS, reactive oxygen species; RT-PCR, reverse transcriptase-PCR; Z-DEVD (or -IETD)-FMK, benzyloxycarbonyl-Asp-Glu-Val-Asp (or Ile-Glu-Thr-Asp)-fluoromethylketone
INTRODUCTION
Doxorubicin (DOX) is an antitumour drug that is being widely used in the treatment of a broad spectrum of cancers [1,2]. The clinical efficacy of this drug is compromised due to the development of a severe form of cardiomyopathy and heart failure [3,4]. One of the proposed mechanisms of DOX-induced cardiotoxicity is generation of ROS (reactive oxygen species) via a redox-cycling mechanism of the semiquinone radical intermediate [5–10]. Several enzymes, including cytochrome P450 reductase, NADH dehydrogenase associated with mitochondrial complex I or endothelial nitric oxide synthase (eNOS), have been proposed to catalyse the reductive metabolism of DOX [8–10]. Selective accumulation of DOX in mitochondria, coupled with increased ROS generation, renders cardiomyocytes more vulnerable to DOX toxicity [11,12]. Studies in vivo and in vitro have shown that DOX stimulates disturbances in cellular calcium homoeostasis and mitochondrial calcium loading that are critical for its cardiotoxic mechanism [13,14]. There is now compelling evidence to show that mitochondria play a central role in regulating both DOX-induced apoptosis and calcium homoeostasis [15]. DOX has been shown to stimulate both intrinsic (mitochondria-mediated) and extrinsic [Fas/Fas L (Fas ligand)-mediated] pathways of apoptosis in cellular and in vivo models [16,17]. However, it still remains unclear whether the two pathways are mechanistically linked, or totally independent of each other. Blocking of the Fas/Fas L pathway of apoptosis with a Fas L neutralizing antibody inhibited DOX-induced toxicity in cardiomyocytes [17,18]; however, the Fas-mediated pathway was not a significant factor in several cancer cells [19,20]. Overall, the mechanism(s) by which Fas/Fas L are controlled by DOX are not fully understood.
Calcineurin or PP2B (protein tyrosine phosphatase 2B) is a calcium-dependent phosphatase that is activated by a sustained elevation in intracellular calcium [21]. NFAT (nuclear factor of activated T-lymphocytes) is a calcium/calcineurin-dependent transcription factor that undergoes dephosphorylation by calcineurin, and translocates into the nucleus [21–23]. Dephosphorylated NFAT subsequently binds to specific consensus sequences in DNA, and increases the transcription of target genes. Although NFAT was initially identified in T-cells, recent reports have indicated that NFAT plays an important role as a transducer of the cardiac hypertrophic response [24,25]. NFAT is also implicated as an important transactivator of the Fas L promoter, which can mediate either paracrine or autocrine apoptosis [26,27]. Identification of NFAT in cardiomyocytes, coupled with its ability to induce cardiac hypertrophy/failure and Fas L expression, makes it a crucial transcription factor in promoting DOX-induced cardiomyocyte apoptosis.
In the present study, we investigated whether DOX-dependent mitochondrial ROS and calcium accumulation stimulate the activation of NFAT and Fas/Fas L-mediated apoptosis in rat cardiac cells. Results show that ROS generated from DOX metabolism in mitochondria result in increased cytosolic calcium levels and activate NFAT signalling, which leads to the initiation of the apoptotic cascade.
MATERIALS AND METHODS
Materials
DPI (diphenyleneiodonium), hydrogen peroxide, GSH (glutathione) ethyl ester, the caspase-3 substrate Ac-DEVD-pNA (N-acetyl-Asp-Glu-Val-Asp p-nitroanilide), the caspase-8 substrate Ac-IETD-pNA (N-acetyl-Ile-Glu-Thr-Asp p-nitroanilide), caspase-3 activity kit, DOX and CsA (cyclosporin A) were purchased from Sigma. BAPTA-AM [bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid tetrakis(acetoxymethyl ester)] and Fluo 3-AM were purchased from Molecular Probes Inc. (Eugene, OR, U.S.A.). The calcium ionophore A23187, the cas-pase-3 inhibitor (C3i) Z-DEVD-FMK (benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone), the caspase-8 inhibitor (C8i) Z-IETD-FMK (benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluorome-thylketone), caspase-9 substrate Ac-LEHD-pNA (N-acetyl-Leu-Glu-His-Asp p-nitroanilide), NFAT-I (NFAT inhibitory peptide) and ionomycin were from Calbiochem Inc. Fas L neutralizing antibody and Fas L antibody for immunoblotting were purchased from Transduction Laboratories (Lexington, KY, U.S.A.). Monoclonal antibody for β-actin was purchased from Chemicon (Temecula, CA, U.S.A.). HRP (horseradish peroxidase)-conjugated rabbit anti-mouse antibody was from Pierce Chemical Co. (Rockford, IL, U.S.A.). Polyclonal antibodies that recognize pro- and active forms of caspases 3 and 8 were purchased from Cell Signaling Inc. (Beverly, MA, U.S.A.). Goat anti-rabbit IgG was from Bio-Rad. The first-strand cDNA synthesis and ECL (enhanced chemiluminescence) detection kits were purchased from Amersham Biosciences. Platinum® PCR SuperMix was from Invitrogen. Mito-Q (a mitochondria-targeted antioxidant consisting of a mixture of mitoquinol and mitoquinone) and Mito-undecanol (Mito-Q without the ubiquinone/ubiquinol moiety) were synthesized according to the procedure published previously [28]. Adenoviruses expressing NFAT-4 [Ad-NF4–GFP (green fluorescent protein)] were prepared by the Harvard Gene Therapy Institute Viral Core (Boston, MA, U.S.A.). Adenoviruses encoding MnSOD (manganese superoxide dismutase) and GPx-1 (glutathione peroxidase 1) were generously given by Dr Larry Oberley (Radiation Research Laboratory, The University of Iowa, Iowa City, IA, U.S.A.).
Culturing of rat H9c2 cardiac cells
Cells from the embryonic rat-heart-derived cell line H9c2, obtained from the American type Cell Collection, were transferred to 75 cm2 filter vent flasks (Costar; Cambridge, MA, U.S.A.), grown in DMEM (Dulbecco's modified Eagle's medium) containing 10% (v/v) FBS (fetal bovine serum), L-glutamine (4 mM), penicillin (100 units/ml), streptomycin (100 μg/ml), 1.5 g/l sodium bicarbonate and incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. For experiments, cells were seeded in 60 mm culture dishes and grown to confluence. The day before the start of treatment, medium was replaced with DMEM containing 10% (v/v) FBS. The above conditions were applied to all of the experiments performed in the present study.
Isolation and culturing of adult rat cardiomyocytes
Male Harlan Sprague–Dawley adult rats were anaesthetized with pentobarbital (60 mg/kg intraperitoneal), and hearts were excised and placed into ice-cold saline solution. Hearts were mounted on aortic cannulas and perfused with a buffer (pH 7.3) containing (in mM): NaCl, 125; Hepes, 25; glucose, 11; creatine, 5; taurine, 20; KCl, 4.7; MgSO4, 1.2; and KH2PO4, 1.2. The perfusion buffer was saturated with 100% oxygen, and supplemented with 1 mM CaCl2. After a 10 min perfusion, the buffer was changed to a Ca2+-free buffer. After a 5 min perfusion with Ca2+-free buffer, the perfusion was continued by recirculation of 40 ml of buffer supplemented with collagenase (type II, 200 units/ml; Life Technologies, Inc.) and CaCl2 (25 μM). After 30 min, the ventricular tissue was minced and incubated for 10 min in a recirculating medium with 1% (w/v) BSA and 20 μg/ml deoxyribonuclease. Cells were released from chunks of tissue by gentle pipetting. The cell suspension was filtered through an 80-mesh screen. The cell suspension was washed twice by gentle centrifugation, and then resuspended in a CaCl2-containing buffer. The concentration of CaCl2 in the buffer was successively increased to 0.2, and then 0.5 mM. To separate myocytes, the cell suspension was layered over a 4% BSA solution in a buffer containing 1 mM CaCl2. Ventricular myocytes were allowed to settle, and then plated on to 60 mm dishes pre-coated with laminin (Life Technologies, Inc.). The culture medium contained M-199 (Sigma) supplemented with 25 mM NaHCO3, 25 mM Hepes, 10% (v/v) FBS, 10 μM cytosine D-arabinofuranoside, 2 mg/ml BSA, 100 units/ml penicillin, and 100 μg/ml streptomycin. Intact cardiomyocytes adhered to the culture plates; damaged cells were washed away during the medium change 2 h after plating. Cardiomyocytes were cultured under these conditions for 7 days before incubation with DOX.
Cell treatments
Unless otherwise stated, all experiments were performed in cells cultured in 60 mm culture dishes. Cells were pre-treated for 2 h individually with the following agents: medium alone, 5 μM C3i, 5 μM C8i, 1 μg/ml Fas L neutralizing antibody, 100 nM CsA, 5 μM BAPTA-AM, 10 μM NFAT-I, 5 μM DPI, 1 μM Mito-Q or 2 mM GSH ethyl ester, followed by the addition of DOX to a final concentration of 1 μM. Following an 8 h or 18 h incubation period, cell-culture medium was aspirated, and cells were washed once with DPBS (Dulbecco's phosphate buffered saline) and collected by gentle scraping. The cell pellets were washed twice with DPBS and used for subsequent assays. For the time-course experiments, cells were treated with 1 μM DOX and collected by gentle scraping at different time points. In some experiments, H9c2 cells were treated with 200 μM H2O2 for 7 h.
Caspase measurements
Caspase-3-, -8- and -9-like activities were determined in cell lysates using Ac-DEVD-pNA, Ac-IETD-pNA and Ac-LEHD-pNA as substrates. After collection, cells were suspended in 100 μl of lysis buffer supplied in the caspase activity kit (Sigma) and passed through a 24-gauge needle 10 times to ensure complete lysis. The lysate was centrifuged at 4 °C for 10 min at 13000 rev./min (16000 g), and 50 μl of the clear supernatant was used for the activity assay, as described by the manufacturer. The increase in absorbance at 405 nm was considered as an index of caspase activity.
Overexpression of NFAT4–GFP
Adenoviral infection with Ad-NF4–GFP in cells was performed in serum-free medium for 1 h at a multiplicity of infection (MOI) of 20 particles/cell, followed by the addition of an equal volume of fresh medium containing 20% (v/v) FBS, and incubation was continued for 24 h. These conditions produced nearly 100% transfection with recombinant adenovirus, as assessed by monitoring the GFP expression. The medium was replaced 24 h after infection with a medium containing 10% FBS. The next day, cells were treated with DOX before and after treatment with antioxidant/antioxidant enzymes.
Determination of Fas L expression and caspase activation by immunoblotting
After terminating the experiment, cells were washed with ice-cold DPBS and resuspended in 100 μl of RIPA buffer [20 mM Tris/HCl (pH 7.4)/0.1 mM EDTA/0.1 mM EGTA/1% Nonidet P40/0.1% sodium deoxycholate/0.1% SDS/100 mM NaCl/10 mM NaF]. To a 10 ml solution of the above, the following agents were added: 1 mM sodium vanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 10 μg/ml pepstatin inhibitors. The cells were homogenized by passing the suspension through a 25-gauge needle (10 strokes). The lysate was centrifuged at 16000 g for 10 min, and the supernatant was used for analysis. Protein concentrations were determined using the Lowry method (Bio-Rad), and 30–40 μg of protein was used for Western blot analysis. Proteins were resolved on an SDS/10% polyacrylamide gel and blotted on to nitrocellulose membranes. Membranes were washed with Tris-buffered saline [140 mM NaCl/50 mM Tris/HCl (pH 7.2)] containing 0.1% Tween 20 and 5% non-fat dried milk (Bio-Rad) to block the non-specific binding. Membranes were incubated either with monoclonal antibodies (1 μg/ml) raised against Fas L (Transduction Laboratories) or β-actin (Chemicon), or with polyclonal antibodies (1 μg/ml) that will detect the pro- and active forms of caspase 8 and 3 (Cell Signalling Technology) in Tris-buffered saline containing 0.1% Tween 20 and 1% non-fat dried milk for 2 h at room temperature, washed 5 times, and then incubated with HRP-conjugated rabbit anti-mouse IgG (Pierce) or goat anti-rabbit IgG (Bio-Rad) for 1.5 h at room temperature. Bands were detected using the ECL method (Amersham Biosciences). Statistical significance was determined using the Student's t-test, employing the Sigmastat software.
RT-PCR (reverse transcriptase-PCR) analysis
Following the termination of experiments, the medium was aspirated, 1 ml of TRIzol® reagent (Invitrogen) was added to the cells in 60 mm culture dishes, and total RNA was isolated according to the manufacturer's protocol (Invitrogen). RNA (5 μg) was used for first-strand cDNA synthesis using the first-strand cDNA synthesis kit (Amersham Biosciences) with random hexamers provided in the kit in a final volume of 15 μl. Aliquots (4 μl) of the cDNA mixture was used to amplify mRNA for Fas L and 18 S rRNA (as a loading control) by PCR using the Platinum® PCR SuperMix (Invitrogen). The primers used for PCR were designed using GCG (Genetics Computer Group, Inc.) software, and the sequences were as follows: for Fas L, forward primer 5′-AAATAGCCAACCCCAGCACACC-3′ and reverse primer 5′-CAACTTCTTCTCCTCCATTAGCACC-3′ (the product length was 302 bp); for 18 S rRNA, forward primer 5′-GCTTAATTTGACTCAACACGGGAAACCTCAC-3′ and reverse primer 5′-AACTAAGAACGGCCATGCACCACCAC-3′ (the product size was 112 bp).
Measurement of NFAT translocation
Following the termination of incubation, cellular GFP was monitored using a Nikon fluorescence microscope equipped with FITC filter settings, and the images were captured using the integrated CCD (charge-coupled-device) digital camera. Ionomycin was added to one of the dishes at a concentration of 100 nM for 1 min, which served as a positive control. The percentage of cells demonstrating nuclear localization of NFAT were manually counted in five or six different areas of view, and averaged.
Intracellular calcium measurement
H9c2 cells were treated with DOX (1 μM) in the presence or absence of Mito-Q (1 μM) or DPI (5 μM). Following an 8 h incubation period, Fluo-3 AM (5 μM) was added to the dishes and incubated for a further 45 min. Later, the cells were washed thrice in DPBS (without calcium), and 2 ml of DPBS was added to the cells, which were immediately photographed using a Nikon fluorescence microscope equipped with FITC filter settings. Cells incubated with the calcium ionophore A23187 (1 μM) for 10 min served as a positive control.
Measurement of oxidative stress
The determination of intracellular oxidant production was based on the oxidation of carboxy-H2DCFDA (carboxy-2′,7′-dichlorodihydrofluorescein diacetate) to the fluorescent product, carboxy-DCF (carboxy-2′,7′-dichlorofluorescein). Following treatment of cells with DOX in the presence or absence of interventions, the medium was aspirated, and the cells were washed twice with DPBS before being placed into 1 ml of cell culture medium without FBS. The carboxy-H2DCFDA was added to a final concentration of 10 μM, and cells were incubated for 20 min. The cells were again washed once with DPBS and maintained in 1 ml of culture medium. Intracellular fluorescence was monitored at wavelengths of 480±30 nm (excitation) and 535±40 nm (emission). The fluorescence intensity values from three different fields of view were calculated using the Metamorph software and averaged.
Statistical significance was obtained using Student's t test employing the SigmaStat software.
RESULTS
DOX-induced nuclear NFAT translocation, up-regulation of Fas L and caspase activation in H9c2 cells: effects of calcium/calcineurin inhibitors
The addition of DOX (1 μM) to H9c2 cells induced a significant nuclear translocation of NFAT (≈35%) after 8 h, as monitored by the fluorescence of the GFP fusion protein (Figure 1A). In the presence of 100 nM CsA, an inhibitor of calcineurin activity that prevents the dephosphorylation of NFAT [29], DOX-induced nuclear translocation of NFAT was suppressed (Figure 1A). Treatment of cells with a well-known calcium ionophore, ionomycin (100 nM), for 1 min caused nuclear translocation of NFAT in nearly 80% of the cells (positive control). The percentage of cells demonstrating nuclear translocation of NFAT is shown in Figure 1(B), indicating that the cellular NFAT–GFP protein is functional and responsive to increased calcium levels in cells. Results from the RT-PCR experiments and the densitometric analysis show that the transcription of Fas L mRNA increased during DOX treatment, reaching a maximum after 8–10 h, before declining at 12 h (Figure 1C). A similar trend was also observed with Fas L protein expression under these conditions (Figure 1D). Fas L protein levels continued to increase until 8 h after DOX treatment, and remained elevated up to 14 h. Although there was a decrease in Fas L mRNA at the 14 h time point, protein levels remained unaffected, possibly due to a longer protein turnover rate.
Figure 1. DOX-induced NFAT translocation, Fas L expression and apoptosis.

(A) NFAT–GFP-overexpressing H9c2 cardiac cells were treated with 1 μM DOX in the presence and absence of CsA for 8 h and the fluorescence of GFP fusion protein is shown in control, DOX-treated, and DOX plus CsA-treated cells. The positive control shows the nuclear translocation of NFAT in the presence of calcium ionophore, ionomycin. (B) Percentage of NFAT-translocated cells, as shown in (A). (C) H9c2 cardiac cells were treated with 1 μM DOX for different time periods. After terminating the incubation after 8 h, total RNA was isolated from the cell lysate and RT-PCR was performed using the gene-specific primers for Fas L and 18 S rRNA. 18 S rRNA was used as a loading control. Band intensities were calculated by the densitometric analysis using Alpha Imager Software. Data shown are the means±S.D. of three different experiments. (D) Conditions same as in (C), except that Fas L protein levels were determined by Western blotting using monoclonal antibodies raised against Fas L, followed by a secondary antibody coupled to HRP. Bands were visualized by the ECL detection method, and the densitometric analysis was performed using Alpha Imager software. (E) Conditions are the same as in (C), except that the caspase-3, -8 and -9 activities were measured as a function of time in the supernatant of cell lysates from cells treated with DOX. Results shown are the means±S.D. for three separate experiments. *P<0.05 for (B), (C) and (D); *P<0.01 for (E).
The addition of DOX (1 μM) to H9c2 cells induced a time-dependent increase in caspase-3, -8 and -9 activation (Figure 1E). A nearly 2-fold induction in caspase-3 and -8 activities was noticeable after only 6–8 h in DOX-treated cells. Although caspase-3 activity continued to increase until the 14 h time point, caspase-8 activity increased only up to 8 h, and remained elevated up to 10 h. The mitochondria-dependent apoptotic pathway is activated at later stages, as the increase in caspase-9 activity became evident, starting from 12 h after DOX treatment (Figure 1E).
DOX-induced nuclear translocation of NFAT was significantly attenuated when cells were pre-treated with BAPTA-AM or NFAT-I (Figures 2A and 2B). NFAT-I (sequence: H-Met-Ala-Gly-Pro-His-Pro-Val-Ile-Val-Ile-Thr-Gly-Pro-His-Glu-Glu-OH) binds to the NFAT recognition site on calcineurin, and inhibits dephosphorylation of NFAT [30].
Figure 2. The effect of calcium chelator and NFAT inhibitors on DOX-induced Fas L expression.

(A) NFAT–GFP-overexpressing cells were treated with 1 μM DOX in the presence or absence of BAPTA and NFAT-I for 8 h, and fluorescent microscopic pictures were obtained as described in the Materials and methods section. Cells demonstrating the nuclear translocation of NFAT were manually counted from five different fields of view, and the values indicated are the means±S.D. for three separate experiments. (B) Percentage of cells showing the extent of NFAT-GFP translocation. (C) Experimental conditions were the same as in (A); Western blot analysis of Fas L protein levels in H9c2 cells treated with 1 μM DOX in the presence or absence of Fas L neutralizing antibody (ab), BAPTA, CsA and NFAT-I for 8 h. Data shown are the means±S.D. for three separate experiments. (D) Same as (C), except that RT-PCR for Fas L and 18 S rRNA was performed. Photographs were captured under a UV-transilluminator, and band intensities were calculated using an Alpha Innotech gel documentation system. Results shown are the means±S.D. for three separate experiments. (E) Caspase-3 and -8 activities were determined from cells treated with 1 μM DOX in the presence or absence of C3i and C8i, BAPTA, CsA and NFAT-I. Results shown are the means±S.D. for three separate experiments. *P<0.05 for (B), (C) and (D); *P<0.01 for (E). #P<0.05 compared with the DOX-treated group for (B), (C) and (D); #P<0.01 for (E).
DOX (1 μM) treatment of H9c2 cells for 8 h caused a nearly 2-fold increase in Fas L protein (Figure 2C) and mRNA levels (Figure 2D). Pre-treatment of cells with 5 μM BAPTA-AM (an intracellular calcium chelator), 100 nM CsA (a potent inhibitor of calcineurin) or 10 μM NFAT-I ablated the increase in Fas L mRNA and protein expression in DOX-treated cells (Figures 2C and 2D). A similar trend was also noticed with respect to activation of caspase-3 and -8 activities. Addition of DOX (1 μM) induced a nearly 2.5-fold increase in caspase-3 and -8 activities after an 8 h incubation period. Pre-treatment of cells with BAPTA-AM, CsA or NFAT-I significantly inhibited DOX-induced caspase-3 and -8 activation (Figure 2E; also see Figures 5C and 5D). Addition of the caspase-3 inhibitor Z-DEVD-FMK (5 μM) inhibited DOX-induced caspase-3 activity without greatly affecting the caspase-8 activity during the 8 h time period; however, the caspase-8 inhibitor, Z-IETD-FMK, inhibited both caspase-3 and -8-activities induced by DOX (Figure 2E; also see Figures 5C and 5D). Pre-incubation of cells with a Fas L neutralizing antibody (1 μg/ml) also reduced DOX-induced caspase-3- and -8-like activities (Figure 2E; also see Figures 5C and 5D). These results imply that DOX treatment induces a calcium/calcineurin-dependent increase in NFAT translocation into the nucleus, transcription and translation of Fas L and, subsequently, the activation of caspase-8 and -3 in DOX-treated H9c2 cells.
Figure 5. Effect of DOX on oxidative stress and caspase activation in H9c2 cells.

(A) H9c2 cardiac cells were treated with 1 μM DOX for different time periods. After terminating the incubation, cells were washed and examined for oxidative stress as described in the Materials and methods section. (B) Mean fluorescence intensity values from three different fields of view obtained using Metamorph software. *P<0.05 as compared with the control; #P<0.05 as compared with the DOX-treated group. (C and D) H9c2 cells were treated with DOX in the presence or absence of caspase inhibitors, Fas L neutralizing antibody, BAPTA, CsA and Mito-Q, as well as in cells overexpressing either GPx1 or MnSOD (E and F). Western blot analysis for caspase-3 and -8 was performed as indicated in the Materials and methods section. These experiments were repeated three times, and similar results were obtained.
Mito-Q inhibits DOX-induced NFAT translocation, Fas L expression and apoptosis
To assess whether DOX-induced mitochondrial ROS is responsible for NFAT translocation, cells were pre-treated with DPI (5 μM) and Mito-Q (1 μM). DPI is a non-specific flavoprotein reductase inhibitor that inhibits the redox cycling of DOX. Mito-Q, as described above, is a mitochondria-targeted antioxidant consisting of a mixture of mitoquinol and mitoquinone [28]. A significant reduction in DOX-mediated nuclear translocation of NFAT was observed in DPI- and Mito-Q-treated cells (Figures 3A and 3B). Pre-treatment of H9c2 cells with DPI (5 μM) and Mito-Q (1 μM) also inhibited DOX-mediated Fas L expression (Figure 3C) and caspase-3 and -8 activities (Figure 3D), suggesting that DOX-induced mitochondrial ROS play a crucial role in activating Fas/Fas L-mediated apoptosis. The addition of Mito-undecanol (Mito-Q without the ubiquinone/ubiquinol moiety) or CoQ (coenzyme Q) at similar concentrations had little or no effect on DOX-induced Fas L or caspase activation (Figures 3C and 3D).
Figure 3. Effect of DPI and Mito-Q on DOX-induced caspase activity, Fas L expression and calcium levels.

(A) NFAT–GFP-overexpressing cells were treated with 1 μM DOX in the presence or absence of Mito-Q (1 μM) and DPI (5 μM) for 8 h, and the fluorescence pictures were captured using a Nikon fluorescence microscope equipped with FITC filter settings. (B) Cells demonstrating the nuclear translocation of NFAT–GFP were manually counted from five different fields of view, and the values shown are the means±S.D. for three separate experiments. (C) H9c2 cells were treated with 1 μM DOX in the presence or absence of 5 μM DPI, 1 μM Mito-Q, 1 μM Mito-undecanol or 2 mM GSH ester for 8 h. The Western blot for Fas L was performed. Densitometric results shown are the means±S.D. for three separate experiments. (D) Caspase-3 and -8 activities were determined. Results shown are the means±S.D. for three separate experiments. (E) H9c2 cells were treated with 1 μM DOX in the presence and absence of Mito-Q and DPI for 8 h. After terminating the incubation, cells were washed and incubated with 5 μM Fluo 3-AM for 30 min. The medium was subsequently aspirated, cells were washed twice with DPBS, and fluorescence images were captured in a Nikon fluorescence microscope equipped with FITC filter settings. (F) Fluorescence intensity was calculated using Metamorph software. Values were averaged from five different fields of view, and the results shown are the means±S.D. for three separate experiments. *P<0.05 for (B) (C) and (F); *P<0.01 for (D). #P<0.05 compared with the DOX-treated group for (B), (C) and (F); #P<0.01 for (D).
DOX treatment elevated intracellular calcium levels to nearly 40% over controls during DOX treatment, as measured by an increase in Fluo-3 staining (Figures 3E and 3F). DOX-induced calcium levels were significantly inhibited in cells pre-treated with Mito-Q (1 μM) and DPI (5 μM), suggesting a causal relationship between mitochondrial ROS and elevated calcium levels (Figures 3E and 3F). A23187 (1 μM for 10 min) was added to the cells as a positive control. Taken together, these results demonstrate that DOX-induced mitochondrial elevation of ROS, and subsequent alterations in calcium levels, lead to the activation of calcineurin, NFAT translocation, and transcriptional up-regulation of Fas L that are responsible for priming the apoptotic cascade in H9c2 cells.
Transfection with antioxidant enzymes inhibits DOX-induced NFAT translocation, Fas L expression and caspase activation
To investigate the role of ROS in DOX-induced NFAT translocation, the levels of GPx-1, an intracellular enzymatic scavenger of H2O2, were varied by adenoviral expression of GPx-1 into H9c2 cells. As shown in Figures 4(A) and 4(B), the NFAT translocation was inhibited in GPx-1-transfected cells treated with DOX. Adenovirus-mediated overexpression of MnSOD into H9c2 cells also inhibited NFAT translocation in response to DOX treatment (Figures 4A and 4B). Both Fas-L expression and caspase activation induced by DOX were mitigated in cells overexpressing GPx-1 and MnSOD (Figures 4C and 4D; also see Figures 5E and 5F). These data indicate that enzymatic detoxification of 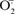 and H2O2 results in the inhibition of DOX-induced NFAT translocation, Fas-L expression and caspase activation.
and H2O2 results in the inhibition of DOX-induced NFAT translocation, Fas-L expression and caspase activation.
Figure 4. Effect of antioxidant enzyme expression on DOX-induced apoptosis and NFAT activation.

H9c2 cells expressing NFAT-GFP were treated with 1μM DOX in the presence or absence of adenoviral overexpression of either MnSOD or GPx1. (A) Following an 8 h incubation, fluorescence pictures were captured using a Nikon fluorescence microscope equipped with FITC filter settings. (B) Cells demonstrating the nuclear translocation of NFAT were manually counted from five different fields of view, and the values shown are the means±S.D. for three separate experiments.(C) Caspase-3 and -8 activities were determined as described in the Materials and methods section. Results shown are the means±S.D. for three separate experiments. (D) Following termination of incubation, cells were collected by gentle scraping, washed thrice with DPBS and lysed in RIPA buffer. Protein (30 μg samples) was resolved with SDS/PAGE (10% gels) and transferred on to a nitrocellulose membrane. Western blot analysis for Fas L was performed as described in Figure 1(D). Results shown are the means±S.D. for three separate experiments. *P<0.01 for (B) and (C); *P<0.05 for (D). #P<0.01 for (B) and (C); #P<0.05 for (D).
Treatment of H9c2 cells with 1 μM DOX induced oxidative stress as early as 6 h, and was significantly increased by 8 h, as evidenced by DCF staining (Figures 5A and 5B). Pre-treatment of cells with either 5 μM DPI or 1 μM Mito-Q, inhibited DOX-induced DCF staining (Figures 5A and 5B).
DOX induces nuclear translocation of NFAT and Fas L up-regulation in adult rat cardiomyocytes
Treatment of primary cultures of cardiomyocytes with 1 μM DOX for 16 h resulted in the nuclear translocation of NFAT in nearly 35–40% of cells, as demonstrated by visualization of NFAT–GFP (Figure 6A). Concomitantly, Fas L expression also increased in a time-dependent manner following DOX treatment (Figure 6B). Pre-treatment of cells with agents that inhibit NFAT activation, NFAT-I, calcium chelator, BAPTA, pharmacological inhibitor of calcineurin, CsA or mitochondrial antioxidant Mito-Q, greatly inhibited DOX-induced Fas L expression (Figure 6C).
Figure 6. Effect of DOX on NFAT translocation and Fas L expression in primary cultures of cardiomyocytes.

(A) NFAT–GFP-overexpressing cardiomyocytes were treated with 1 μM DOX in the presence and absence of CsA for 16 h, and fluorescence photographs were obtained using a Nikon fluorescence microscope equipped with FITC filter settings. (B) Cardiomyocytes were treated with 1 μM DOX for different time intervals and, after terminating the incubation, cells were collected by gentle scraping, washed thrice with DPBS and lysed in RIPA buffer. Protein (30 μg samples) was resolved on SDS/PAGE (10% gels) and transferred on to a nitrocellulose membrane. Western blot analysis for Fas L and β-actin was performed as described in the Materials and Methods section. (C) Treatment conditions were the same as in (B), except that cells were treated with 1 μM DOX either in the absence or presence of CsA (100 nM), NFAT-I (10 μM), BAPTA (5 μM) or Mito-Q (1 μM). These experiments were repeated three times, and similar results were obtained.
Antioxidants and calcium chelator inhibit H2O2-induced NFAT translocation, Fas L expression and apoptosis
We next investigated the effect of exogenously added H2O2. Treatment of H9c2 cells with 200 μM H2O2 for 7 h induced nuclear translocation of NFAT in nearly 30% of cells. Pre-treatment with Mito-Q (1 μM), GSH ester (2 mM) or BAPTA-AM (5 μM) prevented H2O2-induced NFAT translocation into the nucleus (Figures 7A and 7B). A similar trend was observed even in the Fas L expression. H2O2 stimulated nearly 50–60% induction in Fas L protein expression during a 7 h incubation, which was lowered to control levels when cells were pre-incubated with Mito-Q and BAPTA-AM (Figure 7C). These changes were not due to loading differences, as the β-actin levels remained the same under all the conditions (Figure 7C). H2O2-induced caspase-3- and -8-like activities were also reduced to control values upon pre-incubation of cells with Mito-Q and BAPTA (Figure 7D). The results indicate that H2O2 enhances calcium-mediated signalling mechanisms via the mitochondria-dependent pathway, leading to NFAT activation and Fas L expression, and that these effects were mitigated by supplementation with mitochondria-targeted antioxidant and calcium chelator.
Figure 7. Effect of mitochondria-targeted antioxidant and intracellular calcium chelator on H2O2-induced NFAT translocation, Fas L expression and caspase activation.

(A) NFAT–GFP-overexpressing cells were treated with 200 μM H2O2 in the presence or absence of Mito-Q and BAPTA-AM for 7 h, and the fluorescence pictures were captured using a Nikon fluorescence microscope equipped with FITC filter settings. (B) The cells demonstrating nuclear translocation of NFAT–GFP were manually counted from five different fields of view, and the values indicated are the means±S.D. for three separate experiments. H9c2 cells were treated with 200 μM H2O2 in the presence or absence of 1 μM Mito-Q and 5 μM BAPTA-AM for 7 h. After terminating the incubation, cells were collected by gentle scraping, washed thrice with DPBS and lysed in lysis buffer. (C) Protein (30 μg samples) from cells lysed with RIPA buffer were resolved on SDS/PAGE (10% gels) and transferred on to a nitrocellulose membrane. Western blot analysis for Fas L was performed as described in Figure 1(D). Densitometric analysis was performed using an Alpha Innotech gel documentation system. Results shown are the means±S.D. for three separate experiments. (D) Caspase -3 and -8 activities were measured as described earlier. *P<0.01 compared with controls for (D); *P<0.05 for (B) and (C). #P<0.01 compared with the H2O2-treated group for (D) and #P<0.05 for (B) and (C).
DISCUSSION
In the present study, we demonstrated that treatment of rat cardiac cells with DOX activates the transcription factor NFAT, leading to up-regulation of Fas/Fas L-dependent apoptosis through a calcium/calcineurin-signalling pathway. Supplementation of cells with mitochondria-targeted antioxidant, a calcium chelator, or transfection with the antioxidant enzymes GPx-1 and MnSOD, mitigated DOX-induced NFAT signalling, Fas L expression and caspase activation. These results, shown in Scheme 1, provide a mechanistic link between mitochondrial ROS generation and an increase in cytosolic calcium levels in initiating intrinsic and extrinsic pathways of apoptosis during DOX-induced cardiac cell toxicity.
Scheme 1. A schematic representation showing the role of mitochondria, calcium and NFAT in DOX-induced apoptosis in cardiac cells.

DOX undergoes redox cycling, leading to the generation of ROS in mitochondria. Fas L is up-regulated by ROS-dependent calcium/calcineurin-induced translocation of NFAT. NFAT-mediated Fas L signalling leads to activation of caspase-8. Ab, antibody; Cyt C, cytochrome c.
Reports from our and other laboratories showed that intracellular calcium chelator and redox-cycling inhibitor of DOX significantly mitigate DOX-induced apoptosis [31,32]. In the present study, we examined whether BAPTA and DPI could influence DOX-induced Fas L expression and apoptosis. Recent reports indicate that the transcription factor NFAT stimulates Fas L transcription, which is highly sensitive to intracellular levels of calcium [21,26]. Elevated levels of cytosolic calcium activate the calcium-dependent phosphatase, calcineurin, which dephosphorylates NFAT [23]. Dephosphorylated NFAT rapidly translocates to the nucleus and activates target genes, including Fas L ligand [26]. The essential role of mitochondrial ROS in stimulating the cytosolic calcium levels, Fas L expression and apoptosis was inferred from studies using the mitochondria-targeted antioxidant, Mito-Q [28]. Mito-Q, a derivative of ubiquinone, is specifically targeted to mitochondria [28]. Mitochondrial ubiquinone is a respiratory chain component buried within the lipid core of the inner membrane, where it accepts two electrons from complexes I or II, forming the corresponding reduction product (i.e. ubiquinol), which then donates electrons to complex III. The ubiquinone pool in vivo exists largely in the reduced ubiquinol form, acting as an antioxidant and as a mobile electron carrier. Ubiquinol has been reported to function as an antioxidant by donating a hydrogen atom from one of its hydroxyl groups to a lipid peroxyl radical, thereby decreasing lipid peroxidation within the mitochondrial inner membrane [33]. The ubisemiquinone radical formed during this process disproportionates into ubiquinone and ubiquinol [34,35]. The respiratory chain subsequently recycles ubiquinone back to ubiquinol, restoring its antioxidant function. As shown in Figure 3, pre-treatment of cells with Mito-Q greatly mitigated DOX-induced nuclear translocation of NFAT, Fas L expression and caspase-8 and -3 activation. The addition of Mito-undecanol or CoQ had no significant effect on DOX-induced apoptotic signalling pathways, suggesting that the protective effect of Mito-Q is due to its antioxidant property in mitochondria, and not due to its depolarization mechanism.
There is ample evidence in support of the oxidative alterations in mitochondrial calcium regulation as being responsible for cardiomyopathy induced by DOX [11,36]. Recently, DOX-induced decreases in the mitochondrial calcium loading capacity, and the subsequent leakage of calcium from mitochondria, were shown to be responsible for the enhanced susceptibility of cardiomyocytes to DOX [12,36]. However, H2O2 has also been shown to stimulate the Ca2+-release channel from sarcoplasmic reticulum [37]. Reports also suggest that there is a link between endoplasmic reticulum and mitochondrial calcium handling [38,39].
Reports have indicated that both the mitochondria-mediated pathway of apoptosis through caspase-3 and the Fas/Fas L-mediated pathway through caspase-8 are important in DOX-induced cardiotoxicity [16,17]. However, in vivo studies in rats administered DOX indicated a prominent role for the Fas/Fas L-mediated pathway [17]. DOX-induced Fas expression makes cardiomyocytes more vulnerable to Fas/Fas L-mediated apoptosis [17,18,40]. Disturbances in mitochondrial membrane potential alter cellular calcium homoeostasis and initiate Fas/Fas L-mediated apoptosis through NFAT signalling [41]. Although we did not notice a significant elevation in caspase-8 activity in primary cultures of cardiomyocytes under our incubation conditions (results not shown), it is possible that additional regulatory mechanisms, such as those governed by c-FLIP (cellular FLICE-inhibitory protein) [42,43] or matrix metalloproteinases [44], might intervene in myocytes. These factors (e.g. c-FLIP) might be responsible for the increased resistance to DOX-induced apoptosis in primary cardiomyocytes as compared with proliferating myoblasts. Previously, it has been reported that primary cultures of cardiomyocytes are more resistant to Fas L-mediated apoptosis. However, pre-treatment with DOX makes these cells more susceptible to Fas L-mediated apoptosis [18]. Nevertheless, it is evident that DOX induces Fas L through the activation of NFAT in a calcium-dependent mechanism.
Prolonged incubation of H9c2 cells with 1 μM DOX for up to 18 h demonstrated a modest but a significant increase (2.5-fold compared with controls) in caspase-8 activity, compared with a 5–6-fold increase in caspase-3 activity (results not shown). Pre-treatment of cells with BAPTA or Mito-Q significantly reduced caspase-3-, -8- and -9-like activities following DOX-treatment, indicating that calcium and mitochondria play an essential role in mediating apoptosis over extended time periods following treatment with DOX. Pre-treatment of cells with Fas L neutralizing antibody and the caspase-8 peptide inhibitor Z-IETD-FMK significantly reduced DOX-mediated caspase-8 activity. However, caspase-3 activity appeared to be only partially inhibited, and the remaining activity was significantly higher compared with the controls. These results imply that, during longer incubation periods with DOX, although the Fas/Fas L-mediated pathway appears to play a role, a significant proportion of caspase-3 activity may be derived from other mitochondria-dependent mechanisms.
Although Mito-Q supplementation mitigated mitochondria-dependent Fas L expression following treatment with H2O2, the protection did not last over an extended period. Mitochondria may be one of the targets, but not the only target for the extracellularly added oxidants. This scenario is different from the site-specific generation of oxidants with DOX, which is more restricted to mitochondria [11,45]. These results suggest that mitochondrial ROS play a central role in DOX-induced Fas/Fas L-mediated apoptosis through stimulation of NFAT signalling.
Previously, we showed that the transcription factor NF-κB (nuclear factor κB) was activated in response to DOX-induced oxidant stress in endothelial cells and cardiomyocytes [46]. NF-κB activation was shown to be pro-apoptotic under these conditions [46]. In contrast, NF-κB activation by DOX in tumour cells was anti-apoptotic [47]. It was suggested that the dual role of NF-κB in regulating apoptosis could be used to enhance the therapeutic efficacy of DOX. Similarly, calcium-dependent NFAT signalling by DOX may not play a significant role in tumour cells, as these events are induced by mitochondrial ROS generation. These results support our earlier conclusions that the DOX-induced p53-dependent mechanism, but not mitochondrial ROS, plays a major role in tumour cell apoptosis [48]. It is likely that DOX-induced mitochondrial damage is a common denominator for activating several apoptotic signalling pathways, and that the mitochondria-targeted antioxidants may be beneficial in preventing the overall apoptotic cascade.
Acknowledgments
This work was supported by the National Institutes of Health Grant CA77822.
References
- 1.Tan C., Etcubanas E., Wollner N., Rosen G., Gilladoga A., Showel J., Murphy M. L., Krakoff I. H. Adriamycin – an antitumor antibiotic in the treatment of neoplastic diseases. Cancer. 1973;32:9–17. doi: 10.1002/1097-0142(197307)32:1<9::aid-cncr2820320102>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Singal P. K., Iliskovic N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 3.Buzdar A. U., Marcus C., Smith T. L., Blumenschein G. R. Early and delayed clinical cardiotoxicity of doxorubicin. Cancer. 1985;55:2761–2765. doi: 10.1002/1097-0142(19850615)55:12<2761::aid-cncr2820551206>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff D. D., Layard M. W., Basa P., Davis H. L., Jr, Von Hoff A. L., Rozencweig M., Muggia F. M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979;91:710–717. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 5.Bachur N. R., Gordon S. L., Gee M. V. Anthracycline antibiotic augmentation of microsomal electron transport and free radical formation. Mol. Pharmacol. 1977;13:901–910. [PubMed] [Google Scholar]
- 6.Svingen B. A., Powis G. Pulse radiolysis studies of antitumor quinones: radical lifetimes, reactivity with oxygen, and one-electron reduction potentials. Arch. Biochem. Biophys. 1981;209:119–126. doi: 10.1016/0003-9861(81)90263-0. [DOI] [PubMed] [Google Scholar]
- 7.Myers C., Gianni L., Zweier J., Muindi J., Sinha B. K., Eliot H. Role of iron in adriamycin biochemistry. Fed. Proc. 1986;45:2792–2797. [PubMed] [Google Scholar]
- 8.Vasquez-Vivar J., Kalyanaraman B., Martasek P., Hogg N., Masters B. S., Karoui H., Tordo P., Pritchard K. A., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc. Natl. Acad. Sci. U.S.A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalyanaraman B., Joseph J., Kalivendi S., Wang S., Konorev E., Kotamraju S. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol. Cell. Biochem. 2002;235:119–124. [PubMed] [Google Scholar]
- 10.Vasquez-Vivar J., Martasek P., Hogg N., Masters B. S., Pritchard K. A., Jr, Kalyanaraman B. Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry. 1997;36:11293–11297. doi: 10.1021/bi971475e. [DOI] [PubMed] [Google Scholar]
- 11.Wallace K. B. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol. Toxicol. 2003;93:105–115. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhou S., Heller L. J., Wallace K. B. Interference with calcium-dependent mitochondrial bioenergetics in cardiac myocytes isolated from doxorubicin-treated rats. Toxicol. Appl. Pharmacol. 2001;175:60–67. doi: 10.1006/taap.2001.9230. [DOI] [PubMed] [Google Scholar]
- 13.Moore L., Landon E. J., Cooney D. A. Inhibition of the cardiac mitochondrial calcium pump by adriamycin in vitro. Biochem. Med. 1977;18:131–138. doi: 10.1016/0006-2944(77)90084-9. [DOI] [PubMed] [Google Scholar]
- 14.Bachmann E., Zbinden G. Effect of antidepressant and neuroleptic drugs on respiratory function of rat heart mitochondria. Biochem. Pharmacol. 1979;28:3519–3524. doi: 10.1016/0006-2952(79)90394-0. [DOI] [PubMed] [Google Scholar]
- 15.Solem L. E., Heller L. J., Wallace K. B. Dose-dependent increase in sensitivity to calcium-induced mitochondrial dysfunction and cardiomyocyte cell injury by doxorubicin. J. Mol. Cell. Cardiol. 1996;28:1023–1032. doi: 10.1006/jmcc.1996.0095. [DOI] [PubMed] [Google Scholar]
- 16.Kotamraju S., Konorev E. A., Joseph J., Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J. Biol. Chem. 2000;275:33585–33592. doi: 10.1074/jbc.M003890200. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura T., Ueda Y., Juan Y., Katsuda S., Takahashi H., Koh E. Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: In vivo study. Circulation. 2000;102:572–578. doi: 10.1161/01.cir.102.5.572. [DOI] [PubMed] [Google Scholar]
- 18.Yamaoka M., Yamaguchi S., Suzuki T., Okuyama M., Nitobe J., Nakamura N., Mitsui Y., Tomoike H. J. Apoptosis in rat cardiac myocytes induced by Fas ligand: priming for Fas-mediated apoptosis with doxorubicin. J. Mol. Cell Cardiol. 2000;32:881–889. doi: 10.1006/jmcc.2000.1132. [DOI] [PubMed] [Google Scholar]
- 19.Villunger A., Egle A., Kos M., Hartmann B. L., Geley S., Kofler R., Greil R. Drug-induced apoptosis is associated with enhanced Fas (Apo-1/CD95) ligand expression but occurs independently of Fas (Apo-1/CD95) signaling in human T-acute lymphatic leukemia cells. Cancer Res. 1997;57:3331–3334. [PubMed] [Google Scholar]
- 20.Gamen S., Anel A., Lasierra P., Alava M. A., Martinez-Lorenzo M. J., Pineiro A., Naval J. Doxorubicin-induced apoptosis in human T-cell leukemia is mediated by caspase-3 activation in a Fas-independent way. FEBS Lett. 1997;417:360–364. doi: 10.1016/s0014-5793(97)01282-9. [DOI] [PubMed] [Google Scholar]
- 21.Hogan P. G., Chen L., Nardone J., Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 22.Schulz R. A., Yutzey K. E. Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev. Biol. 2004;266:1–16. doi: 10.1016/j.ydbio.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 23.Crabtree G. R., Olson E. N. NFAT signaling: choreographing the social lives of cells. Cell Suppl. 2002;109:S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 24.Wilkins B. J., Dai Y. S., Bueno O. F., Parsons S. A., Xu J., Plank D. M., Jones F., Kimball T. R., Molkentin J. D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2003;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 25.van Rooij E., Doevendans P. A., de Theije C. C., Babiker F. A., Molkentin J. D., de Windt L. J. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J. Biol. Chem. 2002;277:48617–48626. doi: 10.1074/jbc.M206532200. [DOI] [PubMed] [Google Scholar]
- 26.Latinis K. M., Norian L. A., Eliason S. L., Koretzky G. A. Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J. Biol. Chem. 1997;272:31427–31434. doi: 10.1074/jbc.272.50.31427. [DOI] [PubMed] [Google Scholar]
- 27.Su X., Cheng J., Liu W., Liu C., Wang Z., Yang P., Zhou T., Mountz J. D. Autocrine and paracrine apoptosis are mediated by differential regulation of Fas ligand activity in two distinct Jurkat T cell populations. J. Immunol. 1998;160:5288–5293. [PubMed] [Google Scholar]
- 28.Kelso G. F., Porteous C. M., Coulter C. V., Hughes G., Porteous W. K., Ledgerwood E. C., Smith R. A., Murphy M. P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 29.Clipstone N. A., Crabtree G. R. Identification of calcineurin as a key signaling enzyme in T-lymphocyte activation. Nature (London) 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 30.Aramburu J., Yaffe M. B., Lopez-Rodriguez C., Cantley L. C., Hogan P. G., Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- 31.Kalivendi S. V., Kotamraju S., Zhao H., Joseph J., Kalyanaraman B. Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase. Effect of antiapoptotic antioxidants and calcium. J. Biol. Chem. 2001;276:47266–47276. doi: 10.1074/jbc.M106829200. [DOI] [PubMed] [Google Scholar]
- 32.Aoyama M., Grabowski D. R., Dubyak G. R., Constantinou A. I., Rybicki L. A., Bukowski R. M., Ganapathi M. K., Hickson I. D., Ganapathi R. Attenuation of drug-stimulated topoisomerase II-DNA cleavable complex formation in wild-type HL-60 cells treated with an intracellular calcium buffer is correlated with decreased cytotoxicity and site-specific hypophosphorylation of topoisomerase IIalpha. Biochem. J. 1998;336:727–733. doi: 10.1042/bj3360727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagan V. E., Serbinova E. A., Stoyanovsky D. A., Khwaja S., Packer L. Assay of ubiquinones and ubiquinols as antioxidants. Methods Enzymol. 1994;234:343–354. doi: 10.1016/0076-6879(94)34104-4. [DOI] [PubMed] [Google Scholar]
- 34.Ingold K. U., Bowry V. W., Stocker R., Walling C. Autoxidation of lipids and antioxidation by oxidation of human low density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 1993;90:45–49. doi: 10.1073/pnas.90.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Land E. J., Swallow A. J. One-electron reactions in biochemical systems as studied by pulse radiolysis. J. Biol. Chem. 1970;245:1890–1894. [PubMed] [Google Scholar]
- 36.Santos D. L., Moreno A. J., Leino R. L., Froberg M. K., Wallace K. B. Carvedilol protects against doxorubicin-induced mitochondrial cardiomyopathy. Toxicol. Appl. Pharmacol. 2002;185:218–227. doi: 10.1006/taap.2002.9532. [DOI] [PubMed] [Google Scholar]
- 37.Favero T. G., Zable A. C., Abramson J. J. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1995;270:25557–25563. doi: 10.1074/jbc.270.43.25557. [DOI] [PubMed] [Google Scholar]
- 38.Szabadkai G., Rizzuto R. Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood? FEBS Lett. 2004;567:111–115. doi: 10.1016/j.febslet.2004.04.059. [DOI] [PubMed] [Google Scholar]
- 39.Bratton S. B., Cohen G. M. Apoptotic death sensor: an organelle's alter ego? Trends Pharmacol. Sci. 2001;22:306–315. doi: 10.1016/s0165-6147(00)01718-1. [DOI] [PubMed] [Google Scholar]
- 40.Wu S., Ko Y. S., Teng M. S., Ko Y. L., Hsu L. A., Hsueh C., Chou Y. Y., Liew C. C., Lee Y. S. Adriamycin-induced cardiomyocyte and endothelial cell apoptosis: in vitro and in vivo studies. J. Mol. Cell Cardiol. 2002;34:1595–1607. doi: 10.1006/jmcc.2002.2110. [DOI] [PubMed] [Google Scholar]
- 41.Hoth M., Button D. C., Lewis R. S. Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10607–10612. doi: 10.1073/pnas.180143997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nitobe J., Yamaguchi S., Okuyama M., Nozaki N., Sata M., Miyamoto T., Takeishi Y., Kubota I., Tomoike H. Reactive oxygen species regulate FLICE inhibitory protein (FLIP) and susceptibility to Fas-mediated apoptosis in cardiac myocytes. Cardiovasc. Res. 2003;57:119–128. doi: 10.1016/s0008-6363(02)00646-6. [DOI] [PubMed] [Google Scholar]
- 43.Boatright K. M., Deis C., Denault J. B., Sutherlin D. P., Salvesen G. S. Activation of caspases-8 and -10 by FLIP(L) Biochem. J. 2004;382:651–657. doi: 10.1042/BJ20040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wetzel M., Rosenberg G. A., Cunningham L. A. Tissue inhibitor of metalloproteinases-3 and matrix metalloproteinase-3 regulate neuronal sensitivity to doxorubicin-induced apoptosis. Eur. J. Neurosci. 2003;18:1050–1060. doi: 10.1046/j.1460-9568.2003.02838.x. [DOI] [PubMed] [Google Scholar]
- 45.Green P. S., Leeuwenburgh C. Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. Biochem. Biophys. Acta. 2002;1588:94–101. doi: 10.1016/s0925-4439(02)00144-8. [DOI] [PubMed] [Google Scholar]
- 46.Wang S., Kotamraju S., Konorev E., Kalivendi S., Joseph J., Kalyanaraman B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochem. J. 2002;367:729–740. doi: 10.1042/BJ20020752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arlt A., Vorndamm J., Breitenbroich M., Folsch U. R., Kalthoff H., Schmidt W. E., Schafer H. Expression of the NF-kappa B target gene IEX-1 (p22/PRG1) does not prevent cell death but instead triggers apoptosis in Hela cells. Oncogene. 2001;20:69–76. doi: 10.1038/sj.onc.1204061. [DOI] [PubMed] [Google Scholar]
- 48.Wang S., Konorev E. A., Kotamraju S., Joseph J., Kalivendi S., Kalyanaraman B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. Intermediacy of H2O2- and p53-dependent pathways. J. Biol. Chem. 2004;279:25535–25543. doi: 10.1074/jbc.M400944200. [DOI] [PubMed] [Google Scholar]