

Abstract
All 20 cysteine residues are accessible to disulphide reagents in the tubulin dimer, whereas only four are accessible in taxol-stabilized microtubules. Reaction rates with disulphide reagents are a function of the reagent, are decreased by G nucleotides, and increased with increase in pH and urea. With transient (stop-flow) kinetics, DTNB [5,5′-dithiobis-(2-nitrobenzoic acid)] and 2,2′-dithiodipyridine progress curves cannot be fitted by the sum of exponential terms based only on classes of cysteines. The mixed disulphide products react further to form both intra- and intermonomer disulphide bonds that can be reversed by reducing agents. With MMTS (methyl methanethiosulphonate) or ODNB (n-octyl-dithio-2-nitrobenzoate), virtually no protein–protein disulphide bonds are formed and the ODNB reaction can be given as the sum of three exponential terms with pseudo-first-order rate constants of 0.206, 0.069 and 0.010 s−1 at pH 6.5, suggesting three classes of thiol reactivities. Limited cysteine substitution leads to only small changes in tryptophan or CD spectra, whereas complete substitution leads to loss of the helix content. MMTS-induced loss of SH groups leads to progressive increases in the critical concentration and loss of polymerization competence that can be reversed by assembly promoters such as higher protein concentration, taxol or high ionic strength. Under such conditions, the substituted tubulin forms protofilament-based structures such as microtubules, open tubules, sheets and/or bundles.
Keywords: leaving group, mixed disulphide, polymerization, stop-flow kinetics, thiol–disulphide interchange, tubulin
Abbreviations: 2-DTP, 2,2′-dithiodipyridine; DTNB, 5,5′-dithiobis-(2-nitrobenzoic acid); DTT, dithiothreitol; EM, electron microscopy; GdmCl, guanidinium chloride; Mes a.b., Mes assembly buffer; MMTS, methyl methanethiosulphonate; ODNB, n-octyl-dithio-2-nitrobenzoate; TNB, thionitrobenzoate
INTRODUCTION
The tubulin dimer has 20 cysteine residues, of which 12 are in α-tubulin and eight are in β-tubulin. All the cysteine residues except β239 of type III are highly conserved [1] and many appear to be ‘buried’ according to the 3.5 Å (1 Å=10−10 m) electron diffraction structure [2], but are, nevertheless, accessible to reagents. No definitive function has been ascribed to these cysteine residues; no enzymatic redox function is known; cysteines might be transiently involved in folding [3] or oligomerization [4], and it has been suggested that one or two disulphide bonds are required in vivo to promote optimal polymerization [5]. Tubulin can be oxidized by peroxynitrite in vitro [6] and in vivo [7], and these changes can be reversed by the thioredoxin and glutaredoxin systems [8,9]. Nevertheless, cysteine oxidation is usually accompanied by loss of polymerization competence, and it is not clear whether these modifications are only the result of oxidative stress or whether they also function as regulators under normal conditions. Note that fully reduced tubulin is polymerization-competent, forming normal microtubules [10]. Thus a role for the cysteines in tubulin remains unresolved.
In an effort to understand the role of cysteines in tubulin, we have studied their reaction in disulphide interchange reactions. The disulphide reagents DTNB [5,5′-dithiobis-(2-nitrobenzoate], 2-DTP (2,2′-dithiodipyridine), ODNB (n-octyl-dithio-2-nitrobenzoate) and MMTS (methyl methanethiosulphonate) depend on a nucleophilic attack by a protein thiolate on the disulphide bond of the reagent, leading to the formation of mixed disulphides. The reactions studied are depicted in Scheme 1, where PS− is the reacting cysteine residue of the protein and the reaction products are a mixed disulphide of the protein and the second sulphur-containing residue from the reagent. Although many of these reagents contain rather large chromophores, they are more reactive than the alkylating agents, partly because the cleavage products, TNB (thionitrobenzoate) or thiopyridone, are good leaving groups and contribute significantly to reactivity [11–13]. In contrast, differences in the local electrostatic cysteine environments lead to marked differences in their rates of alkylation, even on the protein surface [10]. We have, therefore, asked the following questions: do the factors determining thioether formation in tubulin apply to the formation of disulphide bonds? What is the role of the intrinsic cysteine reactivity when the attacking agent is highly reactive? Are there significant differences among the disulphide reagents? Does charge or hydrophobicity of the disulphide reagent have an effect on reaction rates? And what are the functional consequences of cysteine substitution by disulphide exchange as measured by polymerization competence?
Scheme 1. Thiol–disulphide (‘disulfide’) reactions used in the present study.

EXPERIMENTAL
Materials
DTNB, 2-DTP, ODNB, MMTS, GDP, GTP, ATP and iodoacetamide were purchased from Sigma. Trypsin-TPCK [where TPCK stands for Tos-Phe-CH2Cl (tosylphenylalanylchloromethane)] was obtained from Worthington Biochemical (Freehold, NJ, U.S.A.). Pure (>99%) rat brain tubulin was purified by cycles of warm/cold polymerization as described in [14]. The tubulin concentration was calculated using ε=1.09 mM−1·cm−1 at 276 nm in 6 M GdmCl (guanidinium chloride) by the method of Edelhoch as modified by Pace et al. [15]. The following buffers were used: (i) Mes a.b. (Mes assembly buffer), containing 0.1 M Mes, 1.0 mM MgCl2 and 1.0 mM EGTA (pH 6.9 and pH 6.5 in some experiments); (ii) 0.1 M phosphate buffer, containing 1.0 mM MgCl2 and 1.0 mM EGTA (pH 7.5); and (iii) 0.1 M Tris/HCl, 1.0 mM MgCl2 and 1.0 mM EGTA (pH 8.5). Stock solutions of DTNB (10.0 mM) were prepared in buffer of pH 8.2 and then adjusted to pH 7.0. The concentration was estimated using ε=17.78 mM−1·cm−1 at 324 nm [16]. Stock solutions of ODNB (8.0–9.0 mM) were made in a buffer of pH 6.5 using ε=9.05 mM−1·cm−1 at 338 nm [17]. Stock solutions of 2-DTP (1.5 mM) were prepared in a buffer of pH 6.5 using ε=9.06 mM−1·cm−1 at 281 nm [18].
Methods
For EM (electron microscopy) work, tubulin was modified with MMTS on ice for 90 min at the desired molar ratio (all molar ratios are expressed as mol of reagent/mol of SH group) per SH+10 μM; at these ratios, it was assumed that all of the MMTS had been used up. Polymerization in 0.1 or 0.6 M Mes a.b. and 1 mM GTP was started by the addition of 10% DMSO or 20 μM taxol and 10% DMSO at 37 °C for 10 min. Samples were concentrated on an Airfuge for 5 min at 30 p.s.i. (1 p.s.i.=6.9 kPa) in a prewarmed rotor, supernatants were removed, and pellets were fixed in 2.5% glutaraldehyde and sectioned for EM. For negative staining, 10 μl of samples were placed on grids for 20 s, wicked and washed with 10 μl of Mes a.b., stained with 10 μl of 1% uranyl acetate, wicked and air-dried.
Critical concentrations of control and MMTS-modified tubulin (prepared and polymerized as described above for EM work) were determined in 3 mm light path cells at 350 nm and 37 °C. The best straight line was extrapolated to a D350 (attenuance at 350 nm) of 0.01 for the critical concentration intercept.
Denaturation of tubulin was measured as tryptophan emission after excitation at 295 nm in a PerkinElmer LS50B fluorimeter. Reactions in the presence of increasing concentrations of urea were performed at 4 °C.
CD measurements were made in a Jasco J-715 spectropolarimeter using quartz cuvettes at room temperature (25 °C). In the far-UV region, protein solutions of 200 μg/ml were held in 1 mm path length cuvettes. Spectra were measured from 260 to 190 nm, with four scans at 50 nm/min, time constant=1 s, band width=1 nm and slit width=500 μm. Near-UV spectra were measured with 0.8 mg/ml solutions in 1 cm path length cuvettes, scanning between 320 and 250 nm. The instrument was continually flushed with nitrogen and calibrated with a standard solution of ammonium (−)-10-camphorsulphonate, using the two-point procedure of Chen and Yang [19]. Data were converted into molar ellipticity/residue using the equation:
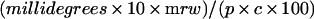 |
1 |
where millidegrees is the measured ellipticity, mrw the mean residue weight of the protein (=111.0), p the path length (cm) and c the protein concentration (mg/ml). Estimates of secondary structure were performed by analysing far-UV data using the CONTIN program [20].
Kinetics
SH modifications of tubulin with DTNB, 2-DTP and ODNB were followed in a Peltier-controlled Cary 300 spectrophotometer at 25 and 4 °C by following the A412 of the product, TNB (ε=14.15 mM−1·cm−1) [21], and, for 2-DTP, by monitoring the A343 of the product, 2-thiopyridone (ε=7.06 mM−1·cm−1) [18]. The reaction was started by adding a small volume (5–10 μl) of tubulin (24–26 μg/μl) to 1.5–3.0 ml of reagent with vigorous stirring and transferred after 15 s to a thermostatically controlled cuvette. As a non-SH-containing control, RNase A was used. DTNB, TNB, 2-DTP and 2-thiopyridone absorbances remained unchanged. ODNB showed a concentration-dependent decrease in A412 in the presence of RNase A, for which corrections were made. All values were given for the SH groups reacted.
Transient (stop-flow) kinetics was measured with an Olis-Rapid Scan spectrophotometer, Olis RSM 1000, at 25 °C under pseudo-first-order conditions with reagent/SH ratios in the range 12–50; the path length was 0.4 cm. Buffers were purged with nitrogen. Spectra from 395 to 545 nm were recorded with scan rates at 1000, 31, 10 and 5 scans/s, depending on the duration of the reactions. Kinetic data at different wavelengths (412, 430, 450 and 470 nm) were extracted and analysed for various fixed excess concentrations of DTNB and various concentrations of tubulin. Each trace was analysed by both OLIS analysis software and SigmaPlot 8.0. ODNB kinetics was measured at ratios of ODNB/SH between 12.5:1 and 25:1.
Estimation of disulphide bonds
Tubulin (10 μM) in Mes a.b. was incubated with 4.0 and 10.0 mM DTNB (20:1 and 50:1 DTNB/SH respectively) at 25 °C under the following conditions: (i) without GdmCl, for 90 min; (ii) with 6 M GdmCl, for 10–15 min; and (iii) with 0.6, 3.0 and 20 mM GTP (60:1, 300:1 and 2000:1 GTP/tubulin), for 2.0 h at 25 °C and 3.0 h at 4 °C. Samples were separated on a Sephadex G25-medium column (25 cm×0.7 cm), and the protein was estimated, adjusted to 2.5 μM with Mes a.b., brought to 6 M GdmCl, and DTT (dithiothreitol) was added to a final concentration of 10 mM. The samples were incubated for 15 min at 25 °C and the A412 was monitored to estimate the number of cysteine residues present as protein/TNB mixed disulphide. Then [20(total SH)−number of SH as protein/TNB mixed disulphide]=number of cysteine residues present as protein–protein disulphide. Samples for CD were prepared as follows: 30 μM tubulin in Mes a.b. at 4 °C was reacted with 12 mM MMTS for 90 min; excess reagent was removed on a Sephadex G25 column equilibrated with 50 mM phosphate buffer (pH 7.0). The ODNB reaction was as for MMTS, but the excess reagent was removed on a Lipidex-1000 column. Fractions were pooled, protein was determined and the pH was adjusted to 7.0 with phosphate buffer.
Gel electrophoresis
Tubulin samples reacted with DTNB, 2-DTP, ODNB and MMTS were separated by SDS/PAGE on NuPAGE 7% Tris/acetate mini-gels (Novex, San Diego, CA, U.S.A.) under non-reducing conditions using the Invitrogen instructions under two different conditions: (1) to quantify the bands due to intermonomer disulphide cross-linking (the higher-molecular-mass bands) by Coomassie Blue staining, we loaded more protein and the electrophoresis was for 40 min; and (2) to see the intramonomer disulphide cross-links, we loaded less protein per lane and electrophoresis was performed for 60 min. Coomassie Blue staining was also adjusted to see the weaker bands; hence the band intensity varied in different gels.
Trypsin digestion and peptide separation
DTNB-modified tubulin samples were digested with trypsin-TPCK (1:20,w/w) at 37 °C for 12–18 h in 0.1 M Tris buffer (pH 8.0) with 5.0 mM CaCl2. To identify the most reactive cysteine residues, tubulin was incubated with DTNB at 4 °C at different molar ratios of DTNB/SH such as 1:20, 3:20 and 5:20 for 45 min. After the DTNB reaction, iodoacetamide was added to 10 mM and incubated for 10 min at 37 °C; then GdmCl was added to a final concentration of 6 M, incubated at 37 °C for 1 h and spun through a P-6 column to remove GdmCl and excess iodoacetamide. The doubly modified tubulin was trypsin-digested and the peptides absorbing at 324 nm were isolated by C-18 reverse-phase HPLC as described in [10]. The 324 nm peaks were concentrated in a Savant Speedvac concentrator and mass spectral analysis and sequencing were performed by the Macromolecular Structure Facility, Michigan State University.
RESULTS AND DISCUSSION
Reagent comparison
Figure 1 depicts a comparison of the reaction of 1.0 μM pure rat brain tubulin (Mes a.b., pH 6.5, at 25 °C) with three disulphide reagents (0.5 mM): ODNB (curve 1), DTNB (curve 2) and 2-DTP (curve 3), at a molar ratio (reagent/SH) of 25:1. The reaction was >95% complete for DTNB by 40 min (curve 2), and for 2-DTP, by 50 min (curve 3), i.e. >19 of the 20 SH cysteine residues have reacted. In contrast, the rate for ODNB is 20 times faster than that for DTNB and 30 times faster than that for 2-DTP. Nevertheless, its reaction with tubulin was not complete over the time span of the experiment, showing that only 18 cysteine equivalents had reacted (curve 1). The remaining two cysteine equivalents of tubulin become instantly accessible in >3 M urea (curve 4). We are inclined to ascribe this high reactivity to the hydrophobicity of the octyl mercaptan moiety [22]. A hydrophobic effect has been demonstrated with the other SH reagents [23,24]. When tubulin was polymerized in the presence of 20 μM taxol, and the washed and dispersed microtubule pellet was exposed to an excess of DTNB over SH groups, only four equivalents of cysteine had reacted by 1 h (curve 5). Other authors have previously found decreased SH accessibility in intact microtubules and N,N′-ethylene bis-iodoacetamide or fluorodinitrobenzene [25,26].
Figure 1. Reaction kinetics of ODNB, DTNB and 2-DTP with tubulin-SH at 25 °C.

Tubulin (1.0 μM=20.0 μM SH) in Mes a.b. (pH 6.5) was treated with 0.5 mM ODNB (curve 1), DTNB (curve 2), 2-DTP (curve 3) or 0.5 mM reagents in the presence of 4 M urea (curve 4). With DTNB and 2-DTP, reactions were 97±1.5% (19.4±0.3 SH) complete, and with ODNB, the reaction was 90±1.5% (18.0±0.3 SH) complete; with 4 M urea, the reaction was 98±1.0% complete within the mixing time. Curve 5, microtubules were assembled in taxol buffer (20 μM taxol and 10% DMSO in Mes a.b.) at 37 °C for 30 min, pelleted, washed twice, resuspended in taxol buffer and then treated with DTNB for different time periods at a DTNB/SH molar ratio of 50:1. Samples were removed at different time points, pelleted and supernatant solutions were assayed at 412 nm.
The apparent biphasic nature of the disulphide exchange reaction of DTNB, namely a fast and a slow phase [27,28], is seen for all three reagents, but, as we shall show below, the kinetics of the reaction is more complex. Attempts to slow the reaction to learn more about the ‘fast’ phase by decreasing the temperature to 4 °C were not successful, even though the concentration of the apparent ‘fast’ cysteines was decreased. The final extent of substitution was not different at 4 °C.
Local denaturation
The possibility that cysteine modification might induce local denaturation was examined first with a known denaturant, urea, on tubulin interactions with disulphide reagents using tryptophan fluorescence [29]. Urea causes a gradual non-linear increase in the rate of reaction of the cysteine residues of tubulin, with 2-DTP starting at concentrations >0.5 M (Figure 2A). An increase in rate already occurs at the concentrations of urea where no significant red shift in tryptophan fluorescence can be detected (ϕ curve). By 4.0 M urea, the reaction with 2-DTP is complete within the mixing time (Figure 2A, inset). The same rate increase occurs with DTNB. Tubulin fully modified with MMTS shows a 2–3 nm red shift in tryptophan emission, comparable with that seen in unmodified tubulin with approx. 2 M urea; this is accompanied respectively by a 23 and 42% decrease in peak fluorescence intensity due either to a change in the tryptophan environment or to static quenching by the newly formed disulphides.
Figure 2. Denaturation and SH reactivity.

(A) Absorption measurements were performed at 4 °C for 5 μM tubulin and 186 μM DTP concentrations in Mes a.b. and results are expressed as initial rates determined graphically. Fluorescence (φ) was measured at room temperature with excitation at 295 nm and 3 mm light path. Open symbols depict absorbance and closed symbols represent fluorescence emission maximum. The inset shows the time course of the DTP reaction in 4 M urea. Reactions were started by tubulin addition and were recorded 20 s later. (B) CD spectra of tubulin in 50 mM phosphate buffer (pH 7.0) at room temperature. ○, control; +, fully modified with ODNB; ———, fully modified with MMTS;········, tubulin in 50 mM Tris/HCl (pH 8.5).
Structural changes resulting from cysteine modification are also revealed by the CD spectra. At neutral pH, tubulin shows a typical α/β protein CD spectrum, with negative peaks at 210 and 222 nm and a positive peak near 190 nm. In the near-UV, a weak negative band at 280 nm indicates an asymmetric environment for some of the aromatic residues in the protein. Increasing the pH of the solution to pH 8.5 produces a conformational change in the protein as shown by a decrease in ellipticity at 222 nm and complete disappearance of the near-UV band (Figure 2B). CD measurements performed on tubulin modified with MMTS to an equivalent of five cysteine residues and ODNB showed only minor changes in conformation as indicated by small changes in their far-UV spectra, which may fall within experimental error. All of the samples maintained most of their negative bands in the 280 nm region (results not shown). CD measurements of tubulin modified at all the 20 cysteine residues with MMTS showed only marginal changes in conformation as suggested by their far-UV spectra. However, tubulin fully modified with the bulkier reagent, ODNB, showed a substantial decrease in secondary structure and lost all the near-UV signal (results not shown). Changes after N-ethylmaleimide treatment (results not shown) were generally similar to those after MMTS treatment.
CONTIN analysis of the CD spectra of the rat brain tubulin controls shown in Figure 2(B) indicates the presence of 28±3% α-helix and 39±11% β-sheet in the unmodified dimer. Chemical modification of up to five cysteine residues did little to perturb its secondary structure. Importantly, the persistence of the near-UV signal in the partially modified proteins indicates that the tertiary structure is also intact. Modification of all SH groups in the protein with MMTS has little effect on the secondary structure, but the introduction of a large number of the bulkier octyl groups results in a loss of the helical structure (CONTIN estimate 21±4%). Most importantly, all of the proteins that are fully modified at the cysteine residues have lost the asymmetric environment of their aromatic residues, indicating changes in tertiary structure. Extensive secondary structure, with no side-chain optical activity, is typical of the molten globule state.
Guanine nucleotides
GTP, required for microtubule assembly under most conditions, is known to interfere with the alkylation of tubulin and cross-linking by bifunctional thiol reagents [30] and can be photo-cross-linked to Cys12 of β-tubulin [31]. Based on the finding that depolymerizing (GDP) microtubules form curved structures or rings [32,33] and yield Raman spectra that show a different α-helix content from GTP-tubulin [34], it has been proposed that the switch from GTP-tubulin to GDP-tubulin leads to significant structural changes. The dimers used in the present study are in the GDP form. Results in Figure 3 show that addition of GTP markedly decreases the rate of reaction of DTNB (curves 3A–3C). GDP is a weaker inhibitor of the reaction of tubulin with DTNB (curves 2A–2C) by a factor of 5–7. This may be due to the extra phosphate on GTP. ATP (1 mM) had no effect (curve 4). A similar preference for GTP over GDP for protection against limited chymotryptic proteolysis of tubulin has been observed [35]. The GTP effect most probably indicates decreased accessibility to a number of cysteines and may be due to structural changes (tightening) of the dimeric structure. However, the fact that added GDP is itself inhibitory, although less so, indicates that additional factors contribute to the G-nucleotide inhibition of the DTNB reaction.
Figure 3. Reaction kinetics of DTNB with tubulin-SH in the presence of GTP, GDP and ATP in Mes a.b. at 25 °C.

Tubulin at 0.5 μM (10.0 μM SH) was reacted with 1.0 mM DTNB in the absence of added nucleotides (curve 1), 30.0 μM GDP (curve 2A), 200 μM GDP (curve 2B) and 1.0 mM GDP (curve 2C); 30.0 μM GTP (curve 3A), 200 μM GTP (curve 3B) and 1.0 mM GTP (curve 3C), or 1.0 mM ATP (curve 4). Tubulin was incubated with nucleotides for 20 min at 20 °C before the addition of DTNB addition.
Reaction rates
The time curve for the DTNB reaction shown in Figure 1, as well as in previous work [27,28], appears to describe two popu-lations of SH groups in tubulin: fast and slowly reacting. In principle, one might expect at least 20 different rates for the formation of the mixed disulphides [10], although they may not be resolved [36,37]. Kinetic analysis assuming a two-cysteine population model yielded unsatisfactory residuals despite correlation coefficients >0.99. Neither an exponential equation with two terms or one with three terms could satisfactorly describe the data.
The primary product of the reaction of DTNB is itself a reactive intermediate for further attack on another thiolate, forming a protein–protein disulphide bond now lacking the chromophore [11,12]. Both inter- and intra-monomer disulphide bond formation are expected to complicate the kinetic analysis; the former would be concentration-dependent, in contrast with the latter. As shown in Figure 4(A), at these tubulin concentrations, an assortment of oligomers is formed as a result of treatment with either DTNB or 2-DTP. At molar ratios of reagent/SH <1.0, disulphide-linked dimers, trimers, tetramers and larger forms are seen in these non-reducing gels. Dimers account for approx. 15% of the total, and higher oligomers for <5%. At a molar ratio of 20, the formation of the disulphide-linked oligomers is decreased but not abolished (Figure 4A, lane 5). Oligomer formation is reversed under reducing conditions (results not shown). Similar results were obtained with 2-DTP. The formation of oligomers occurs early at a molar ratio of DTNB/SH of 20 in the reaction, being detected by 10 s (results not shown).
Figure 4. SDS/PAGE analysis on non-reducing 7% NuPAGE gels (Tris-acetate buffer) of tubulin modified by disulphide reagents.

Coomassie Blue staining. (A) Tubulin (10 μM=200 μM SH) was treated with 2-DTP or DTNB at reagent/SH molor ratios of 0.2, 0.3, 0.5 and 20 in Mes a.b., at pH 6.5 and 25 °C for 60 min; 4 μg of sample/lane was loaded. Lane 1, molecular-mass standards (kDa); lanes 2–5, DTNB-modified tubulin; lanes 6–9, 2-DTP-modified tubulin; lane 10, unmodified tubulin; M, monomer; D, dimer. The broken line and/or * indicates the ideal dimer size. The molar ratios of reagent/SH are listed below the lanes. (B) DTNB-induced mobility changes in monomers as a function of tubulin concentration. Tubulin (0.5, 1 and 2 μM) was treated with 20:1 and 50:1 molar ratios of DTNB/SH in Mes a.b. (pH 6.5) at 25 °C for 60 min and separated. Lane 1, molecular-mass standards; lane 2, untreated control; lanes 3–8, increasing concentrations of tubulin; 0.56 μg of tubulin/lane was loaded. Triplet monomers are labelled 1–3. The arrow indicates control monomer.
Treatment with DTNB or 2-DTP also leads to an increase in monomer electrophoretic mobility. Since intramonomer changes are tubulin-concentration-independent, these side reactions assume great importance in the kinetic studies using low tubulin concentrations. As shown in Figure 4(B), the monomer bands contain triplets, all of which show greater electrophoretic mobility than the control monomers in lane 2. This change is especially marked at the lowest tubulin concentration but persists throughout. It is completely reversed by reduction with DTT.
To learn how many cysteine equivalents are involved in disulphide bond formation, tubulin was reacted with DTNB at reagent/SH molar ratios of 20:1 and 50:1 separated from excess reagent, and the TNB released from tubulin after reduction with DTT was measured (see the Experimental section). At a lower molar ratio approx. 14 TNB/dimer were recovered, and at a molar ratio of 50:1, we found approx. 16 TNB/dimer (Table 1). This suggests that, under the present conditions, a total of three or two disulphide bonds respectively were formed. Although their location has not been identified, the most likely SH pairs calculated from [2] as S–S distances are: in α-tubulin, Cys315–Cys316=3.39 Å and Cys316–Cys376=5.52 Å. In β-tubulin, Cys129–Cys131=5.40 Å and Cys241(239)–Cys356(354)=7.39 Å, except for βIII tubulin. When reactions with DTNB were performed with denatured tubulin, the number of TNB molecules released by DTT was increased by approx. 2, equivalent to a decrease of one disulphide bond, presumably due to an increase in the intercysteine distance in the denatured state. The results also suggest that the conformational changes induced by the nucleotides (Figure 3 and [38–40]) may have displaced some cysteines from ready access for disulphide bond formation. All modifications are reversed with DTT.
Table 1. Estimation of protein disulphide bonds formed by DTNB reaction with tubulin cysteines.
Total number of free cysteine residues in samples 1–5 (estimated by TNB release) was 19.0±0.5. Tubulin (10 μM) was mixed with DTNB and incubated for 60 min at room temperature in Mes a.b. in the absence or presence of 6 M GdmCl or GTP ([GTP]/[tubulin]=60, 300 and 2000); results for all the three GTP levels were identical. (i) After 60 min, the total TNB released was estimated and excess DTNB was separated on a Sephadex G25-medium column. (ii) After estimating the protein in the eluate, excess DTT was added and the total TNB released was estimated. The difference in value between experiments (i) and (ii) gives an estimate of how many disulphide bonds were formed.
| Sample no. | [DTNB]/[tubulin-SH] | No. of free cysteine residues estimated by DTT addition | No. of cysteine residues involved in disulphide bond formation |
|---|---|---|---|
| 1 | 20 | 13.4±0.5 | 6=3 S–S |
| 2 | 50 | 16.1±0.5 | 4=2 S–S |
| 3 | 20 (in GdmCl) | 15.2±0.5 | 4=2 S–S |
| 4 | 50 (in GdmCl) | 17.8±0.5 | 2=1 S–S |
| 5 | 50 (with GTP) | 16.5±0.5 | 3–4=2 S–S |
Next we searched for reagents that are less likely to cause side reactions. The first leaving group resulting from an ODNB reaction with tubulin is TNB; however, the second, octyl mercaptan, now a part of the mixed disulphide, is a poor leaving group that is much less likely to form protein–protein disulphide bonds. Therefore ODNB treatment might yield simpler kinetics. An excellent fit of the data to an equation consisting of the sum of three exponentials
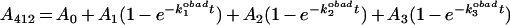 |
2 |
was obtained as shown in Figure 5(A) for pH 6.5. A0 is a possible fast component not resolved here. A1, A2 and A3 are the amplitudes for three different populations of cysteines, and k1, k2, and k3 are the corresponding pseudo-first-order rate constants. The residuals showed no significant deviations from the midline. The means for the A and k values are listed in Table 2. They clearly show three cysteine populations with fast, intermediate and slow kinetics that account for 37, 49 and 12% of the total SH groups of tubulin respectively, or 7, 9 and 2 cysteine residues.
Figure 5. Kinetics of ODNB reaction with tubulin-SH at 25 °C.

Tubulin (1.69 μM=34 μM SH) was mixed with 0.5 mM ODNB in the molar ratio 15:1, in a 4 mm path length cell. The kinetic trace at 412 nm was extracted from scans between 395 and 545 nm. (A) Stop-flow analysis in Mes a.b. (pH 6.5). (B) Stop-flow analysis in 0.1 M Tris/HCl, pH 8.5 (note the change in time scale). Data were fitted to eqn (2) using Olis RSM software; residuals are shown in the insets. Note that the apparently greater noise level is due to the expanded time scale and greater number of points/s.
Table 2. Stop-flow kinetic data for the ODNB reaction with tubulin.
ODNB (1.0 mM) in syringe A and 4 μM tubulin (80 μM SH) in syringe B were mixed in the ratio 1:1 at the onset of the reaction; data were collected at a scan rate of 1/16 ms (scan range 395–545 nm). Data at 412 nm were extracted and fitted to a three-exponential equation using Olis-analysis software. k1, k2 and k3 are first-order rate constants and A1, A2 and A3 are the corresponding amplitudes. The kinetics was measured at 25 °C and at two pH values: pH 6.9 (Mes a.b.) and pH 8.5 (0.1 M Tris/HCl).
| PH | k1 (s−1) | A1 | k2 (s−1) | A2 | k3 (s−1) | A3 |
|---|---|---|---|---|---|---|
| 6.9 | 0.010±0.002 | 0.022±0.002 | 0.069±0.004 | 0.091±0.004 | 0.206±0.01 | 0.068±0.005 |
| 8.5 | 0.096±0.01 | 0.040±0.006 | 0.332±0.02 | 0.084±0.006 | 4.29±0.4 | 0.062±0.004 |
In identical experiments with tubulin and ODNB at pH 8.5 (Figure 5B), all rates were substantially increased, but approximately two cysteine residues were still unavailable. The data were still best-fitted to the equation comprising three exponential terms with essentially flat residuals yielding three larger first-order-rate constants (see Table 2). We did not attempt to calculate pKa values because the rate constants contain factors other than the thiolate concentrations, e.g. tubulin loses its ability for polymerization at pH 8.5 and shows changes in CD [38–40].
From the successful resolution of the ODNB kinetics as the sum of three exponentials, we expected to see fewer side-products than with DTNB. Indeed, there was virtually no oligomer formation at 1 μg of tubulin/μl. An increase in monomer mobility was, nevertheless, still present. Because the octyl mercaptan of the mixed disulphide is a poor leaving group, several arguments have persuaded us that this mobility shift is due to factors other than disulphide bond formation: (i) the dependence on the molar ratio of reagent/SH is the reverse of that seen for the mobility shift with DTNB, i.e. the shift is maximal at maximum substitution. (ii) The ODNB-induced shift was less readily reversed than the DTNB shift. As shown in Figure 6(A), the increase in monomer mobility resulting from ODNB treatment, forming mixed disulphides, was poorly reversed by boiling with 5 mM DTT in contrast with treatment with DTNB, whose variants were instantly decreased by DTT (Figure 6B). (iii) Virtually no mobility shift occurred on treatment of tubulin with MMTS, which adds only a methyl group (results not shown).
Figure 6. Comparison of monomer mobility shifts caused by ODNB and DTNB and their reversal by DTT.

Coomassie Blue staining. Tubulin (2 μM) was treated with 2.0 mM ODNB or DTNB in Mes a.b. at 25 °C for 90 min. (A) ODNB-modified tubulin without (lanes 1–2) or with (lanes 5–9) 5 mM DTT boiled for 0, 1, 3, 5 and 10 min, by loading 2 μg/lane. Lane 1, control; lane 2, ODNB only; lane 3, molecular-mass standards; lane 4, control with DTT. (B) Conditions are the same as for (A) but with DTNB in lanes 2 and 5–9. Large arrows indicate control monomers and small arrows indicate different monomers.
These observations argue against significant protein–protein disulphide bond formation with ODNB and we are inclined to ascribe this mobility shift to the increased hydrophobicity resulting from the addition of octyl groups.
Localization of the reactive cysteine residues
To identify the cysteines that will first react with DTNB, we proceeded as before [10] by using low molar ratios of DTNB/SH as follows: 1, 3 and 5 at 4 °C for 45 min. Unreacted cysteine residues were treated with 10 mM iodoacetamide in the presence of 6 M GdmCl for 1 h at 37 °C; this was followed by removal of excess iodoacetamide and GdmCl on a P-6 column. Although the yields were low, tryptic peptides separated on a reverse-phase C-18 column permitted the identification of three peptides containing cysteine residues Cys347α (α here refers to α-tubulin in this context), Cys241β and Cys356β. Cys347 of α-tubulin was an expected product since it proved to be the most reactive tubulin SH as shown by reaction with chloroacetamide [10] and it is relatively surface-exposed in the dimer. The two β-cysteine residues found above were also seen by alkylation. However, other reactive cysteine residues expected from alkylation reactions, namely Cys305α, Cys315/316α and Cys376α, were lost to disulphide formation by DTNB and could not be reduced without loss of the initial substitution. Radioactive MMTS was too unstable to use in these studies; hence ready identification of the critical cysteine residues with this reagent was not possible.
Effects on the critical concentration
To differentiate the effects of a loss of SH groups from introduction of bulky substituents, we switched from ODNB to MMTS, the smallest disulphide reagent. The functional consequences of cysteine modification by MMTS were judged by their ability to form microtubules. Polymerization competence was measured under three different solvent conditions: 0.1 M Mes a.b. (our standard condition), 0.6 M Mes a.b. and 20 μM taxol in 0.1 M Mes a.b., all at pH 6.9. As shown in Figure 7(A), under standard buffer conditions, tubulin substituted with 1 equivalent of MMTS/mol of dimer showed only a small loss of competence, whereas tubulin with 2 MMTS/dimer polymerized only at higher protein concentrations and exhibited a marked increase in the latent period (Figure 7A, inset). Substitution with 3 MMTS/dimer abolished polymerization competence completely, but tubulin could be polymerized at double the protein concentration (results not shown). This showed that MMTS substitution increases the critical concentration and suggested that agents that lower the critical concentration should promote polymerization of the modified tubulin. The effect of 20 μM taxol or 0.6 M Mes a.b. on unmodified tubulin is depicted in Figure 7(B). Note that the tubulin concentrations were adjusted to facilitate measurement. Taxol promoted assembly even when an equivalent of five cysteine residues had been substituted (Figure 7C), but cold depolymerization (at 4 °C with 5–10 mM calcium) was never complete even when unmodified tubulin was used (results not shown). Hence, for taxol, this criterion was not reliable. This preparation was, nevertheless, fully polymerization-competent after treatment with DTT. When the buffer concentration was increased from 0.1 to 0.6 M, the 3 MMTS/dimer tubulin could now be cold-reversibly polymerized (Figure 7D). With 4 MMTS/dimer tubulin, the observed light scattering in the presence of 0.6 M buffer was only partially reversed by cold exposure, suggesting that non-specific aggregation occurred at the same time as microtubule assembly. At modification levels in excess of 4, only non-specific aggregation was detected by light scattering.
Figure 7. Changes in the critical concentrations produced by MMTS.

Tubulin (TB) was treated with 1–5 mol of MMTS/dimer for 90 min at 4 °C and then polymerized at 37 °C as described in the Methods subsection. (A) Effect of MMTS on tubulin polymerization in 0.1 M Mes a.b. The inset shows typical time courses: A, control; B, 1 MMTS/dimer; C, 2 MMTS/dimer; and D, 3 MMTS/dimer. (B) Effect of polymerization promoters on unmodified tubulin in 0.1 M Mes a.b. without or with 20 μM taxol or 0.6 M Mes a.b. at the critical concentration. (C) Effect of MMTS on tubulin polymerization in 0.1 M Mes a.b. +20 μM taxol. (D) Effect of MMTS on tubulin polymerization in 0.6 M Mes a.b. Note the changes in scale.
Over the limited range of cysteine substitutions examined here, the critical concentration, i.e. the dimer concentration in equilibrium with polymer, controls the polymerization competence of the modified tubulin. As shown in Figure 8, the critical concentrations observed with unmodified tubulin were lowered by 5- and 10-fold with 0.6 M Mes a.b. and 20 μM taxol respectively. Substitution with MMTS increased the critical concentrations as a function of the equivalents of cysteine modified. It was important to check the possibility that the chemical modifications are not equally distributed throughout the tubulin population, i.e. consisting of a mixture of more- and less-modified tubulins, the latter being polymerized by taxol etc. To investigate this, we separated the microtubule pellet from its supernatant solution by warm centrifugation and measured the degree of modification in each fraction. No difference was found. Thus a selective polymerization of the less-modified tubulin is not likely.
Figure 8. Critical concentrations for polymerization of MMTS-modified tubulin.

Data obtained from experiments like those of Figure 9 extrapolated to a D350 of 0.01. Topmost curve, 0.1 M Mes a.b.; middle curve, 0.6 M Mes a.b.; bottom curve, 20 μM taxol in 0.1 M Mes a.b.
It remained to be determined whether or not polymers made from MMTS-modified tubulin were, in fact, microtubules. Under all conditions, normally appearing microtubules were present as shown by negative staining. However, other protofilament-derived structures were also present, including microtubule bundles and protofilament sheets. Polymers were collected by centrifugation in an Airfuge, fixed with glutaraldehyde and sectioned. The protein concentrations were adjusted to achieve roughly similar D350 values. As shown in Figures 9(A) and 9(B), microtubules made under standard buffer conditions from 2 MMTS/dimer tubulin were similar to microtubules made from unmodified tubulin. The presence of taxol yielded microtubules plus a great abundance of sheets and bundles that did not align well in the centrifugal field (Figure 9C); a better alignment was observed after slower rates of polymerization. To our surprise, pretreatment with 4 MMTS/dimer before taxol showed a much larger proportion of normal microtubules (Figure 9D) approaching the distribution seen in untreated tubulin (Figure 9A). Similar findings were observed after treatment with 0.6 M Mes a.b. In unmodified tubulin, there was a preponderance of sheets (Figure 9E). Again, pretreatment with 3 MMTS/dimer changed the distribution, yielding a higher concentration of normally appearing microtubules (Figure 9F). With 5 MMTS/dimer in 0.6 M Mes a.b., no microtubules were formed and only aggregates were seen. These results suggest that the light-scattering data should be interpreted with caution.
Figure 9. Effect of polymerization promoters on microtubule assembly of MMTS-modified tubulin.

(A, C, E) unmodified tubulin; (B, D, F) MMTS-modified tubulin. (B) 2 MMTS/dimer; (D) 4 MMTS/dimer; (F) 3 MMTS/dimer. (A, B) In 0.1 M Mes a.b.; (C, D) in 20 μM taxol in 0.1 M Mes a.b.; (E, F) in 0.6 M Mes a.b. All magnifications are×123000.
Whatever the mechanism behind these changes in critical concentration and structure, it is clear that tubulin, no longer able to polymerize under standard conditions, can still form microtubules and protofilament-derived structures when the critical concentration is decreased by taxol or high buffer concentrations.
Comments
The overall rate of reaction of tubulin cysteine residues can be considered as the product of two component reactivities: that of the thiol/thiolate and that of the reagent. Thiol reactivity is determined by the pH (i.e. ∼104 to 105 times higher reactivity of the thiolate anion compared with the thiol), the electrostatics of the local environment, the accessibility of the cysteines, the nature of the G-nucleotide present (for tubulin), the native status of the protein and possible local denaturation effects. Reagent reactivity is a function of its leaving group and hence the possibility of side reactions, size and its effect on the structure, polarity, and charge compatibility between the approaching reagent and the local electrostatic field around the cysteine. The relative contribution of these factors to the total rate depends on the reactivity of the attacking reagent. Thus, for ‘slow’ reagents such as chloro- or iodo-acetamide, the local environment contributes a larger fraction to the total rate than for ‘fast’ reagents used in disulphide interchange [10]. The success of a productive interaction with a reagent depends, partly, on the fraction of total time that structural fluctuations present the so-called ‘open’ state, a highly active reagent such as DTNB may be successful, whereas a less reactive reagent, e.g. iodoacetamide, is probably not.
The difference in the kinetics of DTNB and ODNB appears to result from at least two factors: the nature of the mixed disulphide product and the rate of the reaction. Since the reactivity of disulphides is a function of the pKa values of the component thiols that form them [11–13], the mixed alkyl/alkyl disulphide product of the ODNB reaction with tubulin will be much more stable than that formed by DTNB (alkyl/aryl). The protein/thionitrobenzoate or the protein/thiopyridone disulphides are very reactive intermediates retaining a good leaving group. This promotes interaction with other protein thiolates under favourable conditions, leading to the formation of either intra- or inter-protein disulphide bonds (Figure 4). In contrast, the n-octyl mercaptan of the tubulin/octyl mixed disulphide is a poor leaving group and is stable enough such that virtually no oligomers are formed. This, in turn, permits kinetic analysis.
The choice of the reagent is important not only for the differing propensities to form intra- and inter-protein–protein disulphide bonds, but also, e.g., for the functional consequences (polymerization competence) of reacting limited numbers of cysteines in tubulin with reagents of different size, e.g. as seen in the CD spectrum and the greater damage done by N-ethylmaleimide than MMTS (P. J. Britto and P. McPhie, unpublished work). This is further supported by a comparison of thiol–disulphide interchange with cysteine oxidation. Khan and Ludueña [41] first proposed that modification of certain cysteine residues of tubulin might regulate the dimer/microtubule equilibrium and that the thioredoxin system might, in turn, regulate this equilibrium. The fact that disulphide bond formation after oxidative stress is reversible confirms this to be a possible regulatory function, although oxidation of other proteins must occur simultaneously [6–9,42]. Despite a greater degree of cysteine modification produced by peroxynitrite [6], it induces a smaller loss of polymerization competence than do alkylating or disulphide interchange reagents.
The present results show that substitution of the cysteine residues of tubulin increases the critical concentration and eventually yields tubulin that is not capable of assembling into microtubules. However, conditions that decrease the critical concentration, such as taxol, high ionic strength, or increases in dimer concentration, can restore protofilament formation and microtubule assembly of the modified tubulin. Glutathiolation of tubulin, which we (L. Knipling and J. Wolff, unpublished work) and others [6,7,42] have observed, may be the natural mediator for such thiol–disulphide interchange and can, in turn, regulate the dimer–polymer equilibrium by its effect on the critical concentration.
Acknowledgments
We thank Dr E. Tekle and Dr B. Chock (National Heart Lung and Blood Institute, National Institutes of Health, Bethesda, MD, U.S.A.) for the use of, and help with, the stop-flow instrument and Dr D. Sackett (National Institute of Child Health and Human Development, National Institutes of Health) and Dr B. Chock for a critical reading of this paper.
References
- 1.Cowan N. Tubulin genes and the diversity of microtubule function. Oxford Surveys on Eukaryotic Genes. In: Maclean N., editor. Oxford, U.K.: University of Oxford Press; 1984. pp. 26–60. [PubMed] [Google Scholar]
- 2.Lowe J., Li H., Downing K. H., Nogales E. Refined structure of alpha beta-tubulin at 3.5 A resolution. J. Mol. Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 3.Robinson A. S., King J. Disulphide-bonded intermediate on the folding and assembly pathway of a non-disulphide bonded protein. Nat. Struct. Biol. 1997;4:450–455. doi: 10.1038/nsb0697-450. [DOI] [PubMed] [Google Scholar]
- 4.Li P. P., Nakanishi A., Clark S. W., Kasamatsu H. Formation of transitory intrachain and interchain disulfide bonds accompanies the folding and oligomerization of simian virus 40 Vp1 in the cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1353–1358. doi: 10.1073/pnas.032668699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaudhuri A. R., Khan I. A., Ludueña R. F. Detection of disulfide bonds in bovine brain tubulin and their role in protein folding and microtubule assembly in vitro: a novel disulfide detection approach. Biochemistry. 2001;40:8834–8841. doi: 10.1021/bi0101603. [DOI] [PubMed] [Google Scholar]
- 6.Landino L. M., Hasan R., McGaw A., Cooley S., Smith A. W., Masselam K., Kim G. Peroxynitrite oxidation of tubulin sulfhydryls inhibits microtubule polymerization. Arch. Biochem. Biophys. 2002;398:213–220. doi: 10.1006/abbi.2001.2729. [DOI] [PubMed] [Google Scholar]
- 7.Lind C., Gerdes R., Hamnell Y., Schuppe-Koistinen I., von Lowenhielm H. B., Holmgren A., Cotgreave I. A. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch. Biochem. Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 8.Landino L. M., Iwig J. S., Kennett K. L., Moynihan K. L. Repair of peroxynitrite damage to tubulin by the thioredoxin reductase system. Free Radical Biol. Med. 2004;36:497–506. doi: 10.1016/j.freeradbiomed.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 9.Landino L. M., Moynihan K. L., Todd J. V., Kennett K. L. Modulation of the redox state of tubulin by the glutathione/glutaredoxin reductase system. Biochem. Biophys. Res. Commun. 2004;314:555–560. doi: 10.1016/j.bbrc.2003.12.126. [DOI] [PubMed] [Google Scholar]
- 10.Britto P. J., Knipling L., Wolff J. The local electrostatic environment determines cysteine reactivity of tubulin. J. Biol. Chem. 2002;277:29018–29027. doi: 10.1074/jbc.M204263200. [DOI] [PubMed] [Google Scholar]
- 11.Parker A. J., Kharasch N. Derivatives of sulfenic acids. XXXVI. The ionic scission of the sulfur-sulfur bond. Part 1. J. Am. Chem. Soc. 1960;82:3071–3075. [Google Scholar]
- 12.Pryor W. New York, NY: McGraw-Hill Book Co.; 1962. Mechanisms of Sulfur Reactions. [Google Scholar]
- 13.Gilbert H. F. Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. Relat. Areas Mol. Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 14.Wolff J., Knipling L., Sackett D. L. Charge-shielding and the ‘paradoxical’ stimulation of tubulin polymerization by guanidine hydrochloride. Biochemistry. 1996;35:5910–5920. doi: 10.1021/bi9527395. [DOI] [PubMed] [Google Scholar]
- 15.Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riddles P. W., Blakeley R. L., Zerner B. Ellman's reagent: 5,5′-dithiobis(2-nitrobenzoic acid) – a reexamination. Anal. Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- 17.Faulstich H., Tews P., Heintz D. Determination and derivatization of protein thiols by n-octyldithionitrobenzoic acid. Anal. Biochem. 1993;208:357–362. doi: 10.1006/abio.1993.1061. [DOI] [PubMed] [Google Scholar]
- 18.Brocklehurst K., Little G. Reactions of papain and of low-molecular-weight thiols with some aromatic disulphides. 2,2′-dipyridyl disulphide as a convenient active-site titrant for papain even in the presence of other thiols. Biochem. J. 1973;133:67–80. doi: 10.1042/bj1330067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen G. C., Yang J. T. Two-point calibration of circular dichrometer with d-10-camphorsulfonic acid. Anal. Lett. 1977;10:1195–1207. [Google Scholar]
- 20.Provencher S. W., Glockner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 21.Eyer P., Worek F., Kiderlen D., Sinko G., Stuglin A., Simeon-Rudolf V., Reiner E. Molar absorption coefficients for the reduced Ellman reagent: reassessment. Anal. Biochem. 2003;312:224–227. doi: 10.1016/s0003-2697(02)00506-7. [DOI] [PubMed] [Google Scholar]
- 22.Rossi R., Barra D., Bellelli A., Boumis G., Canofeni S., Di Simplicio P., Lusini L., Pascarella S., Amiconi G. Fast-reacting thiols in rat hemoglobins can intercept damaging species in erythrocytes more efficiently than glutathione. J. Biol. Chem. 1998;273:19198–19206. doi: 10.1074/jbc.273.30.19198. [DOI] [PubMed] [Google Scholar]
- 23.Robinson G. W., Bradshaw R. A., Kanarek L., Hill R. L. The thiol groups of fumarase. J. Biol. Chem. 1967;242:2709–2718. [PubMed] [Google Scholar]
- 24.Kveder M., Krisko A., Pifat G., Steinhoff H. J. The study of structural accessibility of free thiol groups in human low-density lipoproteins. Biochim. Biophys. Acta. 2003;1631:239–245. doi: 10.1016/s1388-1981(03)00022-2. [DOI] [PubMed] [Google Scholar]
- 25.Ludueña R. F., Roach M. C. Interaction of tubulin with drugs and alkylating agents. 1. Alkylation of tubulin by iodo[14C]acetamide and N,N′-ethylenebis(iodoacetamide) Biochemistry. 1981;20:4437–4444. doi: 10.1021/bi00518a031. [DOI] [PubMed] [Google Scholar]
- 26.Lee Y. C., Yaple R. A., Baldridge R., Kirsch M., Himes R. H. Inhibition of tubulin self-assembly in vitro by fluorodinitrobenzene. Biochim. Biophys. Acta. 1981;671:71–77. doi: 10.1016/0005-2795(81)90095-7. [DOI] [PubMed] [Google Scholar]
- 27.Roychowdhury M., Sarkar N., Manna T., Bhattacharyya S., Sarkar T., Basusarkar P., Roy S., Bhattacharyya B. Sulfhydryls of tubulin. A probe to detect conformational changes of tubulin. Eur. J. Biochem. 2000;267:3469–3476. doi: 10.1046/j.1432-1327.2000.01369.x. [DOI] [PubMed] [Google Scholar]
- 28.Di Simplicio P., Tiezzi A., Moscatelli A., Bianco M. T., Cresti M. The SH-SS exchange reaction between the Ellman's reagent and protein containing SH groups as a method for determining conformational states: tubulin. Ital. J. Biochem. 1989;38:83–90. [PubMed] [Google Scholar]
- 29.Sackett D. L., Bhattacharyya B., Wolff J. Local unfolding and the stepwise loss of the functional properties of tubulin. Biochemistry. 1994;33:12868–12878. doi: 10.1021/bi00209a019. [DOI] [PubMed] [Google Scholar]
- 30.Ludueña R. F., Roach M. C. Tubulin sulfhydryl groups as probes and targets for antimitotic and antimicrotubule agents. Pharmacol. Ther. 1991;49:133–152. doi: 10.1016/0163-7258(91)90027-j. [DOI] [PubMed] [Google Scholar]
- 31.Shivanna B. D., Mejillano M. R., Williams T. D., Himes R. H. Exchangeable GTP binding site of beta-tubulin. Identification of cysteine 12 as the major site of cross-linking by direct photoaffinity labeling. J. Biol. Chem. 1993;268:127–132. [PubMed] [Google Scholar]
- 32.Melki R., Carlier M. F., Pantaloni D., Timasheff S. N. Cold depolymerization of microtubules to double rings: geometric stabilization of assemblies. Biochemistry. 1989;28:9143–9152. doi: 10.1021/bi00449a028. [DOI] [PubMed] [Google Scholar]
- 33.Muller-Reichert T., Chretien D., Severin F., Hyman A. A. Structural changes at microtubule ends accompanying GTP hydrolysis: information from a slowly hydrolyzable analogue of GTP, guanylyl (alpha,beta)methylenediphosphonate. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3661–3666. doi: 10.1073/pnas.95.7.3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Audenaert R., Heremans L., Heremans K., Engelborghs Y. Secondary structure analysis of tubulin and microtubules with Raman spectroscopy. Biochim. Biophys. Acta. 1989;996:110–115. doi: 10.1016/0167-4838(89)90102-7. [DOI] [PubMed] [Google Scholar]
- 35.Maccioni R. B., Seeds N. W. Limited proteolysis of tubulin: nucleotide stabilizes an active conformation. Biochemistry. 1983;22:1567–1572. doi: 10.1021/bi00276a007. [DOI] [PubMed] [Google Scholar]
- 36.Banas T., Banas B., Wolny M. Kinetic studies of the reactivity of the sulfhydryl groups of glyceraldehyde-3-phosphate dehydrogenase. Eur. J. Biochem. 1976;68:313–319. doi: 10.1111/j.1432-1033.1976.tb10790.x. [DOI] [PubMed] [Google Scholar]
- 37.Connors K. New York, NY: VCH Publishers; 1990. Chemical Kinetics. [Google Scholar]
- 38.Olmsted J. B., Borisy G. G. Characterization of microtubule assembly in porcine brain extracts by viscometry. Biochemistry. 1973;12:4282–4289. doi: 10.1021/bi00745a037. [DOI] [PubMed] [Google Scholar]
- 39.Gaskin F., Cantor C. R., Shelanski M. L. Turbidimetric studies of the in vitro assembly and disassembly of porcine neurotubules. J. Mol. Biol. 1974;89:737–755. doi: 10.1016/0022-2836(74)90048-5. [DOI] [PubMed] [Google Scholar]
- 40.Regula C. S., Pfeiffer J. R., Berlin R. D. Microtubule assembly and disassembly at alkaline pH. J. Cell Biol. 1981;89:45–53. doi: 10.1083/jcb.89.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan I. A., Ludueña R. F. Possible regulation of the in vitro assembly of bovine brain tubulin by the bovine thioredoxin system. Biochim. Biophys. Acta. 1991;1076:289–297. doi: 10.1016/0167-4838(91)90280-d. [DOI] [PubMed] [Google Scholar]
- 42.Cumming R. C., Andon N. L., Haynes P. A., Park M., Fischer W. H., Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. J. Biol. Chem. 2004;279:21749–21758. doi: 10.1074/jbc.M312267200. [DOI] [PubMed] [Google Scholar]