

Abstract
The cytokine macrophage migration inhibitory factor (MIF) has emerged to be an important regulator of the inflammatory response and is critically involved in the development of septic shock, arthritis, and glomerulonephritis. Although the biological activities of MIF are presumed to require a receptor-based mechanism of action, the protein is also a tautomerase and has a catalytically active N-terminal proline that is invariant in structurally homologous bacterial isomerases. This observation raises the possibility that MIF may exert its biological action via an enzymatic reaction. Physiologically relevant substrates for MIF have not been identified, nor have site-directed mutagenesis studies consistently supported the requirement for a functional catalytic site. Small molecule inhibitors of MIF's isomerase activity also have been developed, but none have been shown yet to inhibit MIF biological activity. We report herein that the iminoquinone metabolite of acetaminophen, N-acetyl-p-benzoquinone imine (NAPQI), inhibits both the isomerase and the biological activities of MIF. The reaction between NAPQI and MIF is covalent and produces a NAPQI-modified MIF species with diminished cell binding activity and decreased recognition by anti-MIF mAb. These data are consistent with a model by which the NAPQI reacts with the catalytic Pro-1 of MIF to disrupt the integrity of epitope(s) critical to MIF's biological activity and point to the importance of the catalytic domain, but not the catalytic activity per se, in MIF function. These results also point to a powerful approach for the design of small molecule inhibitors of MIF based on interaction with its catalytic site and constitute an example of a pharmacophore capable of irreversibly inhibiting the action of a proinflammatory cytokine.
Macrophage migration inhibitory factor (MIF) was defined originally as an activity responsible for the inhibition of random macrophage migration during the delayed-type hypersensitivity response (1, 2). With the cloning and preparation of pure, recombinant MIF (3, 4), several new biological functions for this mediator have emerged. MIF is released by the anterior pituitary gland as a consequence of the systemic stress response (5, 6) and is expressed by macrophages (7), T cells (1, 2, 8), and eosinophils (9) after immune/inflammatory activation. MIF has the capacity to override, or counterregulate, the immunosuppressive effects of glucocorticoids (5, 6, 8, 10), which accounts in part for the proinflammatory role of MIF in conditions such as septic shock (5, 6, 11–13), arthritis (14–16), and glomerulonephritis (17). Additional biological activities for MIF include the regulation of macrophage and T cell activation (7, 8), IgE synthesis (18), insulin release (19) and carbohydrate metabolism (20, 21), cell growth and apoptosis (22, 23), and tumor angiogenesis (24).
MIF was discovered to tautomerize the nonphysiological substrates D-dopachrome and L-dopachrome methyl ester (ref. 25; Fig. 1). Elucidation of the three-dimensional crystal form of MIF revealed it to be a homotrimer and related structurally to the small bacterial isomerases 4-oxalocrotonate tautomerase, 5-carboxymethyl-2-hydroxymuconate, and chorismate mutase (26). More recently, phenylpyruvic acid, p-hydroxyphenylpyruvic acid, 3,4-dihydroxyphenylaminechrome, and norepinephrinechrome also have been found to be MIF substrates (27). Michaelis constant (Km) values suggest, however, that reactions involving these substrates are unlikely to comprise the natural function for MIF (27–29).
Figure 1.

(A) Structures of dopachrome methyl ester (1), a chromogenic substrate for the measurement of MIF enzymatic activity, and its tautomerized, colorless product (2). (B) Structures of acetaminophen (3) and the oxidation products N-acetyl-p-benzoquinone imine (NAPQI, 4) and N-acetyl-3-hydroxy-p-benzoquinone imine (HQ, 6), which are related structurally to 1.
A crystal structure for the complex formed between human MIF and p-hydroxyphenyl-pyruvic acid revealed that the substrate binds to a hydrophobic cavity at the N terminus and interacts with Pro-1, Lys-32, and Ile-64 in one subunit and with Tyr-95 and Asn-97 in an adjacent subunit (30). Similar interactions between murine MIF and (E)-2-fluoro-p-hydroxycinnamate have been reported (31), and solution NMR studies support a direct interaction between model substrates and the Pro-1 located within the hydrophobic cavity (30). Of note, the environment of this cavity reduces the pKa of Pro-1 by almost 4 pH units relative to a proline amide (32), suggesting that this residue functions as a catalytic base to effect substrate tautomerization (33, 34). Site-directed mutagenesis studies also have established that the N-terminal proline is involved in the catalytic function of MIF (35). Substitution of Ser for Pro-1 produces a protein (P1S MIF) that is catalytically inactive (35). Nevertheless, P1S MIF shows full glucocorticoid counterregulatory activity and, similar to the P1F MIF mutant, is capable of inhibiting monocyte chemotaxis (35, 36). In contrast to these results, the catalytically inactive P1G MIF mutant was impaired greatly in its ability to stimulate superoxide generation in activated neutrophils (29). The precise connection between the enzymatic and biological activities of MIF remains open to question.
Recently completed clinical trials have affirmed that inhibition of proinflammatory cytokine action with mAbs is a powerful means for treating diseases such as rheumatoid arthritis and inflammatory bowel disease (37, 38). In particular, antibodies that bind to MIF have been shown to exert significant effects on its biological activities in various animal studies (5–8, 11–17, 21). Accordingly, investigations into the enzyme activity of MIF are of high interest not only to provide a potentially powerful approach for the design of selective, low molecular weight MIF inhibitors but also to resolve fundamental questions regarding the structure-function relationships of the protein. We noted that D-dopachrome and its tautomerized product bear significant structural similarities to acetaminophen and some of its active metabolites (Fig. 1). Here, we show that the acetaminophen metabolites and several related chemotypes irreversibly inhibit both the enzymatic and in vitro biological activities of MIF.
Materials and Methods
Reagents.
Unless otherwise indicated, all chemicals including compounds 3, 12, 13, 15, 17, and 18 were from Aldrich or Sigma and were of the highest grade commercially available. Compounds 4, 5, 8, 9, and 10 were prepared according to previously reported methods and had 1H NMR characteristics that were identical to the known compounds (39–45). Compound 4, which is known to be unstable (39), was characterized by GC/MS and stored as a solid at −70°C. Compounds 6, 11, 14, and 16 were prepared from their corresponding dihydroquinones (20 mM in DMSO) through the addition of 1 vol of NaIO4 at 18 mM in H2O (0.9 equivalents) at 4°C. After 5 min, the solutions were diluted in 50% aqueous DMSO at 4°C and then were added to recombinant mouse or human MIF at the concentrations indicated. Dopachrome methyl ester (1) was prepared similarly to previously published procedures (34, 35). Briefly, to an aqueous 4 mM solution of L-dopa methyl ester was added NaIO4 to a final concentration of 6 mM. The solution was placed on ice immediately. Assays were initiated at a time when the absorbance at 475 nm reached a maximal value, signifying that the limiting reagent, NaIO4, was consumed. Recombinant human and mouse MIF was expressed in Escherichia coli and purified as reported previously (4). The mouse mAb against MIF used in these studies binds to an unknown epitope and has been described (10, 13, 17, 21).
Treatment of MIF with Inhibitors.
MIF samples (0.72–600 μg/ml as indicated in the figure legends and text) in 50 mM sodium phosphate at pH 6.6 (with or without serum samples or BSA as indicated in the text) were treated with varying concentrations of the inhibitors for 5–20 min (exact time specified in the text) at room temperature. The modified proteins then were analyzed for enzyme activity by using the dopachrome tautomerase assay. Protein concentrations were determined by using the micro BCA assay (Pierce).
Dopachrome Tautomerase Assays.
To a room temperature solution (0.7 ml) of recombinant mouse or human MIF samples (0.72 μg/ml or at the concentrations indicated in the text) in PBS at pH 7.2 (with or without serum or BSA as indicated in the text) was added 1 (0.3 ml at 4 mM, prepared in situ according to refs. 34 and 35). The sample was monitored immediately for loss in absorbance at 475 nm compared with untreated MIF solutions and to 1 without the addition of MIF.
Fructose-2,6-bisphosphate (F2,6BP) Assay.
As described earlier for this assay (21), L6 rat myoblasts (CRL-1458, American Type Culture Collection) were cultured in DMEM containing 10% FBS at a density of 2 × 104 cells per ml. Differentiation was induced by treatment with bovine insulin (0.3 μM, GIBCO) in DMEM containing 1% horse serum. The cells were stimulated 3 days later with different MIF samples (each at 3 nM) for 24 h, and the F2,6BP then was extracted from cells and measured (15). NAPQI- and HQ-modified MIF were prepared by treating 60 nM MIF with 167 μM NAPQI and 33 μM HQ for 15 min, respectively, followed by extensive dialysis against PBS (modification resulted in 100% inhibition of MIF tautomerase activity). The experiments were performed independently three times.
Inhibition of Tumor Necrosis Factor α (TNFα) Production.
The ability of various MIF proteins to regulate glucocorticoid suppression of TNFα production in monocytes was assayed as described (35). Monocytes were purified from human peripheral blood by adherence, and 1 × 106 cells per well were preincubated for 1 h with dexamethasone (10−8) or dexamethasone plus MIF (3 nM native MIF, P1S MIF, or NAPQI-MIF) before the addition of 0.5 μg/ml lipopolysaccharide (E. coli 0111:B4, Sigma). NAPQI-MIF was prepared by treatment of MIF with 167 μM NAPQI for 15 min (resulting in 96% inhibition of enzyme activity) and then dialyzed overnight against PBS to remove low molecular weight products. Cell culture supernatants were collected after 16 h, and the secreted TNFα was quantified by ELISA. NAPQI-MIF did not affect cell viability, as assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide reduction (46).
Binding Studies.
MIF was labeled with the fluorescent dye Alexa-488 following the manufacturer's protocol (Molecular Probes). The average dye/MIF homotrimer ratio was 1:3 as calculated by matrix-assisted laser desorption ionization (MALDI)-MS. By using a previously described assay (35), Alexa-MIF was shown to bind to human microvascular endothelial cells (Clonetics–Biowhittaker, Walkerville, MD) in a saturable and competable manner and to initiate activation of the extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase cascade over the same dose range as unlabeled recombinant MIF. Alexa-MIF (3 μM) was reacted with NAPQI (66 μM) or incubated in PBS as control and dialyzed overnight against PBS. Control MIF or NAPQI-MIF (each now labeled fluorescently) then were added at 0.2 μM to 2 × 105 endothelial cells that had been washed previously and resuspended in 0.1 ml of cold DMEM containing 1% FCS. After incubation for 1 h at 4°C, the cells were washed three times and analyzed for bound MIF by flow cytometry using CELLQUEST software (FACSCalibur, Becton Dickinson). At least 10,000 cells were analyzed per experiment, and the data shown are the means ± SD of triplicate samples.
In Vivo Experiments.
C57BL/6 mice (8 weeks old, n = 3 per group) were injected i.p. with 100 or 200 mg/kg (300 and 600 mg/m2, respectively) of acetaminophen dissolved in 0.3 ml of sterile PBS. Control mice received PBS alone. After 8 h, the mice were killed by CO2 asphyxiation, the livers were resected immediately and homogenized in Tris-buffered saline, and the insoluble material was removed by centrifugation (1,000 × g) for 10 min at 4°C. The supernatant was filtered (0.45-μm filter) and subjected to Mono Q/FPLC anion exchange chromatography followed by C8-SepPak fractionation to isolate MIF as described (4). MIF purity was assessed by SDS/PAGE, and 200-ng aliquots were tested for dopachrome tautomerase activity. Mass spectral data on liver-purified MIF were acquired on an Agilent Technologies 1100 liquid chromatography (LC)/mass spectrometer detector using electrospray ionization (ES). LC separation was performed on a Zorbax 300SB-C8 reverse-phase column, 2.1 × 150 mm, with a flow rate of 0.5 ml/min at a temperature of 36°C. Gradient elution was performed by using 0.5% (vol/vol) acetic acid in deionized water (solvent A) to 0.5% (vol/vol) acetic acid in acetonitrile (solvent B) over a 10-min period. ES was carried out with a drying gas temperature of 350°C and a flow rate of 12 liters/min. The mass range scanned was between 500 and 2,000 atomic mass units with the fragmenter set to 150 V and the capillary tip set at 3,500 V. A tune file with a peak width of 1.0 atomic mass units was used.
Results and Discussion
Various dopachrome analogs have been shown to reversibly inhibit MIF tautomerase activity, but the effects on biological function were not reported (47, 48). We sought compounds that bound to the MIF catalytic site and, because of close proximity to the nucleophilic Pro-1, would react covalently so as to irreversibly inhibit enzymatic activity. Interestingly, the tautomerization of dopachrome (1) by MIF leads to a dihydroxyindole product (2), which bears a strong structural relationship to the analgesic drug acetaminophen (3) and some of its metabolites (Fig. 1). NAPQI (4) for instance has the iminoquinone functionality of L-dopachrome methyl ester and is known to be an electrophile and alkylating agent (39, 40, 43, 44).
The tautomerization of L-dopachrome methyl ester by recombinant MIF was measured in the presence of increasing concentrations of acetaminophen (3), NAPQI (4), and structurally related analogs. Acetaminophen (3) was very weakly inhibitory (IC50 = 10 mM; Fig. 2). NAPQI (4) was significantly more potent with an IC50 value (40 μM) that was 250-fold lower than that of acetaminophen. The most potent compound in this series was the hydroxyquinone (HQ, 6), also a known acetaminophen metabolite (43, 44), which had an IC50 of 0.7 μM, corresponding to >715-fold more inhibition than dihydroxyacetaminophen (5). Of ≈60 related quinones tested, the most active compounds were observed to contain an ortho- or para-quinoid structure similar to that found in dopachrome 1 (Table 1). The most potent agents were HQ (6) and the cinnamate derivative 16, which had submicromolar inhibitory activities.
Figure 2.

Dose-dependent inhibition of the enzymatic activity of MIF by acetaminophen (3) and its metabolites (4–6). MIF (60 nM) was incubated with test compounds for 5 min at room temperature, and the tautomerase activity was measured by using dopachrome methyl ester (1) as a substrate (25). Recombinant human MIF was expressed in E. coli and purified as described (35).
Table 1.
Selected compounds tested as inhibitors of the tautomerase activity of MIF
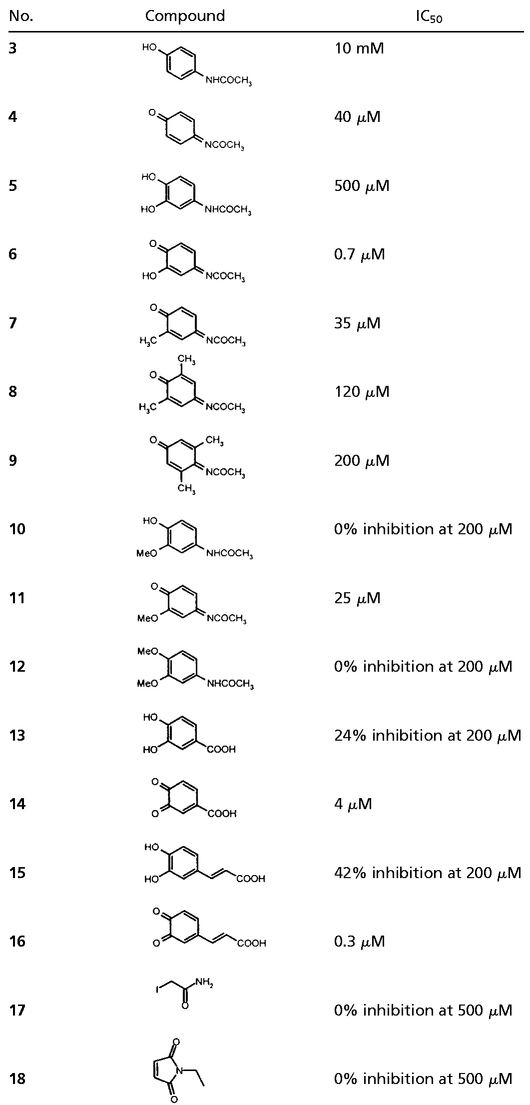 |
Recombinant human MIF was treated with various concentrations of the compounds for 5 min, and residual enzyme activity was determined using dopachrome methyl ester (1) as a substrate. IC50 values (50% MIF inhibition) were determined based on the activity of untreated MIF.
NAPQI and HQ are known to react with nucleophiles, particularly thiols (43, 44). It is noteworthy that no inhibition of MIF enzymatic activity was obtained with the thiol-reactive agents iodoacetamide (17) and N-ethylmaleimide (18), suggesting that NAPQI and HQ are reacting at sites other than free cysteines present in MIF. The efficiency of MIF enzyme inhibition by the quinones varied according to the concentration of MIF present in the reaction. When the reactions were performed at higher MIF concentrations (3 μM), HQ (6) and 16 virtually eliminated MIF enzymatic activity at doses that were equimolar with the monomer component of the MIF homotrimer. All the tautomerization activity of MIF was lost when the protein was treated with 1.7 equivalents per monomer of either HQ (6) or NAPQI (4), and no activity could be recovered by exhaustive dialysis to remove reversibly bound, low molecular weight products. Under these reaction conditions, the inhibitory activity of these compounds is exceedingly efficient and consistent with irreversible, covalent modification of MIF. We considered that dopachrome-related quinones may comprise a general class of irreversible MIF enzyme inhibitors and focused our attention on NAPQI, which is the major metabolite of acetaminophen (43, 44).
To provide insight into the specificity of the reaction, NAPQI and dilute solutions of MIF (0.72 μg/ml, 19 nM) were combined in the presence of high concentrations of whole serum or serum albumin. Albumin is a frequent target of electrophilic attack in vivo because of the complement of hydrophobic domains and numerous reactive side chains (59 lysines and 1 cysteine) on the molecule as well as to its high circulating concentration (49). Only when the concentration of serum albumin exceeded 10 mg/ml (0.15 mM) was there a detectable loss in the inhibitory activity of NAPQI. A residual inhibitory activity of 31% was still evident when the albumin concentration was 20 mg/ml. Thus, in the presence of a very large molar excess of a nontarget protein with chemically reactive residues, NAPQI still exerts significant inhibitory activity on MIF tautomerase activity. These results support the hypothesis that NAPQI binds to MIF via a specific interaction with the protein.
NAPQI in high concentrations can induce the crosslinking of certain proteins in vitro (43). Incubation of MIF with 190 μM of NAPQI did not produce covalent crosslinking as detected by either SDS/PAGE (Fig. 3 A and B) or MALDI-MS (Fig. 3 C and D). Direct evidence for the formation of a covalent adduct between NAPQI and MIF was obtained by MS, which indicated a new molecular ion with m/z = 12,491, corresponding to an MIF monomer with one bound molecule of NAPQI (theoretical mass of 12,494 Da). A minor peak at m/z = 12,645 Da also was observed, which likely corresponds to MIF modified by two molecules of NAPQI (theoretical mass of 12,643 Da). It is noteworthy that a significant portion (≈2/3) of native (unmodified) MIF (m/z = 12,343) also was detected under these incubation conditions, which were performed with a concentration of NAPQI that produced 96% inactivation of enzyme activity. These results suggest that covalent modification of each of the monomeric units in MIF by NAPQI may not be required for full inhibition of tautomerase activity.
Figure 3.

Reducing (A) and nonreducing (B) SDS/PAGE analysis of human MIF treated with NAPQI. MIF (3 μM) was either untreated (lane 1) or incubated for 5 min with NAPQI at 1 μM (lane 2, 0% enzyme inhibition), 49.5 μM (lane 3, 37% enzyme inhibition), 190 μM (lane 4, 94% enzyme inhibition), and 500 μM (lane 5, 100% enzyme inhibition). Proteins were visualized by Coomassie blue staining. The molecular mass of standard proteins (in kDa) are shown adjacent to A. (C) MALDI mass spectrum of human MIF (predicted molecular mass, 12,345 Da). (D) MALDI mass spectrum of human MIF (3 μM) incubated for 5 min with NAPQI (200 μM, 96% enzyme inhibition). The newly detected mass ions are within 3 atomic mass units of the predicted masses for the 1:1 (12,494 Da) and 1:2 (12,643 Da) covalent MIF/NAPQI adducts. Samples were analyzed with a Perceptive Voyager DE MALDI-MS (DHB matrix). (E) LC-ESMS analysis of MIF extracted from the liver of control (saline-treated) mice. ES produces multiply charged analyte ions with mass-to-charge (m/z) ratios within the range of the quadrupole MS. Data were processed by using the Agilent CHEMSTATION software to determine the measured molecular mass that was calculated to be 12,375 Da (predicted mass for mouse MIF, 12,372 Da). (F) LC-ESMS analysis of MIF that was extracted and purified from the liver of mice treated with 200 mg/kg acetaminophen. The measured molecular mass was 12,524 Da, which is within 3 atomic mass units of the predicted molecular mass of a NAPQI-MIF adduct (149 Da [NAPQI] + 12,372 Da [MIF] = 12,521 Da).
To provide evidence for the specific reaction of NAPQI with the N-terminal proline of MIF, we analyzed by MS the products of the reaction between P1S MIF (27) and NAPQI. In contrast to the mass spectra obtained with native MIF incubated with NAPQI (Fig. 3D), we were unable to identify any new mass ions as the result of this reaction (data not shown). These data are consistent with the notion that the N-terminal proline of MIF serves as the major nucleophilic center for covalent reaction with NAPQI.
It was of interest to examine whether MIF undergoes covalent modification with NAPQI in vivo. The liver is a major site of acetaminophen metabolism into NAPQI (39), and this tissue also has been shown to be a rich source of MIF for biochemical and structural studies (35). Mice were treated with either saline or single, tolerated doses of acetaminophen at 100 or 200 mg/kg (representing 300 and 600 mg/m2, respectively), and the livers were removed 8 h later for MIF purification (4). MIF from mice treated with acetaminophen showed a 45 ± 4% and 72 ± 3% decrease in dopachrome tautomerase activity at the 100 and 200 mg/kg doses, respectively (P < 0.001), compared with MIF isolated from the livers of the saline-treated controls. In addition, LC-ESMS analysis revealed molecular ions corresponding to NAPQI-modified MIF in the mice that were treated with acetaminophen. The MIF from mice treated with saline had a molecular mass of 12,375 Da, which is within 3 atomic mass units of the expected mass of 12,372 Da for native mouse MIF (Fig. 3E). Purification of liver MIF by reversed-phase chromatography from acetaminophen-treated mice led to two forms of the protein in proportions related to the relative enzymatic activity compared with the saline-treated controls. The lower molecular weight form was identical to native MIF both in terms of enzymatic activity and LC-ESMS (data not shown). The higher molecular weight MIF species had a molecular mass of 12,524 Da (Fig. 3F), which corresponds to a NAPQI mono-adduct (expected mass of 12,521 Da). These results demonstrate that acetaminophen, through its metabolites, modulates the enzymatic activity of MIF in vivo through the formation of a covalent bond. It is important to note that these effects were obtained by using clinically relevant doses of acetaminophen, which typically are 175–590 mg/m2.
MIF exerts profound immunoregulatory effects in vivo, in part as a result of its ability to counterregulate the immunosuppressive effects of glucocorticoids on proinflammatory cytokine production (5, 6, 8, 10). We asked whether NAPQI-modified MIF could override the inhibitory effects of dexamethasone on TNFα production by human monocytes. As shown in Fig. 4A, NAPQI significantly inhibited the ability of MIF to override the immunosuppressive effect of dexamethasone on endotoxin- (lipopolysaccharide) stimulated TNFα production by monocytes. In agreement with prior studies (27), both native and the catalytically inactive P1S MIF showed full glucocorticoid counterregulatory activity in the assay.
Figure 4.

(A) NAPQI-modified MIF (NAPQI-MIF) shows decreased glucocorticoid-regulating activity on activated monocytes. The ability of various MIF proteins to regulate glucocorticoid suppression of TNFα production in monocytes was assayed as described (35). Monocytes from human peripheral blood were preincubated for with dexamethasone (10−8) or dexamethasone plus MIF (3 nM native MIF, P1S MIF, or NAPQI-MIF) before the addition of 0.5 μg/ml lipopolysaccharide (LPS). NAPQI-MIF was prepared by treatment of MIF with 167 μM NAPQI for 15 min (resulting in 96% inhibition of enzyme activity) and then dialyzed overnight against PBS to remove low molecular weight products. The data shown are mean ± SD of triplicate wells in experiments that were repeated twice. (B) Effect of various MIF proteins on F2,6BP production in differentiated L6 myotubes. L6 rat myoblasts were stimulated with different MIF samples (each at 3 nM) for 24 h, and the F2,6BP then was extracted from cells and measured (15). NAPQI-MIF was prepared by exposing MIF to NAPQI (167 μM) for 5 min. The data shown are mean ± SD and are representative of three independently performed experiments. Con, control. (C) NAPQI-MIF shows decreased immunoreactivity by ELISA. Various MIF proteins (1.4 nM), prepared as described above, were captured with an anti-MIF mAb (10, 13, 17, 21) and quantified by sandwich ELISA (37). (D) NAPQI-MIF shows decreased cell surface binding to human microvascular endothelial cells. Alexa-MIF was formed by reacting MIF with the fluorescent dye Alexa-488. Binding to human microvascular endothelial cells as determined by flow cytometry was compared with Alexa-MIF, which was modified further with NAPQI. The data shown are the mean ± SD of triplicate samples.
A recently described biological action for MIF outside the immune system is its ability to stimulate glucose metabolism by up-regulating the production of the allosteric regulator of phosphofructokinase-1, F2,6BP (21). To test the effects of the chemically modified forms of MIF in this model, cultured L6 myoblasts were exposed to MIF or MIF that was pretreated with NAPQI or HQ. Both these compounds produced a significant decrease in the ability of MIF to increase F2,6BP levels when compared with native MIF or P1S MIF (Fig. 4B).
In vitro and in vivo studies that have relied on antibody neutralization (5–8, 11–17, 21) or on activation of the mitogen-activated protein kinase signaling cascade (4) strongly suggest that the biological actions of MIF are mediated by a high-affinity interaction between MIF and a cell surface receptor. The retention of biological activity by the P1S MIF (35) and P1F MIF (36) contrasts sharply with our observations that NAPQI inactivates both the enzymatic and biological activities of MIF. We hypothesize that the covalent modification of MIF by NAPQI imparts a change in the conformational integrity of the protein and disrupts a region critical for receptor binding. This region may include the Pro-1-containing hydrophobic pocket that, when irreversibly modified by NAPQI, inhibits productive interaction with a receptor or downstream effector proteins. Thus, although the inhibitory effect of NAPQI on MIF biological activity is not necessarily linked to the inhibition of the catalytic properties of the protein, we have shown that it is possible to conscript irreversible enzyme inhibitors for the inhibition of the biological activities of MIF.
Conformational alterations in NAPQI-modified MIF were explored by first examining the reactivity of NAPQI-modified MIF in an MIF capture ELISA using an anti-MIF mAb that neutralizes MIF biological activity (10, 13, 17, 21). As shown in Fig. 4C, there was an 80% decrease in the recognition of NAPQI-modified MIF with this anti-MIF antibody when compared with native MIF or P1S MIF. A second method to establish conformational changes involved binding of a fluorescently tagged MIF species (Alexa-MIF) to microvascular endothelial cells that have been shown previously to respond to MIF (24). The retention of biological activity of the Alexa-MIF conjugate was verified in a p44/p42 mitogen-activated protein kinase stimulation assay (ref. 50 and data not shown). Cells were treated with either Alexa-MIF or Alexa-MIF that was treated with NAPQI. Flow-cytometry analysis revealed that NAPQI-modification produced a 36% reduction in binding activity when compared with unmodified MIF (Fig. 4D).
We conclude that NAPQI irreversibly modifies the MIF catalytic site and disrupts the conformational integrity of an epitope that is critically involved in MIF biological activity. The N-terminal proline of MIF resides in a hydrophobic pocket and displays an unusually low pKa (≈5.6; refs. 22–25). This renders the proline amine nucleophilic and allows it to effect the tautomerization of model substrates. Our MS data are consistent with alkylation of Pro-1 by NAPQI, because we could detect no covalent reaction with the P1S MIF species. Elucidation of the precise conformational perturbation in MIF induced by NAPQI may be revealed more definitively by two-dimensional NMR or x-ray crystallography studies. NAPQI-modified MIF nevertheless displays diminished cell binding activity and decreased recognition by mAb. These data emphasize the importance of the MIF catalytic domain but not the catalytic activity per se in MIF function and are consistent with the high conservation of this region across evolution (29).
NAPQI is an oxidative metabolite of acetaminophen, a widely used drug, the mechanism of action of which continues to be debated (51). It is not yet known whether the active form of acetaminophen is the parent drug or one of its reactive metabolites, such as NAPQI or HQ. Acetaminophen inhibits prostaglandin H synthase in vitro but at concentrations that are probably not clinically relevant (45). Whether inhibition of MIF by the reactive metabolites of acetaminophen is a mechanism of action for certain pharmacological properties of this drug warrants further study. Given the powerful inhibitory action of anti-MIF antibodies in septic shock (11–13), autoimmune arthritis (15, 16), glomerulonephritis (17), and tumor angiogenesis (24), the therapeutic potential of pharmacological inhibitors of MIF is high. The presence within MIF of a catalytic site offers advantages for the design of molecules that can be targeted to this cytokine, as we have demonstrated here. Our design of potential MIF inhibitors capitalized on structural similarities of the MIF substrate/product pair with quinone-based metabolites of acetaminophen. Whether NAPQI or related quinone compounds find therapeutic application must await further preclinical evaluation. Nevertheless, these compounds constitute the first examples of low molecular weight agents that are targeted to a cytokine and capable of irreversibly inhibiting its proinflammatory action.
Acknowledgments
We thank Drs. Ingegerd Hellström, Karl Erik Hellström, and Donald Malins of the Pacific Northwest Research Institute for their support of the study. This work was supported by National Institutes of Health Grant 1R01 AI 42310 (to R.B.), the Manning Foundation, funding from Cytokine Pharmasciences, Inc. (to R.B., P.D.S., and G.J.T.), and Grant SP01GM 32165 (to S.D.N.).
Abbreviations
- MIF
macrophage migration inhibitory factor
- NAPQI
N-acetyl-p-benzoquinone imine
- F2,6BP
fructose-2,6-bisphosphate
- HQ
N-acetyl-3-hydroxy-p-benzoquinone imine
- TNFα
tumor necrosis factor α
- MALDI
matrix-assisted laser desorption ionization
- ES
electrospray ionization
- LC
liquid chromatography
References
- 1.Bloom B R, Bennett B. Science. 1966;153:80–82. doi: 10.1126/science.153.3731.80. [DOI] [PubMed] [Google Scholar]
- 2.David J. Proc Natl Acad Sci USA. 1966;56:72–77. doi: 10.1073/pnas.56.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiser W Y, Temple PA, Witek-Giannotti J S, Remold H G, Clark S C, David J R. Proc Natl Acad Sci USA. 1989;86:7522–7526. doi: 10.1073/pnas.86.19.7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernhagen J, Mitchell R A, Calandra T, Voelter W, Cerami A, Bucala R. Biochemistry. 1994;33:14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- 5.Bernhagen J, Calandra T, Mitchell R A, Martin S B, Tracey K J, Voelter W, Manogue K R, Cerami A, Bucala R J. Nature (London) 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 6.Calandra T, Bernhagen J, Metz C N, Spiegel L A, Bacher M, Donnelly T, Cerami A, Bucala R T. Nature (London) 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 7.Calandra T, Bernhagen J, Mitchell R A, Bucala R. J Exp Med. 1994;179:1895–1902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bacher M, Metz C N, Calander T, Mayer K, Chesney J, Lohoff M, Gemsa D, Donnelly T, Bucala R. Proc Natl Acad Sci USA. 1996;93:7849–7854. doi: 10.1073/pnas.93.15.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossi A G, Haslett C, Hirani N, Greening A P, Rahman I, Metz C N, Bucala R, Donnelly S C. J Clin Invest. 1998;101:2869–2874. doi: 10.1172/JCI1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly S C, Haslett C, Reid P T, Grant I S, Wallace W A, Metz C N, Bruce L J, Bucala R. Nat Med. 1997;3:320–323. doi: 10.1038/nm0397-320. [DOI] [PubMed] [Google Scholar]
- 11.Calandra T, Spiegel L A, Metz C N, Bucala R. Proc Natl Acad Sci USA. 1998;95:11383–11388. doi: 10.1073/pnas.95.19.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bozza M, Satoskar A R, Lin G, Lu B, Humbles A A, Gerard C, David J R. J Exp Med. 1999;189:341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calandra T, Echtenacher B, Roy D L, Pugin J, Metz C N, Hultner L, Heumann D, Mannel D, Bucala R, Glauser M P. Nat Med. 2000;6:164–170. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 14.Mikulowska A, Metz C N, Bucala R, Holmdahl R. J Immunol. 1997;158:5514–5517. [PubMed] [Google Scholar]
- 15.Leech M, Metz C, Santos L, Peng T, Holdsworth S R, Bucala R, Morand E F. Arthritis Rheum. 1998;41:910–917. doi: 10.1002/1529-0131(199805)41:5<910::AID-ART19>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 16.Leech M, Metz C, Hall P, Hutchinson P, Gianis K, Smith M, Weedon H, Holdsworth S R, Bucala R, Morand E F. Arthritis Rheum. 1999;42:1601–1608. doi: 10.1002/1529-0131(199908)42:8<1601::AID-ANR6>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 17.Lan H Y, Bacher M, Yang N, Mu W, Nikolic-Paterson D J, Metz C, Meinhardt A, Bucala R, Atkins R C. J Exp Med. 1997;185:1455–1465. doi: 10.1084/jem.185.8.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mikayama T, Nakano T, Gomi H, Nakagawa Y, Liu Y C, Sato M, Iwamatsu A, Ishii Y, Weiser W Y, Ishizaka K. Proc Natl Acad Sci USA. 1993;90:10056–10060. doi: 10.1073/pnas.90.21.10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waeber G, Calandra T, Roduit R, Haefliger J A, Bonny C, Thompson N, Thorens B, Temler E, Meinhardt A, Bacher M, Metz C N, Nicod P, Bucala R. Proc Natl Acad Sci USA. 1997;94:4782–4787. doi: 10.1073/pnas.94.9.4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakaue S, Nishihira J, Hirokawa J, Yoshimura H, Honda T, Aoki K, Tagami S, Kawakami Y. Mol Med. 1999;5:361. [PMC free article] [PubMed] [Google Scholar]
- 21.Benigni F, Atsumi T, Calandra T, Metz C, Echtenacher B, Peng T, Bucala R. J Clin Invest. 2000;106:1291–1300. doi: 10.1172/JCI9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudson J D, Shoaibi M A, Maestro R, Carnero A, Hannon G J, Beach D H. J Exp Med. 1999;190:1375. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes F J, Roger T, Calandra T, Kapurniotu, et al. Nature (London) 2000;408:211–216. doi: 10.1038/35041591. [DOI] [PubMed] [Google Scholar]
- 24.Chesney J, Metz C, Bacher M, Peng T, Meinhardt A, Bucala R. Mol Med. 1999;5:181–191. [PMC free article] [PubMed] [Google Scholar]
- 25.Rosengren E, Bucala R, Aman P, Jacobsson L, Odh G, Metz C N, Rorsman H. Mol Med. 1999;2:143–149. [PMC free article] [PubMed] [Google Scholar]
- 26.Sun H W, Bernhagen J, Bucala R, Lolis E. Proc Natl Acad Sci USA. 1996;93:5191–5196. doi: 10.1073/pnas.93.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosengren E, Aman P, Thelin S, Hansson C, Ahlfors S, Bjork P, Jacobsson L, Rorsman H. FEBS Lett. 1997;417:85–88. doi: 10.1016/s0014-5793(97)01261-1. [DOI] [PubMed] [Google Scholar]
- 28.Matsunaga J, Sinha D, Pannell L, Santis C, Solano F, Wistow G J, Hearing V J. J Biol Chem. 1999;274:3268–3271. doi: 10.1074/jbc.274.6.3268. [DOI] [PubMed] [Google Scholar]
- 29.Swope M D, Lolis E. Rev Physiol Biochem Pharmacol. 1999;139:1–32. doi: 10.1007/BFb0033647. [DOI] [PubMed] [Google Scholar]
- 30.Lubetsky J B, Swope M, Dealwis C, Blake P, Lolis E. Biochemistry. 1999;38:7346–7354. doi: 10.1021/bi990306m. [DOI] [PubMed] [Google Scholar]
- 31.Taylor A B, Johnson W H, Jr, Czerwinski R M, Li H S, Hackert M L, Whitman C P. Biochemistry. 1999;38:7444–7454. doi: 10.1021/bi9904048. [DOI] [PubMed] [Google Scholar]
- 32.Stivers J T, Abeygunawardana C, Mildvan A S, Hajipour G, Whitman C P. Biochemistry. 1996;35:814–823. doi: 10.1021/bi9510789. [DOI] [PubMed] [Google Scholar]
- 33.Stamps S L, Fitzgerald M C, Whitman C P. Biochemistry. 1998;37:10195–10202. doi: 10.1021/bi9806955. [DOI] [PubMed] [Google Scholar]
- 34.Swope M, Sun H W, Blake P R, Lolis E. EMBO J. 1998;17:3534–3541. doi: 10.1093/emboj/17.13.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bendrat K, Al-Abed Y, Callaway D J, Peng T, Calandra T, Metz C N, Bucala R. Biochemistry. 1997;36:15356–15362. doi: 10.1021/bi971153a. [DOI] [PubMed] [Google Scholar]
- 36.Hermanowski-Vosatka A, Mundt S S, Ayala J M, Goyal S, Hanlon W A, Czerwinski R M, Wright S D, Whitman C P. Biochemistry. 1999;38:1284–12849. doi: 10.1021/bi991352p. [DOI] [PubMed] [Google Scholar]
- 37.Weinblatt M E, Kremer J M, Bankhurst A D, Bulpitt K J, Fleischmann R M, Fox R I, Jackson C G, Lange M, Burge D J. N Engl J Med. 1999;340:253–259. doi: 10.1056/NEJM199901283400401. [DOI] [PubMed] [Google Scholar]
- 38.O'Dell J R. N Engl J Med. 1999;340:310–312. doi: 10.1056/NEJM199901283400411. [DOI] [PubMed] [Google Scholar]
- 39.Dahlin D C, Nelson S D. J Med Chem. 1982;25:885–886. doi: 10.1021/jm00350a001. [DOI] [PubMed] [Google Scholar]
- 40.Chen W, Koenigs L L, Thompson S J, Peter R M, Rettie A E, Trager W F, Nelson S D. Chem Res Toxicol. 1998;11:295–301. doi: 10.1021/tx9701687. [DOI] [PubMed] [Google Scholar]
- 41.Rashed M S, Nelson S D. J Chromatogr. 1988;474:209–222. doi: 10.1016/s0021-9673(01)93916-0. [DOI] [PubMed] [Google Scholar]
- 42.Holme J A, Hongslo J K, Bjoerge C, Nelson S D. Biochem Pharmacol. 1991;42:1137–1142. doi: 10.1016/0006-2952(91)90299-k. [DOI] [PubMed] [Google Scholar]
- 43.Albano E, Rundgren M, Harvison P J, Nelson S D, Moldéus P. Mol Pharmacol. 1985;28:306–311. [PubMed] [Google Scholar]
- 44.Chen W, Shockcor J P, Tonge R, Hunter A, Gartner C, Nelson S D. Biochemistry. 1999;38:8159–8166. doi: 10.1021/bi990125k. [DOI] [PubMed] [Google Scholar]
- 45.Harvison P J, Egan R W, Gale P H, Christian G D, Hill B S, Nelson S D. Chem Biol Interact. 1988;64:251–266. doi: 10.1016/0009-2797(88)90101-9. [DOI] [PubMed] [Google Scholar]
- 46.Mosmann T. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki M, Sugimoto H, Tanaka I, Nishihira J. J Biochem. 1997;122:1040–1045. doi: 10.1093/oxfordjournals.jbchem.a021844. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Bucala R. Bioorg Med Chem Lett. 1999;9:3193–3198. doi: 10.1016/s0960-894x(99)00561-2. [DOI] [PubMed] [Google Scholar]
- 49.Korch-Weser J, Sellers E M. N Engl J Med. 1976;294:311–316. doi: 10.1056/NEJM197602052940605. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell R, Metz C N, Peng T, Bucala R. J Biol Chem. 1999;274:18100–18106. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 51.Raffa R B, Codd E E. Life Sci. 1996;59:37–40. doi: 10.1016/0024-3205(96)00273-1. [DOI] [PubMed] [Google Scholar]