

Abstract
Background
Oncogene-Induced Senescence (OIS) is a form of senescence that occurs as a consequence of oncogenic overstimulation and possibly infection by oncogenic viruses. Whether senescence plays a role in the pathogenesis of cervical cancer (CC) is not well understood. Moreover, whether cervical epithelial cells that are part of the premalignant cervical intraepithelial neoplasia (CIN), exhibit markers of OIS in Human Papillomavirus (HPV)-infected tissue, has not been investigated.
Methods
We utilized a set of patient-derived premalignant and malignant tissue samples to investigate the protein (Ki67 and Lamin B1) and gene (TP53, IL1A, CCL2, and MMP9) expression of several OIS-associated biomarkers using immunohistochemistry (IHC) and qRT-PCR, respectively. Furthermore, we characterized the HPV status of all tissue samples.
Results
Most of the CC samples (34/37) were positive for HPV, mainly HPV-16 which was observed in 62.2% of the CC samples. Among CINs, HPV infection was found in 60.2% of the 32 samples with HPV-16 as the dominant genotype in 58.5% of the CINs. IHC analysis revealed a significant increase in the expression levels of both Ki67 and Lamin B1 proteins in CC tissue compared to CIN. On average, 93% of tumor cells were positive for Ki67 in comparison to only 25% of premalignant cells in CIN samples. Similarly, Lamin B1 expression was observed in 89% of tumor cells in malignant tissue on average, compared to 60% in CIN samples. Importantly, Lamin B1 expression was elevated in nonmalignant cervical tissue suggesting that its downregulation is more predominant in the premalignant state. Furthermore, RT-PCR revealed a significant decrease in the expression of TP53, IL1a, CCL2, and MMP9 markers in CC samples compared to CINs. Specifically, 84% of CC samples showed reduced TP53 expression, 90% showed reduced IL1a expression, 74% showed reduced CCL2 expression, and 76% showed reduced MMP9 expression when compared with their premalignant baseline. Infection of HPV was confirmed in 61% of the tumor tissues while only 25% of the CINs were positive for HPV.
Conclusion
This work shall provide an opportunity to further examine the role of OIS in the process of HPV-driven CC development.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12885-025-13499-0.
Keywords: Oncogene-induced senescence; SASP; Cervix; CIN, cancer; HPV
Introduction
Oncogene-Induced Senescence (OIS) is a form of senescence that emerges as a consequence of hyperproliferation-inflicted DNA damage arising from oncogenic overstimulation [1]. Although oncogene overexpression does not necessarily lead to senescence, as for example in healthy keratinocytes [2], in many models, OIS is often precipitated when a somatic cell is subject to oncogene activation (or tumor suppressor gene inactivation) including Ras [3], BRAFV600E [4], Akt [5], E2F [6], PTEN [7], and others [8]. OIS enforces a durable growth arrest on oncogene-harboring cells which accounts for its classical tumor suppressor function [9–11]. Subsequently, oncogene-induced senescent cells accumulate in a wide variety of premalignant lesions including colonic adenomas [12], premalignant skin nevi [13], prostatic intraepithelial neoplasia [14], and oral leukoplakia [15].
OIS is critical in mediating anti-tumor immune responses by driving cell-mediated immunosurveillance against premalignant cells, thus directly interfering with malignant transformation [16]. However, OIS has been described to have a double-edged sword effect on tumor progression largely due to its effect on the tumor environment mediated by the Senescence-associated Secretory Phenotype (SASP) [17, 18]. The SASP is characterized by the secretion of various bioactive and inflammatory molecules that can, in a context-dependent fashion, promote tumor growth [19]. In addition, escape of OIS, and its associated irreversible growth arrest, is implicated in malignant transformation [20–23]. This was shown through the paradoxical observation that overexpression of certain oncogenes, such as Akt, can facilitate the escape from Ras-induced OIS, a phenomenon expected to occur as part of cancer development [24].
In addition to oncogenes, viral infections have been also shown to induce senescence [25]. Whether senescence plays a tumor suppressor role against oncogenic viruses is not yet elucidated. A special connection arises between the human papillomavirus (HPV), a primary cause of cervical cancer (CC), and senescence. Oncogenic HPVs are double-stranded DNA viruses that infect anogenital epithelial cells. While HPVs are likely to remain asymptomatic and are frequently eliminated by the immune system, only a minority of infected individuals develop dysplastic changes such as cervical intraepithelial neoplasia (CIN). It was demonstrated that the oncogenic proteins E6 and E7 of HPV contribute to the ability of human diploid fibroblast cells to escape senescence [26, 27]. On the other hand, the ectopic expression of viral E2 protein in HPV-positive tumor cells can, in fact, induce senescence marked by p21Cip1 upregulation, a phenotype resembling OIS [28, 29]. Whether senescence plays a role in the pathogenesis of CC is not well understood. Moreover, whether cervical epithelial cells that are part of the premalignant CIN exhibit markers of OIS has not been investigated.
In this work, we utilized a set of both patient-derived premalignant and malignant tissue samples to investigate the protein and gene expression of several OIS-associated biomarkers. Moreover, we have characterized the HPV status of all tissue samples. This work provides proof-of-principal evidence that markers of OIS are expressed in cervical premalignant lesions and their resolution is a feature of malignant transformation. Lastly, this work paves the way towards the possible utilization of senescence-eliminating pharmacotherapy for the interference with CC progression [30, 31].
Methods
Tissue samples
The sample comprised formalin-fixed paraffin embedded (FFPE) tissue blocks of 32 premalignant cervical lesions, 37 CC samples, and 12 cervical samples with active chronic cervicitis, which were obtained from Jordanian Royal Medical Services (JRMS) and Prince Hamza Hospital (PHH), Amman, Jordan. The inclusion criteria for sample collection included: all cervical premalignant and malignant histopathological subtypes, regardless of their HPV status, and women over the age of 18 years. The exclusion criteria include CC patients whose FFPEs blocks were not available, and patients whose age was less than 18 years. All research activities under this work were conducted based on ethical approval obtained by Institutional Review Boards (IRB) at the Hashemite University (Protocol No. 11/9/2021/2022), JRMS (Protocol No. 10/2022/10), and PHH (Protocol No. HH/Research/5284) in accordance with the ethical standards as laid down in the Declaration of Helsinki. The requirement for informed consent was waived by the IRB at the primary institution of the principal investigator that approved the study (the Hashemite University) due to its nature that strictly includes the use of surplus tumor tissue samples. This waiver was further confirmed by the IRBs of the two clinical centers where samples were collected, namely, JRMS and PHH.
Sample processing and RNA extraction
From each FFPE block, a 20 μm thick section was obtained for RNA extraction using RNeasy FFPE Kit (QIAGEN, Germany) as described previously [32]. RNA concentration was measured using the Qubit 3.0 fluorometer (Thermo Fisher Scientific, USA). Then, the samples were stored at -80℃ for subsequent use.
Reverse transcriptase and real-time quantitative PCR
The RNA was converted to cDNA using the QuantiTect Reverse Transcription Kit (QIAGEN, Germany) according to the manufacturers’ recommendations. QuantiTect SYBR Green PCR Kit (QIAGEN, Germany) was utilized to amplify the target DNA for each sample using the primers shown in Additional file 1: Table S1 under the following conditions: initial denaturation at 95ºC for 15 min, followed by 40 cycles at 94ºC for 15 s, 57.5ºC for 30 s, and 72ºC for 30 s, with a final extension step of 72 ºC for 5 min. All PCR runs included negative, positive, and internal controls. PCR products were electrophoresed on a 2% agarose gel stained with ethidium bromide and visualized using UV transillumination (Alpha Innotech, USA). Real-time quantitative PCR (BIOER, China) was used to measure gene expression, aiming to assess alterations in the expression levels of specific genes, whether upregulated or downregulated. Gene expression levels were determined through the ∆∆Ct (cycle threshold) formula [33]. Initially, we conducted a comparative threshold cycle analysis (∆Ct) for CIN samples. The ∆Ct values were derived using the formula:
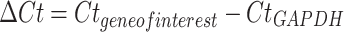 |
where (CtGAPDH) serves as the reference housekeeping gene. We then computed the average ∆Ct for the CIN samples to establish a baseline reference value which was termed “reference CIN samples”. For malignant specimens, we performed a similar ∆Ct calculation between the gene of interest and GAPDH. These values were then normalized to the reference ∆Ct of the CIN samples, yielding the double delta Ct (∆∆Ct):
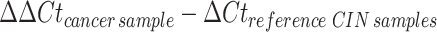 |
Subsequently, we calculated the relative quantification (RQ) of gene expression using the equation:
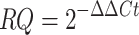 |
To facilitate comparison across all samples and genes, we applied a logarithmic transformation to the RQ values. It is important to note that instances where a gene’s Ct value was zero prompted a re-assay of the respective sample and gene. Should the Ct value remain zero upon re-assay, the gene was deemed non-quantifiable, and the corresponding sample for that gene was excluded from further analysis.
DNA extraction and HPV detection
FFPE tissue blocks from each patient were acquired from the pathology departments of both centers. For each patient, one section of 20 μm thickness was collected and DNA was extracted using the QIAamp DNA tissue kit (Qiagen, Germany) following the provided instructions. DNA was eluted in 200 µl AE buffer and stored at -20ºC for subsequent processing. HPV detection and genotype identification were performed using the Real-time PCR (RT-PCR) thermocycler from Bioer, China as previously described [34]. The REALQUALITY RQ-Multi HPV detection kit, an in vitro diagnostic kit (AB ANALITICA, Italy) was utilized for this purpose. This kit can identify 28 HPV types, including 14 high-risk (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68),, 6 potential high-risk (-26, -53, -67, -70, -73, -82), and 8 low-risk (6, 11, 40, 42, 43, 44, 55, 83) genotypes. If a sample tested positive for HPV 16, HPV 18, or any other high-risk or potentially high-risk genotype (received a single signal), it is classified as a single-genotype infection. If two signals were detected, it was considered a two-genotype infection, and if three signals were detected, it was classified as a three-genotype infection. The amplification targets a region in the HPV genome encompassing E6 and E7, with the genotypes detected collectively except for HPV-16 and HPV-18, which are individually identified. The kit incorporates an internal, negative, and positive controls. The thermocycling conditions involved an initial step of UNG activation at 50 °C for 2 min, followed by initial denaturation at 95 °C for 10 min, and then 45 cycles consisting of denaturation at 95 °C for 15 s, annealing at 60 °C for 60 s, and extension at 72 °C for 60 s. Data were interpreted according to the manufacturers’ guidelines as detailed previously [35].
Immunohistochemistry and immunofluorescence
Immunohistochemical (IHC) and immunofluorescence (IF) analyses were performed as previously described [36–39]. Briefly, multiple 4 μm tissue sections were deparaffinized using xylene and rehydrated using graded ethanol jars (100%, 95%, and 70%). The heat-induced epitope retrieval for Lamin B1 and HPV-16 E6/HPV-18 E6 antibodies was performed with sodium citrate buffer solution (pH 6.0), and with Tris-EDTA buffer (pH 9.0) for Ki67 antibody. Following washing twice with phosphate-buffered saline (PBS), incubation with 3% hydrogen peroxide for 10 min (IHC only), and 5% bovine serum albumin (BSA) blocking, slides were incubated at room temperature for 2 h for Lamin B1 (1:600, Novus biologicals, Cat. No. NBP2-59783, clone 4001, USA), Ki67 (1:600, ab279653, Abcam, Cambridge, United Kingdom), and HPV-16 E6 + HPV-18 E6 antibodies (1/400, C1P5, sc-460, Santa Cruz Biotechnology, USA). For IHC, the tissue sections were washed three times in PBS and treated with ultraVIEW Universal HRP Multimer (Ventana Medical Systems, Inc., Roche, USA), then washed twice using PBS and incubated in a dark place with 3, 3´-diaminobenzidine chromogen as substrate buffer for 10 min, then washed and counterstained with hematoxylin. Finally, dehydrated, cleared, slides were mounted with DPX and visualized under a light microscope (Olympus Optical, Tokyo, Japan). For IF, slides were incubated with fluorescent secondary antibodies 1:500 (Alexa Flour 488, A21121 and Alexa Flour 546, A11035, Invitrogen) for 1 h at room temperature. Subsequently, slides underwent a 15-minute PBS wash. To visualize cell nuclei, DAPI staining (Thermo Scientific, product number 62248, diluted 1:1000) was applied for 3 min at ambient temperature, followed by an additional 3-minute PBS rinse. Anti-fade fluorescence mounting medium (Abcam, catalog number ab104135) was applied and slides were stored in light-protected containers at 4 °C. Images were acquired using a Zeiss LSM780 confocal microscope system (Carl Zeiss AG, Jena, Germany).
Histopathological evaluation and immunofluorescence quantification
Senescence-associated biomarkers (Lamin B1 and Ki67) were assessed according to the median percentage of positively stained tumor cells for each marker by two independent pathologists in The Department of Pathology at JRMS (co-author: AAR) and The Department of Pathology, Microbiology and Forensic Medicine/Jordan University Hospital (co-author: NAS) using light microscopy (Olympus Optical, Tokyo, Japan) under 20 × and 40 × objective lenses and as described previously [37, 38]. HPV-16 E6 + HPV-18 E6 staining was evaluated as positive (any evidence of staining in tumor or non-tumor cells) or negative (complete absence of staining).
For immunofluorescence, ten random cell fields were acquired at 63x magnification for each sample. Quantification of cellular markers was performed using QuPath software [40]. For each field, we manually counted cells positive for E6 alone, E6 co-localized with Ki67 (E6+/Ki67+), E6 co-localized with Lamin B1 (E6+/Lamin B1+). The total cell count per field was determined using DAPI nuclear staining. The proportion of cells in each category was calculated as a percentage of the total cell count for each field. This analysis was performed separately for CIN and CC samples.
Statistical analysis
Data were initially stored on excel sheets which later were transformed into SPSS (version 25) for further analysis. Shapiro–Wilk showed that our data do not appear to be normally distributed, therefore non-parametric test were used (Mann–Whitney U test) was performed. All reported p values were two-tailed and p ≤ 0.05 was considered statistically significant.
Results
Identification of HPV subtypes in premalignant (CIN) and malignant (CC) tissue samples
Among the 37 CC specimens, there were 34 (91.9%) that tested positive for HPV, with 100% showing positivity for high-risk HPV (HR-HPV). HPV-16 and − 18 were detected in 62.2% and 18.9% of the cases, respectively. HPV infections in CC samples were presented as a single genotype infection in 13 cases (35.1%), two-genotype infections in 14 cases (37.8%), and three-genotype infections in 7 cases (18. 9%). In the 32 CIN samples, two thirds of the cases (60.7%) were HPV positive, with the majority (94.1%) being HR-HPV positive. HPV-16 was the dominant subtype (58.8%), followed by HPV-18 (35.3%). HPV infections in CIN samples were observed as a single genotype infection in 10 cases (31.3%), two-genotype infections in 8 cases (25.0%), and three-genotype infections in 2 cases (6.3%). The distribution of HR-, possibly HR- and LR-HPV infections among the studied samples are detailed in Additional file 1: Table S2.
A protein expression profile consistent with OIS is evident in CIN tissue samples in comparison to their CC tissue counterparts
Following the assessment of HPV status in CIN and CC samples, we examined the protein expression levels of two OIS markers within the same 32 CIN and 37 CC tissue samples. The two examined markers were Lamin B1, a nuclear lamina protein whose downregulation is strongly reflective of a senescent status both in vitro and in vivo [41, 42], and Ki67, a proliferation marker that is reduced under the senescence-associated growth arrest [43, 44]. Our IHC scores of Lamin B1 and Ki67% of positive cells showed that both markers exhibit a relatively downregulated expression in CIN when compared to CC (Fig. 1). For both markers, the IHC scores of the percentage of neoplastic cells with nuclear positivity were significantly lower in CIN samples than in CC samples (Fig. 1A and B). The median expression of Lamin B1 among CIN samples was 60%, while it remarkably increased to a median of 89% among the 37 CC samples (p = 0.031) (Fig. 1A). This increase was more evident for Ki67 where its protein expression level increased from a median of 25% positive cells in CIN samples to 93% among CC samples (p < 0.001) (Fig. 1B). Figure 2 shows a representative image of Lamin B1 and Ki67 IHC staining within CINs and CC. This data suggests a protein expression profile consistent with OIS in CIN tissue samples that resolves upon progression to a malignant state. We also compared the expression profile of Lamin B1 and Ki67 in non-malignant cervical tissue (n = 12). Our analysis revealed a significantly higher Lamin B1 protein expression within the endocervix (positivity of 95% of observed cells) relative to ectocervix (18% of cells were positive) (Additional file 2: Figure S1A-C). Conversely, Ki67 protein expression was higher within ectocervix (10% positive cells) compared to just minimal positivity in endocervix (Additional file 2: Figure S1D-E). This data confirms that Lamin B1 downregulation is most significantly observed in premalignant lesions in comparison to non-malignant or malignant tissue. This also confirms our previous findings and findings of others that Lamin B1 is a reliable senescence indicator in vivo [45–47].
Fig. 1.

Protein expression levels of Lamin B1 and Ki67in CC and CIN tissue samples. (A) Lamin B1. (B) Ki67. Protein expression was assessed by the percentage of DAP-positive cells within CIN (n = 32) and CC (n = 37) tissue samples. Statistical difference was calculated using Mann-Whitney test. *: pvalues < 0.05; ***: p values < 0. 001. Dots represent samples and line represent the median
Fig. 2.

Representative immunohistochemistry of Lamin-B1 and Ki67 staining in CC and CIN tissue samples. For both markers, protein expression was assessed by calculating the percentage of neoplastic cells exhibiting nuclear positivity. (A) and (B) demonstrate an increase in the median percentage of Lamin B1 positive tumor cells in CC samples compared to CIN samples, along with corresponding representative IHC images showing an increase in the percentage of positive neoplastic cells in a CC sample compared to a CIN sample, where positivity was confined to the neoplastic cells in the basal layers of the lesion. (C) and (D) demonstrate a more prominent increase in the median percentage of Ki67-positive tumor cells in CC samples compared to CIN samples, along with representative IHC images showing an increase in the percentage of cells exhibiting Ki67 nuclear positivity in a CC sample compared to a CIN sample showing limited basal positivity. IHC: immunohistochemistry
Restoration of Lamin B1 and Ki67 expression in CC tissue samples co-localizes with E6 expression
To confirm the connection between HPV infection and the downregulation of Lamin B1 and Ki67 in CIN tissue samples, we performed co-immunofluorescent staining to measure the expression of these two markers relative to the expression of the HPV oncoprotein, E6 (Fig. 3). Our data indicates that E6 is expressed in approximately 30% of premalignant cells within CIN samples, while E6+/Ki67 + and E6+/Lamin B1 + co-staining was detected in about 10% and 5% of cells, respectively. These observations suggest that HPV-infected cells might be in an OIS-like status (Fig. 3and Additional file 2: Figure S2). In comparison, cancer samples exhibited higher positivity across all markers. E6 expression was found in roughly 50% of malignant cells, while E6+/Ki67 + co-expression was present in about 30% of cells, and E6+/Lamin B1 + co-expression was observed in approximately 25% of cells (Fig. 4and Additional file 2: Figure S2). These data support the contention that progression into cervical malignancy might require escape from HPV-associated OIS.
Fig. 3.

Expression of Lamin B1 and Ki67 in co-localization with E6 in CIN tissue samples. Representative co-immunofluorescence staining fields showing few positively stained cells with Ki67 (red) and Lamin B1 (red) protein levels from E6 (green)-expressing cells in CIN tissue samples. Analysis of co-immunofluorescence staining is shown in (Additional file 2, Figure S2)
Fig. 4.

Protein expression of E6 co-localized with Lamin B1 and Ki67 in CC tissue samples. Representative co-immunofluorescence staining fields showing wide positive staining of Ki67 (red) and Lamin B1 (red) protein levels from E6 (green)-expressing cells in CIN tissue samples Analysis of co-immunofluorescence staining is shown in (Additional file 2, Figure S2)
Upregulation of TP53 and SASP markers upon progressing from CIN to CC lesions
Since the assessment of senescence in vivo requires the investigation of several associated markers [48], we next examined the differences in relative gene expression of several senescence-associated genes using qRT-PCR (Fig. 5). The chosen genes represent various components of the senescent phenotype, and their change is indicative of either senescence development or escape. Namely, TP53, a major regulator of DNA damage repair response and the senescence-associated growth arrest [3], and IL1A, CCL2, and MMP9, all components of the SASP [49]. With the exception of TP53, which could also reflect a quiescent state, the other SASP markers, if increased, are likely to reflect a senescent state [50]. Following the analysis of relative expression of the above-mentioned genes, we found that most CC samples exhibited a notable decrease in their expression relative to their CIN counterparts. For example, 84% (16/19) of the samples had a notable decline in TP53 relative expression compared to CIN samples (Additional file 2: Figure S3A), while 89% (16/18) had a similar trend in IL1A expression (Additional file 2: Figure S3B). Likewise, 74% (17/23) and 72% (21/29) of the samples exhibited a decrease in CCL2 and MMP9, respectively, among CC samples compared to CIN samples (Fig. 5 and Additional file 2: Figure S3C-D). Again, this data suggests the resolution of a senescence-related signature upon cervical malignant transformation.
Fig. 5.

Analysis of relative expression of senescence associated genes in CC relative to CIN tissue samples using RT-qPCR. Mean ΔCt of CIN samples was used as reference point to quantify the corresponding gene expression among CC samples TP53 (p53), IL1a (IL-1a), CCL2 (CCL2) and MMP9 (MMP9). The formula used to calculate the relative expression is detailed in the Methods. Negative values indicate that the corresponding gene was higher within precancerous (CIN) samples, while positive values suggest higher expression within the malignant (CC) tissues
Discussion
HPV is the most frequently known sexually-transmitted infection in females worldwide [51], and has an established role in the pathogenesis of CC [52]. Among the fifteen HR-HPV genotypes, HPV-16 and − 18 are the most prevalent, accounting for around 70–75% of cancer-related lesions in humans [53]. In this work, 91.9% of the tested CC samples were positive for HPV, all of them were of HR variants. HPV-16 and − 18 were detected in 62.2% and 18.9% of the cases, respectively. In comparison, 60.7% of the CIN samples were positive for HPV, with the majority being of high-risk, which is consistent with local and global rates [54–57]. For example, in the Eastern Mediterranean Region, HPV-16 is consistently the most common HPV subtype identified within malignant and premalignant cervical lesions [58].
The tumor suppressive role of cellular senescence arises from imposing a stable growth arrest in cells suffering from DNA damage or oncogenic stress [9, 10], however, similar roles in antagonizing virus-induced transformation is yet to be determined. Certainly, senescence is identified in virus-infected cells leading to notable growth stagnation of the host cells [59], although the replication of certain viruses was enhanced within senescent cells [60]. In cervical tissue, the connection between HPV infection and senescence remains inconclusive. First, and against expectation, p53 upregulation is frequently observed in HPV positive cells [61]. Second, the expression of HPV-16 E6 and E7 is necessary for human diploid fibroblasts to escape cellular senescence [26, 62]. Interestingly, replicative senescence can be escaped in HPV-infected fibroblasts through E6-mediated p53 inactivation [27], indicating that E6 and E7 might be essential for premalignant cells to escape senescence [63]. Third, exogenous expression of E2 in HPV-positive cancer cells culminates in a senescent growth arrest marked by p21Cip1 induction, another reliable senescence biomarker and a downstream cell cycle regulator to p53 [28, 29]. Importantly, these studies were performed in HPV-positive immortalized CC cells in vitro [64–66], while no studies have examined the induction of senescence in cervical tissue samples in vivo. Thus, these findings largely stem from experiments conducted on immortalized cell lines previously infected with HPV (probably “addicted to HPV for their own growth and proliferation), where cellular reliance on the inhibition of p53 and Rb by E6 and E7 was already established.
In contrast, when HPV-naïve cells are infected with HPV, senescence would occur as a form of OIS. This suggests that even cells immortalized by HPV through E6 or E7 can re-enter senescence when these oncoproteins are suppressed. Additionally, virus-free cells may undergo senescence upon HPV infection due to E7’s ability to trigger OIS. This contradiction is more elucidated when carefully examining work by the Shay group demonstrating that HPV E6/E7 would reverse SA-β-gal expression in Ras-induced senescent cells [67], and work by Rodier et al.. which showed that exogenous E7 expression induces senescence in HPV-naïve human fibroblasts in a similar fashion to Ras-induced senescence marked by significant SA-β-gal upregulation and DNA damage [68].
It is understandable that our proposition that HPV infection (accompanied by E6 expression within cervical epithelial cells) would trigger OIS is against previous literature that suggests that E2, rather than oncogenic E6 or E7, is what could possibly mediate HPV-induced senescence. This proposition is supported by the observations that shRNA-mediated knockdown of E6 and E7 and the pharmacological inhibition of E6-mediated degradation of p53 precipitate a senescence-mediated growth arrest in HeLa cells [69–71]. Similarly, CRISPR/Cas9-mediated knockout of E6 or E7 also induces senescence in HeLa cells as identified by the expression of SA-β-gal and Lamin B1 degradation [72]. Moreover, it is well established that E6 and E7 drive the immortalization of epithelial cells and thus are expected to facilitate overcoming tumor suppressive growth arrest mechanisms such as senescence [73–75].
However, other evidence suggests a more complicated role of E6 and E7 in the immortalization of cervical epithelial cells. For example, HPV-infected cancer-associated fibroblasts in CC showed senescence-like features including high IL-6 secretion, which was largely driven by E6 [76]. Furthermore, cervical smears showing elevated p16INK4a and p14ARF levels support our finding that HPV-infected epithelial cells tend to enter senescence early following infection [77]. Similarly, Feng et al. further confirmed this by finding increased senescence markers like p14ARF and p16INK4a in CIN II-III lesions [78]. Although HPV status was not assessed, it is likely these lesions were HPV-positive, given the virus’s key role in CC. Our proposal here suggests that for full immortalization, E6 and E7 might need to overcome tumor suppressor barriers such as OIS, although this remains to be proven.
In this work, we examined the expression level of the OIS-associated markers Lamin B1 and Ki67 proteins in cervical precancerous and cancerous samples. In CIN samples, both Ki67 and Lamin B1 median positive expression was significantly higher than in CC samples providing evidence suggestive for an OIS signature in premalignant lesions. Several studies have identified markers of OIS in premalignant lesions, suggesting its role as a barrier against tumorigenesis, not only through halting the proliferation capacity of premalignant cells but also by enabling the immune clearance of these affected cells via the secretion of anti-tumorigenic SASP [79]. Importantly, cervical Pap smears show high levels of p16INK4a, suggesting that early during HPV infection, cervical epithelial cells are in a senescent state [77]. More specifically, the protein expression levels of p14ARF, p15INK4b, p16INK4a, p53 and Ki67 in 20 CIN samples were significantly upregulated compared to normal cervical epithelium [78]. However, the HPV infection status in the CIN samples was not examined. In this work, we were able to establish a connection between the expression of OIS-biomarkers and HPV infection in premalignant cervical lesions. This is the first work to provide proof-of-principle observations that OIS is a component of cervical premalignant lesions. Collectively, our data suggests that markers of OIS are more predominant in the premalignant status rather than the malignant status of the cervix which confirms the involvement of OIS in suppressing tumor progression and supports previous findings in human colon and urinary bladder cancer where OIS markers are resolved as premalignant lesions progress into cancer [80].
Lastly, we have also examined the gene expression of TP53 and three SASP-related genes, namely, IL1A, CCL2, and MMP9 in both premalignant and malignant tissue samples. Our data shows that TP53 expression is relatively higher in premalignant lesions when compared to their malignant counterparts. However, several samples showed loss of TP53 transcript expression in the premalignant or malignant stages of CC. This can be explained by the possibility of acquiring somatic mutations during transformation [81]. These mutations are not solely caused by HPV infection [82], but possibly follow a Vogelsteinian pattern of tumor progression. Additionally, TP53 expression in CC can be epigenetically suppressed through acetylation leading to loss of transcript expression [83].
We have only measured the gene expression level of p53, rather than the protein expression which could be affected by E6-targeted degradation. Evidently, the detection of p53 using IHC in CC has shown considerable inconsistency across studies, highlighting several issues with the technique and inconclusive findings for this protein. For example, Raju et al. reported that most malignant and premalignant cervical lesions were negative for p53 as expected, assuming that most of these lesions are HPV-infected and E6 producing [84]. Interestingly, Inoue et al. found only 22% and 26% positivity rates in cervical cell squamous (SCC) and adenocarcinomas, respectively, indicating no significant changes in p53 protein expression level upon malignant transformation [85]. This aligns with Dimitrakakis’s et al. findings of 29% positivity rate in SCC [86]. While these studies still show lower p53 protein expression level as expected, conversely, Haensgen et al., found that 86% of SCCs to be p53-positive [87]. Similarly, Grace et al. also reported relatively higher positivity rate for p53 in all 60 SCC samples analyzed [88]. Highlighting this complexity, a systematic review found higher p53 expression in 4 out of 9 studies examined, indicating that in around 50% of the time, p53 expression might be higher than expected in CC lesions [89], as other research reported positivity in 96 out of 203 samples [90]. This variability in p53 protein expression level certainly underscores the challenges in reliably detecting p53 using IHC as the primary method in cervical lesions, and potentially its low relevance for senescence in this cancer type.
An alternative explanation for our data might be that premalignant lesions contained a preponderance of quiescent, rather than senescent, cells, based on the fact that both phenotypes represent growth arrest with reduced Ki67 expression. Quiescence is a transient, reversible state often triggered by mitogen deprivation or contact inhibition, regulated by p27Kip1 and p21Cip1. Senescence, however, is a durable form of growth arrest triggered often by telomere dysfunction or DNA damage, involving p16INK4a, and key regulators like p53 and Rb. Given HPV involvement in our study, p16INK4a was not a reliable marker to be used for the distinction between quiescence and senescence. Instead, we used Lamin B1 degradation, a hallmark of senescence (but not quiescence) [42], and demonstrated its downregulation alongside reduced Ki67 in premalignant lesions. Furthermore, SASP markers such as IL-1α, CCL2, and MMP9 were significantly elevated, reflecting a senescent rather than a quiescent state. It is noteworthy that an ultimate distinction between the two phenotypes in vivo remains challenging due to the lack of definitive markers [91, 92].
Our study has several limitations. First, we have not utilized the classical senescence marker SA-β-gal to examine senescence induction since all samples were formalin-fixed, while the detection of SA-β-gal requires the utilization of frozen samples [93–95]. Despite this issue, staining for SA-β-gal is not always conclusive for senescence since it must be performed as soon as possible after sample resection [96–98]. However, we utilized other markers of OIS that could be a reliable surrogate for senescence and as identified in other gynecological neoplastic lesions (up to our knowledge, senescence was not investigated in malignant and premalignant cervical tissue samples). For example, SA-β-gal expression was somewhat reliably coupled with measuring the immunostaining of p16INK4a, Ki67, HMGA1, and HMGA2 to identify senescence in uterine leiomyoma [99]. We have previously utilized a three-marker TIS signature of Lamin B1, Ki67 and p16INK4a to evaluate senescence in breast cancer samples, where we confirmed that the downregulation of Lamin B1 [41, 100], reduced Ki67 [101], and p16INK4a upregulation [102] were reliable for senescence evaluation in clinical cancer samples [36–38, 46, 92]. Second, we acknowledge the small sample size of our study. This interfered with our ability to test whether markers of OIS are more predominant in late dysplastic or carcinoma in situ lesions (for example comparing CIN I vs. CIN II vs. CIN III) as reported previously in oral or epidermal epithelial dysplasia [103]. Third, HPV genotyping was performed using the REALQUALITY RQ-Multi HPV detection kit, which is designed to identify HPV 16 and HPV 18 individually, a pool of 12 other HR-HPV, a pool of 6 possibly HR-HPV, a pool of 2 LR-HPV [6, 11], and a pool of 6 other LR-HPV. This is largely due to overall low number of diagnosed CC and the relatively lower prevalence of HPV infection in Jordan where this study was conducted [34]. We ensure that all cervical samples that met our inclusion and exclusion criteria at the indicated period were collected from two centers to insure adequate representation. Nevertheless, our results should only be viewed as proof-of-concept and that further investigation of senescence in CC is essential. These results also confirm the complexity of detecting senescence markers in clinical cancer samples and the requirement for the identification of better surrogate markers [92].
Conclusions
Collectively, our study provides proof-of-principle model suggesting that cervical epithelial cells infected with HPV might develop markers of OIS. Moreover, this study provides evidence that markers of OIS expressed in premalignant cervical lesions resolve following full malignant transformation, somewhat confirming the hypothesis that escape from OIS is a prerequisite for full malignant transformation. Further investigation into the role of senescence in the pathogenesis of HPV-associated CC is required. Lastly, the accumulation of senescent cells in CIN lesions might provide an avenue whereby pharmacological therapy can be utilized to selectively target cervical senescent cells and prevent or delay malignant progression [31].
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
All authors are highly thankful to the Researchers Supporting Project number (RSPD2025R786), King Saud University, Riyadh, Saudi Arabia. We are also thankful for the Cell Therapy Center at the University of Jordan for granting access for their microscopy unit.
Abbreviations
- OIS
Oncogene-Induced Senescence
- CC
Cervical Cancer
- CIN
Cervical Intraepithelial Neoplasia
- HPV
Human Papillomavirus
- IL1α
Interleukin 1 alpha
- CCL2
C-C motif chemokine ligand 2
- MMP9
Matrix Metalloprotease 9
- qRT-PCR
Quantitative Real Time-Polymerase Chain Reaction
- SASP
Senescence-associated Secretory Phenotype
- FFPE
Formalin-fixed Paraffin Embedded
- JRMS
Jordanian Royal Medical Services
- PHH
Prince Hamza Hospital
- IRB
Institutional Review Board
- GAPDH
Glyceraldehyde 3-phosphate Dehydrogenase
- RQ
Relative Quantification
- Ct
Cycle threshold
- UNG
Uracil-N-Glycosylase
- PBS
Phosphate-buffered Saline
- DPX
Dibutylphthalate Polystyrene Xylene
- IHC
Immunohistochemistry
- HR-HPV
High-risk Human Papillomavirus
- LR-HPV
Low-risk Human Papillomavirus
- shRNA
Short-hairpin RNA
Author contributions
AIK led the project and supervised and contributed to HPV evaluation experiments along with AAG and NH. SAS conducted data analysis, fluorescent microscopy, figure design and wrote the results. AAR, NAS, and OAAK all performed pathological evaluation of cervical lesions and the immunohistochemical expression of the studied protein markers. MES and AA were responsible for tissue processing and immunohistochemical staining. SK and BS conducted RNA extraction and measured gene expression levels under supervision from NH. SAM, MAS, SAD, RT, and MN were all responsible for sample collection, organization, and clinical data assessment. MRA edited and revised the manuscript. MYM revised and analyzed gene expression data and assisted with data analysis. RB performed confocal microscopy and quantification. FA and RK obtained ethical approval. TS was responsible for conceptualization, supervised the work, obtained funding, and wrote the manuscript.
Funding
All research activities conducted as part of this work were supported by the internal grant funding provided by the Deanship of Scientific Research, The Hashemite University (465/83/2019) directed to Dr. Tareq Saleh laboratory.
Data availability
All data analyzed during this study are included in this published article and its supplementary information files, while all raw data generated as part of this work are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
All research activities under this work were conducted based on ethical approval obtained by Institutional Review Boards (IRB) at the Hashemite University (Protocol No. 11/9/2021/2022), JRMS (Protocol No. 10/2022/10), and PHH (Protocol No. HH/Research/5284) in accordance with the ethical standards as laid down in the Declaration of Helsinki. The requirement for informed consent was waived by the IRB at the primary institution of the principal investigator that approved the study (the Hashemite University) due to its nature that strictly includes the use of surplus tumor tissue samples. This waiver was further confirmed by the IRBs of the two clinical centers where samples were collected, namely, JRMS and PHH.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mohammed El-Sadoni and Ahmad Alhesa contributed equally to this work.
Ala’ Abu Ghalioun, Suzan Khawaldeh and Bayan Shawish contributed equally to this work.
References
- 1.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444(7119):638–42. [DOI] [PubMed] [Google Scholar]
- 2.Henrard DR, Thornley AT, Brown ML, Rheinwald JG. Specific effects of ras oncogene expression on the growth and histogenesis of human epidermal keratinocytes. Oncogene. 1990;5(4):475–81. [PubMed] [Google Scholar]
- 3.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. [DOI] [PubMed] [Google Scholar]
- 4.Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, Van Der Horst CMAM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720–4. [DOI] [PubMed] [Google Scholar]
- 5.Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 2004;23(1):212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dimri GP, Itahana K, Acosta M, Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14ARF tumor suppressor. Mol Cell Biol. 2000;20(1):273–85. [DOI] [PMC free article] [PubMed]
- 7.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene. 2008;27(20):2801–9. [DOI] [PubMed]
- 9.Yaswen P, Campisi J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell. 2007;128(2):233–4. [DOI] [PubMed] [Google Scholar]
- 10.Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001;11(11):27–31. [DOI] [PubMed] [Google Scholar]
- 11.Braig M, Schmitt CA. Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res Cancer Res. 2006;66:2881–4. [DOI] [PubMed] [Google Scholar]
- 12.Tateishi K, Ohta M, Kanai F, Guleng B, Tanaka Y, Asaoka Y, et al. Dysregulated expression of stem cell factor Bmi1 in precancerous lesions of the gastrointestinal tract. Clin Cancer Res. 2006;12(23):6960–6. [DOI] [PubMed] [Google Scholar]
- 13.Gray-Schopfer VC, Cheong SC, Chong H, Chow J, Moss T, Abdel-Malek ZA, et al. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer. 2006;95(4):496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14(2):146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bascones-Martínez A, López-Durán M, Cano-Sánchez J, Sánchez-Verde L, Díez-Rodríguez A, Aguirre-Echebarría P, et al. Differences in the expression of five senescence markers in oral cancer, oral leukoplakia and control samples in humans. Oncol Lett. 2012;3(6):1319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–51. [DOI] [PubMed] [Google Scholar]
- 17.Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31(2):172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15(8):978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malaquin N, Vercamer C, Bouali F, Martien S, Deruy E, Wernert N, et al. Senescent fibroblasts enhance early skin carcinogenic events via a paracrine MMP-PAR-1 Axis. Eckert RL, editor. PLoS ONE. 2013;8(5):e63607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martínez-Zamudio RI, Stefa A, Nabuco Leva Ferreira Freitas JA, Vasilopoulos T, Simpson M, Doré G, et al. Escape from oncogene-induced senescence is controlled by POU2F2 and memorized by chromatin scars. Cell Genomics. 2023;3(4):100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Wu Z, Chanal M, Guillaumond F, Goehrig D, Bachy S, et al. Oncogene-induced senescence limits the progression of pancreatic neoplasia through production of activin a. Cancer Res. 2020;80(16):3359–71. [DOI] [PubMed] [Google Scholar]
- 22.Kolodkin-Gal D, Roitman L, Ovadya Y, Azazmeh N, Assouline B, Schlesinger Y, et al. Senolytic elimination of Cox2-expressing senescent cells inhibits the growth of premalignant pancreatic lesions. Gut. 2021;71(2):345–55. [DOI] [PMC free article] [PubMed]
- 23.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9(5):493–505. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011;42(1):36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature. 2021;599(7884):283–9. [DOI] [PubMed] [Google Scholar]
- 26.Litaker JR, Pan J, Cheung YC, Zhang DK, Liu Y, Wong SCH, et al. Expression profile of senescence-associated beta-galactosidase and activation of telomerase in human ovarian surface epithelial cells undergoing immortalization. Int J Oncol. 1998;13(5):951–6. [DOI] [PubMed] [Google Scholar]
- 27.Filatov L, Golubovskaya V, Hurt JC, Byrd LL, Phillips JM, Kaufmann WK. Chromosomal instability is correlated with telomere erosion and inactivation of G2 checkpoint function in human fibroblasts expressing human papillomavirus type 16 E6 oncoprotein. Oncogene. 1998;16(14):1825–38. [DOI] [PubMed] [Google Scholar]
- 28.Wells SI. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21CIP-dependent pathways. EMBO J. 2000;19(21):5762–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wells SI, Aronow BJ, Wise TM, Williams SS, Couget JA, Howley PM. Transcriptome signature of irreversible senescence in human papillomavirus-positive cervical cancer cells. Proc Natl Acad Sci U S A. 2003;100(12):7093–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saleh T, Khasawneh AI, Himsawi N, Abu-Raideh J, Ejeilat V, Elshazly AM, et al. Senolytic therapy: a potential Approach for the elimination of Oncogene-Induced senescent HPV-Positive cells. Int J Mol Sci. 2022;23(24):15512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saleh T, Carpenter VJ. Potential use of senolytics for pharmacological targeting of precancerous lesions. Mol Pharmacol. 2021;100(6):580–7. [DOI] [PubMed]
- 32.Linton KM, Hey Y, Dibben S, Miller CJ, Freemont AJ, Radford JA, et al. Methods comparison for high-resolution transcriptional analysis of archival material on Affymetrix Plus 2.0 and exon 1.0 microarrays. Biotechniques. 2009;47(1):587–96. [DOI] [PubMed] [Google Scholar]
- 33.Regier N, Frey B. Experimental comparison of relative RT-qPCR quantification approaches for gene expression studies in poplar. BMC Mol Biol. 2010;11:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khasawneh AI, Himsawi N, Abu-Raideh J, Salameh M, Abdullah N, Khasawneh R, et al. Prevalence of Human Papillomavirus Associated with Head and Neck Squamous Cell Carcinoma in Jordanian patients. Open Microbiol J. 2020;14(1):57–64. [Google Scholar]
- 35.Khasawneh AI, Himsawi N, Sammour A, Al Shboul S, Alorjani M, Al-Momani HH, et al. Association of Human Papilloma Virus, Cytomegalovirus, and Epstein–Barr virus with breast Cancer in Jordanian women. Med (B Aires). 2024;60(5):699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saleh T, Alhesa A, Al-Balas M, Abuelaish O, Mansour A, Awad H, et al. Expression of therapy-induced senescence markers in breast cancer samples upon incomplete response to neoadjuvant chemotherapy. Biosci Rep. 2021;41(5):BSR20210079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Sadoni M, Shboul S, Al, Alhesa A, Shahin NA, Alsharaiah E, Ismail MA, et al. A three-marker signature identifies senescence in human breast cancer exposed to neoadjuvant chemotherapy. Cancer Chemother Pharmacol. 2023;91(4):345–60. [DOI] [PubMed] [Google Scholar]
- 38.Al Shboul S, El-Sadoni M, Alhesa A, Shahin NA, Abuquteish D, Abu O, et al. NOXA expression is downregulated in human breast cancer undergoing incomplete pathological response and senescence after neoadjuvant chemotherapy. Sci Rep. 2023;13:15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al Shboul S, Curran OE, Alfaro JA, Lickiss F, Nita E, Kowalski J, et al. Kinomics platform using GBM tissue identifies BTK as being associated with higher patient survival. Life Sci Alliance. 2021;4(12):e202101054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27(16):1787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freund A, Laberge R-MRM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23(11):2066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehners N, Ellert E, Xu J, Hillengass J, Leichsenring J, Stenzinger A, et al. Oncogene-induced senescence: a potential breakpoint mechanism against malignant transformation in plasma cell disorders. Leuk Lymphoma. 2018;59(11):2660–9. [DOI] [PubMed] [Google Scholar]
- 44.Fan DNY, Schmitt CA. Detecting markers of Therapy-Induced Senescence in Cancer cells. Methods Mol Biol. 2017;1534:41–52. [DOI] [PubMed] [Google Scholar]
- 45.Wang AS, Ong PF, Chojnowski A, Clavel C, Dreesen O. Loss of lamin B1 is a biomarker to quantify cellular senescence in photoaged skin. Sci Rep. 2017;7(1):15678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saleh T, Alhesa A, El-Sadoni M, Shahin NA, Alsharaiah E, Shboul S, Al, et al. The expression of the Senescence-Associated Biomarker Lamin B1 in human breast Cancer. Diagnostics (Basel Switzerland). 2022;12(3):609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lv T, Wang C, Zhou J, Feng X, Zhang L, Fan Z. Mechanism and role of nuclear laminin B1 in cell senescence and malignant tumors. Cell Death Discov. 2024;10(1):269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D, González-Gualda E, Baker AG, et al. A guide to assessing Cellular Senescence in Vitro and in vivo. FEBS J. 2021;288(1):56–80. [DOI] [PubMed] [Google Scholar]
- 49.Buhl JL, Selt F, Hielscher T, Guiho R, Ecker J, Sahm F, et al. The senescence-associated secretory phenotype mediates Oncogene-induced senescence in Pediatric Pilocytic Astrocytoma. Clin Cancer Res. 2019;25(6):1851–66. [DOI] [PubMed] [Google Scholar]
- 50.Purcell M, Kruger A, Tainsky MA, Purcell M, Kruger A, Gene MAT, et al. Gene expression profiling of replicative and induced senescence gene expression profiling of replicative and induced senescence. Cell Cycle. 2014;13(24):3927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.CL Shannon and JD Klausner. The growing epidemic of sexually transmitted infections in adolescents: a Neglected Population. Curr Opin Pediatr. 2019;30(1):137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clifford GM, Tully S, Franceschi S. Carcinogenicity of human papillomavirus (HPV) types in HIV-positive women: a meta-analysis from HPV infection to cervical cancer. Clin Infect Dis. 2017;64(9):1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Tang D, Wang K, Wang J, Zhang Z, Chen Y, et al. HPV genotype prevalence and distribution during 2009–2018 in Xinjiang, China: baseline surveys prior to mass HPV vaccination. BMC Womens Health. 2019;19(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ouh YT, Min KJ, Cho HW, Ki M, Oh JK, Shin SY et al. Prevalence of human papillomavirus genotypes and precancerous cervical lesions in a screening population in the Republic of Korea, 2014–2016. J Gynecol Oncol. 2018;29(1). [DOI] [PMC free article] [PubMed]
- 55.Bel Haj Rhouma R, Ardhaoui M, El Fehri E, Marzougui A, Laassili T, Guizani I et al. Distribution of human papillomavirus in precancerous and cancerous cervical neoplasia in Tunisian women. Infect Agent Cancer. 2021;16(1). [DOI] [PMC free article] [PubMed]
- 56.Ma X, Yang M. The correlation between high-risk HPV infection and precancerous lesions and cervical cancer. Am J Transl Res. 2021;13(9):10830. [PMC free article] [PubMed] [Google Scholar]
- 57.Correa RM, Baena A, Valls J, Colucci MC, Mendoza L, Rol M, et al. Distribution of human papillomavirus genotypes by severity of cervical lesions in HPV screened positive women from the ESTAMPA study in Latin America. PLoS ONE. 2022;17(7):e0272205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shoja Z, Farahmand M, Hosseini N, Jalilvand S. A Meta-analysis on human papillomavirus type distribution among women with cervical neoplasia in the WHO Eastern Mediterranean Region. Intervirology. 2019;62(3–4):101–11. [DOI] [PubMed] [Google Scholar]
- 59.Baz-Martínez M, Da Silva-Álvarez S, Rodríguez E, Guerra J, El Motiam A, Vidal A, et al. Cell senescence is an antiviral defense mechanism. Sci Rep. 2016;6:37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim JA, Seong RK, Shin OS. Enhanced viral replication by cellular replicative senescence. Immune Netw. 2016;16(5):286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Velasco JA, Stevens CW, Esteban JM, Ruthsatz MKJ, Ramsamooj P, Dritschilo A, et al. Modulation of proliferation and tumorigenic potential of cervical-carcinoma cells by the expression of sense and antisense p53. Int J Oncol. 1995;7(4):883–8. [DOI] [PubMed] [Google Scholar]
- 62.Holt S, Gollahon L, Willingham T, Barbosa M, Shay J. p53 levels in human mammary epithelial cells expressing wild-type and mutant human papillomavirus type 16 (HPV-16) E6 proteins. Int J Oncol. 1996;8(2):263–70. [DOI] [PubMed] [Google Scholar]
- 63.Helt A-M, Funk JO, Galloway DA. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J Virol. 2002;76(20):10559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goodwin EC, DiMaio D. Induced senescence in HeLa cervical carcinoma cells containing elevated telomerase activity and extended telomeres. Cell Growth Differ. 2001;12(11):525–34. [PubMed] [Google Scholar]
- 65.Lee CJ, Suh EJ, Kang HT, Im JS, Um SJ, Park JS, et al. Induction of senescence-like state and suppression of telomerase activity through inhibition of HPV E6/E7 gene expression in cells immortalized by HPV16 DNA. Exp Cell Res. 2002;277(2):173–82. [DOI] [PubMed] [Google Scholar]
- 66.DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77(2):1551–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, et al. Absence of cancer–associated changes in human fibroblasts immortalized with telomerase. Nat Genet. 1999;21(1):115–8. [DOI] [PubMed] [Google Scholar]
- 68.Rodier F, Muñoz DP, Teachenor R, Chu V, Le O, Bhaumik D, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124(1):68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gu W, Putral L, Hengst K, Minto K, Saunders NA, Leggatt G, et al. Inhibition of cervical cancer cell growth in vitro and in vivo with lentiviral-vector delivered short hairpin RNA targeting human papillomavirus E6 and E7 oncogenes. Cancer Gene Ther. 2006;13(11):1023–32. [DOI] [PubMed] [Google Scholar]
- 70.Celegato M, Messa L, Goracci L, Mercorelli B, Bertagnin C, Spyrakis F, et al. A novel small-molecule inhibitor of the human papillomavirus E6-p53 interaction that reactivates p53 function and blocks cancer cells growth. Cancer Lett. 2020;470:115–25. [DOI] [PubMed] [Google Scholar]
- 71.Braun JA, Herrmann AL, Blase JI, Frensemeier K, Bulkescher J, Scheffner M, et al. Effects of the antifungal agent ciclopirox in HPV-positive cancer cells: repression of viral E6/E7 oncogene expression and induction of senescence and apoptosis. Int J Cancer. 2020;146(2):461–74. [DOI] [PubMed] [Google Scholar]
- 72.Inturi R, Jemth P. CRISPR/Cas9-based inactivation of human papillomavirus oncogenes E6 or E7 induces senescence in cervical cancer cells. Virology. 2021;562:92–102. [DOI] [PubMed] [Google Scholar]
- 73.Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380(6569):79–82. [DOI] [PubMed] [Google Scholar]
- 74.Yugawa T, Kiyono T. Molecular mechanisms of cervical carcinogenesis by high-risk human papillomaviruses: novel functions of E6 and E7 oncoproteins. Rev Med Virol. 2009;19(2):97–113. [DOI] [PubMed] [Google Scholar]
- 75.Gewin L, Myers H, Kiyono T, Galloway DA. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004;18(18):2269–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ren C, Cheng X, Lu B, Yang G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment. Eur J Cancer. 2013;49(18):3889–99. [DOI] [PubMed] [Google Scholar]
- 77.Von Keyserling H, Kühn W, Schneider A, Bergmann T, Kaufmann AM. p16INK4a and p14ARF mRNA expression in pap smears is age-related. Mod Pathol. 2012;25(3):465–70. [DOI] [PubMed] [Google Scholar]
- 78.Feng W, Xiao J, Zhang Z, Rosen DG, Brown RE, Liu J, et al. Senescence and apoptosis in carcinogenesis of cervical squamous carcinoma. Mod Pathol. 2007;20(9):961–6. [DOI] [PubMed] [Google Scholar]
- 79.Takasugi M, Yoshida Y, Hara E, Ohtani N. The role of cellular senescence and SASP in tumour microenvironment. FEBS J. 2023;290(5):1348–61. [DOI] [PubMed] [Google Scholar]
- 80.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444(7119):633–7. [DOI] [PubMed] [Google Scholar]
- 81.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–12. [DOI] [PubMed]
- 82.Andersson S, Hellström AC, Ren ZP, Wilander E. The carcinogenic role of oncogenic HPV and p53 gene mutation in cervical adenocarcinomas. Med Oncol. 2006;23(1):113–9. [DOI] [PubMed] [Google Scholar]
- 83.Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001;20(6):1331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raju GC, Teh M, Wee A. An immunohistochemical study of p53 protein in cervical intraepithelial neoplasia and squamous cell carcinoma. Pathology. 1996;28(1):17–9. [DOI] [PubMed] [Google Scholar]
- 85.Inoue M, Fujita M, Enomoto T, Morimoto H, Monden T, Shimano T, et al. Immunohistochemical analysis of p53 in gynecologic tumors. Am J Clin Pathol. 1994;102(5):665–70. [DOI] [PubMed] [Google Scholar]
- 86.Dimitrakakis C, Kymionis G, Diakomanolis E, Papaspyrou I, Rodolakis A, Arzimanoglou I, et al. The possible role of p53 and bcl-2 expression in cervical carcinomas and their premalignant lesions. Gynecol Oncol. 2000;77(1):129–36. [DOI] [PubMed] [Google Scholar]
- 87.Haensgen G, Krause U, Becker A, Stadler P, Lautenschlaeger C, Wohlrab W, et al. Tumor hypoxia, p53, and prognosis in cervical cancers. Int J Radiat Oncol Biol Physicsi. 2001;50(4):865–72. [DOI] [PubMed] [Google Scholar]
- 88.Grace VMB, Shalini JV, Lekha TTS, Devaraj SN, Devaraj H. Co-overexpression of p53 and bcl-2 proteins in HPV-induced squamous cell carcinoma of the uterine cervix. Gynecol Oncol. 2003;91(1):51–8. [DOI] [PubMed] [Google Scholar]
- 89.Silva DC, Gonçalves AK, Cobucci RN, Mendonça RC, Lima PH, Cavalcanti G. Immunohistochemical expression of p16, Ki-67 and p53 in cervical lesions - a systematic review. Pathol Res Pract. 2017;213(7):723–9. [DOI] [PubMed] [Google Scholar]
- 90.Wu J, Li XJ, Zhu W, Liu XP. Detection and pathological value of papillomavirus DNA and p16INK4A and p53 protein expression in cervical intraepithelial neoplasia. Oncol Lett. 2014;7(3):738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15(7):397–408. [DOI] [PubMed] [Google Scholar]
- 92.Saleh T, Bloukh S, Hasan M, Al Shboul S. Therapy-induced senescence as a component of tumor biology: evidence from clinical cancer. Biochim Biophys Acta-Reviews Cancer. 2023;1878:188994. [DOI] [PubMed] [Google Scholar]
- 93.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci. 1995;92(20):9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell. 2006;5(2):187–95. [DOI] [PubMed] [Google Scholar]
- 95.Kurz DJDJ, Decary S, Hong Y, Erusalimsky JDJD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113(20):3613–22. [DOI] [PubMed] [Google Scholar]
- 96.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging. 2017;9(8):1867–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matacchione G, Perugini J, Di Mercurio E, Sabbatinelli J, Prattichizzo F, Senzacqua M, et al. Senescent macrophages in the human adipose tissue as a source of inflammaging. GeroScience. 2022;44(4):1941–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kopp HG, Hooper AT, Shmelkov SV, Rafii S. Beta-galactosidase staining on bone marrow. The osteoclast pitfall. Histol Histopathol. 2007;22(9):971–6. [DOI] [PubMed] [Google Scholar]
- 99.Laser J, Lee P, Wei JJ. Cellular senescence in usual type uterine leiomyoma. Fertil Steril. 2010;93(6):2020–6. [DOI] [PubMed] [Google Scholar]
- 100.Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, et al. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015;10(4):471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sliwinska MA, Mosieniak G, Wolanin K, Babik A, Piwocka K, Magalska A, et al. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech Ageing Dev. 2009;130(1–2):24–32. [DOI] [PubMed] [Google Scholar]
- 102.Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibrolasts. Oncogene. 1998;16(9):1113–23. [DOI] [PubMed] [Google Scholar]
- 103.Natarajan E, Saeb M, Crum CP, Woo SB, McKee PH, Rheinwald JG. Co-expression of p16INK4A and laminin 5 γ2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am J Pathol. 2003;163(2):477–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data analyzed during this study are included in this published article and its supplementary information files, while all raw data generated as part of this work are available from the corresponding author on reasonable request.