

ABSTRACT
Background and Objective
Oral bacteria can translocate to the intestine, and their colonization efficiency is influenced by the gastrointestinal tract pH. Understanding how oral bacteria resist acidic environments is crucial for elucidating their role in gut health and disease.
Methods
To investigate the mechanisms of acid resistance in oral bacteria, an in vitro gastrointestinal tract Dynamic pH Model was established. This model was used to simulate the acidic conditions encountered by bacteria during their translocation from the mouth to the intestine.
Results
Fusobacterium nucleatum exhibited the highest survival rate in an acidified fluid mimicking the stomach pH (pH 1.5). The survival was significantly increased in the presence of erucic acid C22:1(n9) in cell membranes. Phylogenetic tree analysis revealed that C22:1(n9) synthesis was significantly associated with FnFabM gene expression in F. nucleatum at pH 1.5. Inhibition of FnFabM expression by cerulenin reduced the C22:1(n9) content and decreased the colonization efficiency of F. nucleatum in the stomach and jejunum of mice.
Conclusions
Oral F. nucleatum translocate to the intestine by resisting the acidic environment owing to the presence of erucic acid in its cell membrane, which is regulated by FnFabM. These results provide novel insights into the mechanisms underlying the oral bacteria survival in acidic environments and their potential to colonize the intestine; thus, shedding light on the oral-gut axis and its implications on human health.
KEYWORDS: Bacterial acid resistance, Fusobacterium nucleatum, FabM, erucic acid C22:1(n9), in vitro GI tract dynamic ph model
Key messages
Fusobacterium nucleatum possesses extraordinary acid resistance among the simulated oral flora owing to the presence of erucic acid in its cell membrane; this leads to its frequent transfer to the intestine and potential colonization.
The content of monounsaturated fatty acid, erucic acid C22:1(n9), is associated with high acid resistance under extreme environmental acid stress.
FnFabM, an enoyl-CoA hydratase-related protein, has a regulatory importance on erucic acid.
Introduction
Oral microbiota comprises over 700 diverse bacterial species [1]. It plays crucial roles in maintaining oral health and the development of various diseases [2,3]. Extensive research has investigated the correlation between oral bacteria and systemic diseases, with specific bacterial groups linked to conditions including Alzheimer’s disease [4], diabetes [5,6], atherosclerosis [7,8], and cancer [9–12]. Pathogenic bacteria, such as Porphyromonas gingivalis, are implicated in chronic inflammation [13–17] and have been detected in the postmortem brain tissue of patients with Alzheimer’s disease [16,18–20]. Furthermore, oral bacteria have been observed in atherosclerotic plaques [17,21], adverse pregnancy-related outcomes [22–24], rheumatoid arthritis [25,26], and specific cancers [9–12], indicating the potential transfer of bacteria to distant organs via the bloodstream [27,28].
In particular, disruption of oral microbiota is associated with inflammatory bowel disease [29] and colorectal cancer [9–11]. The oral and gut microbiota are interconnected through the gastrointestinal tract, although their composition can be differentiated by factors such as gastric and bile acids [30,31]. Prolonged use of proton pump inhibitors, which reduce the levels of gastric acid, can change the gut microbiome and reduce its diversity [30]. Animal studies have demonstrated that the oral microbiota can infiltrate the gut and reshape the gut microbiota [32,33]. This interaction between oral and gut microbiota can lead to the translocation of oral bacteria to the gut, which in turn contributes to the development and progression of various diseases. The impaired intestinal barrier function resulting from diseases [33–36], therapeutic measures, or administration of antibiotics and proton pump inhibitors [37,38] can facilitate the colonization of oral bacteria in the intestines.
The highly acidic environment of the stomach serves as a natural barrier against exogenous bacteria [39,40], thus, decreasing the gastric acidity level may permit the entry of harmful bacteria into the gastrointestinal tract [39]. Additionally, pathogenic oral bacteria can potentially enter the intestine through the saliva [33], further influencing the gut microbiota and systemic health. Taken together, the oral-gut axis is an emerging research area, with evidence suggesting that interactions between the oral and gut microbiota can influence disease development through inflammation, immune regulation, and metabolites. Understanding how oral bacteria traverse the gastric acid barrier is crucial to comprehend their broad implications on human health. Therefore, this study aimed to explore the mechanisms by which oral bacteria traverse the gastric acid barrier and their implications on human health.
Materials and methods
The used bacterial strains
Table 1 The bacterial strains used in this study
Table 1.
The bacterial strains used in this study.
| Species | Strain | Origin |
|---|---|---|
| Prevotella denticola | KCOM1525 | QIBEBT, CAS* |
| Prevotella maculosa | KCOM2290 | QIBEBT, CAS |
| Gemella morbillorum | This study | |
| Rothia dentocariosa | ATCC17931 | QIBEBT, CAS |
| Veillonella parvula | This study | |
| Lactobacillus renteri | This study | |
| Streptococcus cristatus | This study | |
| Streptococcus mutans | UA140 | QIBEBT, CAS |
| Streptococcus mutans | UA159 | QIBEBT, CAS |
| Streptococcus agalactiae | This study | |
| Streptococcus mutans | KCOM2966 | QIBEBT, CAS |
| Actinomyces naeslundii | ATCC12104 | QIBEBT, CAS |
| Fusobacterium nucleatum | ATCC25586 | Purchase from ATCC |
| Fusobacterium varium | pm-7 | This study |
| Fusobacterium varium | 36.1 | This study |
| Fusobacterium varium | 84 < 2> | This study |
| Fusobacterium mortiferum | 811 | This study |
| Fusobacterium nucleatum | 612 | This study |
| Fusobacterium necrophorum | M530 | This study |
*Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT, CAS).
Several bacterial strains used in this study were isolated in Microbiome Medicine Center, Division of Laboratory Medicine, Zhujiang Hospital, Southern Medical University, Guangzhou, China, in 2018. This work was as part of a previous research project that received ethics approval from the Ethical Review Committee of the Chinese Center for Disease Control and Prevention (Approval ID: 201519-A) [41].
Bacterial were isolated from fecal samples collected from randomly selected local individuals with no self-reported diseases at the Physical Examination Center of Zhujiang Hospital, Guangzhou, Guangdong, China (PEC).
Fusobacterium Selective Agar (FSA) was used as an enriched selective medium for the isolation and presumptive identification of Fusobacterium species. Columbia Blood Agar (Dijing, Guangdong, China) was used for isolating other strains. Briefly, fecal samples were quickly inoculated into the culture medium using a cotton swab immediately after collection. Samples were then anaerobically transferred to an anaerobic workstation (Don Whitley, Bradford, UK) for subsequent culture. The isolated strains were characterized using MALDI-TOF mass spectrometry (Quan TOF, IntelliBio, Qingdao, China) and 16S rRNA full gene sequencing (Forward Primer : AGAGTTTGATCMTGGCTCAG, Reverse Primer : TACGGYTACCTTGTTACGACTT) for taxonomic identification. After taxonomic identification, bacterial isolates were cryopreserved in liquid nitrogen for long-term storage. For future experiments, frozen bacterial cultures were resuscitated and propagated in appropriate growth media, with a subset selected for the current study.
Growth conditions and bacterial culture
All the bacterial strains were maintained in Columbia Blood Agar except for Lactobacillus renteri which was grown on MRS Agar (Huankai, Guangdong, China) at 37°C in A35 anaerobic workstation with an atmosphere containing 5% CO2, 10% H2, and 85% N2 (v/v). BHI broth supplemented with 0.05% L-cysteine HCl (w/v), 0.05% sodium thioglycolate (w/v), 0.001% hemin, and 0.001% vitamin K (w/v) was used as the growth medium. The basic steps for preparing a liquid medium suitable for anaerobes were previously described [42].
The bacterial strains listed in Table 1 were cultured on Columbia Blood Agar or MRS Agar and then transferred into BHI or MRS broth (L. renteri) in the anaerobic workstation in an atmosphere containing 5% CO2, 10% H2 and 85% N2 cultured at 37°C for 12–24 h to reach the logarithmic growth phase.
Survival of simulated oral flora in the in vitro GI tract dynamic pH model
Each bacterial strain was diluted using BHI broth to reach an optical density (OD) of 0.1. Afterwards, 1 mL of each cultured medium was mixed and centrifuged at 10,000 × g for 5 min at 4°C, and the cells in the pellet were suspended using the same volume of 0.9% NaCl and washed twice. After washing, the cells were resuspended in 100 mL simulated salivary fluid (pH 6.8; Biochemazone, Alberta, Canada). Each organism contained 1 × 107 colony-forming units (CFUs)/mL, and 19 organisms were prepared and mixed together.
A sterilized, ready-to-use, artificial gastric fluid that simulates the composition and pH of gastric juice was prepared (pH 1.5; Biochemazone, Alberta, Canada); it was mainly composed of 47.2 mm sodium chloride, 6.9 mm potassium chloride, 15.6 mm hydrochloric acid, 10 U/mL pepsin, and other components. The simulated duodenal fluid was prepared using 6.8 g potassium dihydrogen phosphate (Rhawn, Shanghai, China) dissolved in 500 mL distilled water; the pH was adjusted to 6.5 using 0.4% (w/v) NaOH. A total amount of 10 g trypsin (Merck, Darmstadt, Germany) was added to the solution and stirred overnight at 4°C; the volume was completed to 1 L followed by sterilization using 0.22-μm filter.
A modified in vitro GI-tract Dynamic pH Model that simulated the human upper gastrointestinal tract was set up as previously described [43]; it consisted of 0.5 and 1.0 L jacketed glass beakers with inlets and outlets (Loikaw, Shanghai, China) representing the stomach and duodenum, respectively (Figure 1). A cover was designed to accommodate a pH electrode integrated with a temperature probe and entry ports for the delivery of simulated saliva and artificial gastric fluid into the stomach reactor, and an emptying port for the stomach reactor. For the duodenal reactor, there were four entry ports for the stomach digesta, acid-neutralized NaOH, and simulated duodenal fluid plus a pH electrode. A magnetic stir bar was placed inside each vessel, and the agitation was controlled using a magnetic plate. The temperature inside the reactors was controlled by circulating water at 37°C through the jacketed beakers [43]. Multi-channel peristaltic pumps with controller (LHZWKJ, Guangdong, China) were used to control the delivery rate of the products to be added as well as the emptying rate of the stomach reactor into the duodenum reactor. A signal recorder (Enlai, Fujian, China) connected to a pH controller (Enlai) was used to monitor pH changes. Devices for monitoring and controlling the flow rate, pH, and temperature were integrated into a control station. The emptying rate of the stomach reactor was controlled to closely mimic the conditions in the stomach following yoghurt consumption and saliva secretion in humans [43–45]. The quantity of saliva-oral flora mixture remaining in the stomach reactor was calculated by considering the rate of addition of each solution and rate of emptying from the stomach vessel. Artificial gastric fluid addition to the stomach vessel was controlled to reproduce a pH of 1.5 similar to that in humans [46,47].
Figure 1. .
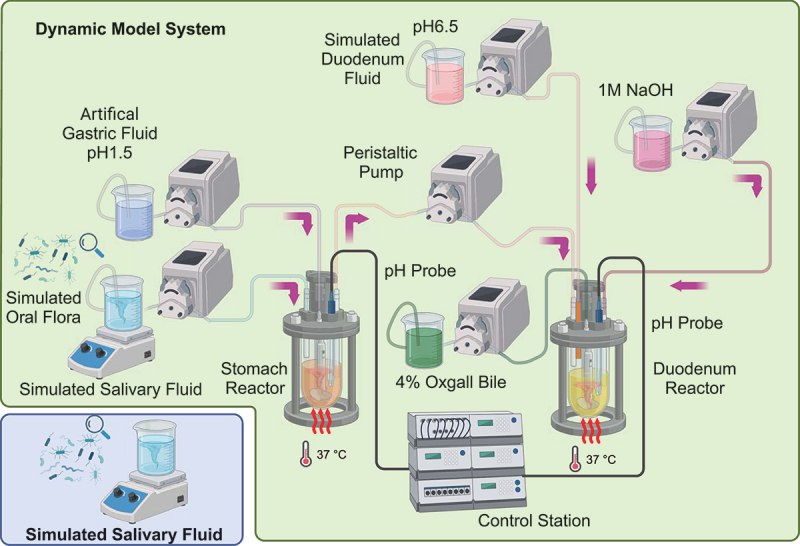
Schematic diagram of the human upper GI tract Model system (in vitro GI tract dynamic pH model) for acid resistant screening of simulated oral flora. Peristaltic pumps were used for the model system input and output. Jacketed glass beakers represented the stomach and duodenum reactors. pH probes were used to monitor the pH and temperature of the system; a control station was used to control the pH and the peristaltic pumps on line. Temperature control of the reactors was achieved using a water bath with circulating water.
Throughout the experiment, the pH of the duodenal vessels was maintained at a pH of 6.5. Initially, the stomach reactor contained 17.5 mL of artificial gastric fluid [48]. The rate of artificial gastric fluid delivery was set at 3.5 mL/min until the pH of the stomach reactor reached a pH of 1.5. Subsequently, the delivery rate was reduced to 0.9 mL/min to simulate gastrin inhibition. To maintain the pH of 6.5 in the duodenal reactor, 1 M NaOH was added at a rate of 0.65 mL/min. Salivary fluid with the same amount of simulated oral flora was prepared and used as a control (Figure 1).
Bacterial viability was determined using LIVE/DEAD BacLight Bacterial Viability kit (Thermo Fisher Scientific, Massachusetts, USA) in a 1 mL sample taken from stomach reactor at 0, 15, 30, 60, 90 min, and from the duodenum reactor at 2, 3, 3.5, 4.5, and 5.5 h to simulate the stomach accommodation and emptying. Samples were also collected from the control group at corresponding time points. The experiment was performed three times.
Survival of simulated oral flora at different pH of acid stress
To explore the survival of oral flora at different pH of acid stress, a series of solutions with pH values of 1.5, 2.5, 3.5, 4.5, 5.5, 6.5 and 7.2 without enzymes were prepared as previously described with some modifications [49]. Briefly, 3.1 g NaCl, 1.1 g KCl, 0.15 g CaCl2 and 0.6 g NaHCO3 were dissolved in 1 L distilled water; 0.1 M HCl and 0.4% (w/v) NaOH was added to the water to adjust the pH to the desired values. The bacterial mixture was prepared as previously described and resuspended with the series of solutions having different pH values. Then, it was incubated at the anaerobic workstation in an atmosphere of 5% CO2, 10% H2 and 85% N2 and cultured for the corresponding timepoints (0, 0.5, 1, 1.5, 3, and 6 h) at 37°C. Bacterial viability was determined using the LIVE/DEAD BacLight Bacterial Viability kit. A control pH of 7.2 was set and the experiment was performed three times.
Fluorescent stains for bacterial viability determination and flow cytometry sorting
A two-color bacterial viability assay was performed using the LIVE/DEAD BacLight Bacterial Viability kit (Thermo Fisher Scientific, Massachusetts, USA) to assess the viability of bacterial populations as a function of cell membrane integrity, following the manufacturer’s protocol with slight modifications. Briefly, 1 mL sample was centrifuged at 10,000 × g for 3 min. The supernatant was removed and the pellet was resuspended in 1 mL of 0.22-μm prefiltered 0.9% NaCl; the centrifugation was repeated and the pellet was resuspended in 1 mL of 0.9% NaCl. The bacterial suspension was diluted (100×); subsequently, 60 μL of the bacterial suspension were added to a 120-μL prediluted CYTO9 (400×). This was followed by incubation at 25°C in the dark for 15 min. Afterwards, 120 μL prediluted propidium iodide (40,000×) were added in the microfuge tube for another 5 min. The stained bacterial suspension was transferred to a flow cytometry tube using a CytoFLEX benchtop flow cytometer (Beckman Coulter, Inc., California, USA), and standard filters were set at 480/500 nm for SYTO9 and 490/635 nm for propidium iodide. Cells with a compromised membrane were considered dead or dying and were stained red, whereas cells with an intact membrane were stained green.
MoFlo XDP Cell Sorter (Beckman Coulter, Inc., California, USA) was used for sorting the stained bacteria collected from acid survival tests using a 50-μm CytoNozzle. The bacteria staining protocol was the same as that for the microbial viability analysis, where 50,000 PI staining negative bacteria and 100,000 PI staining positive bacteria were collected in 1.5 mL centrifuge tubes and stored at −80°C until DNA extraction.
Genomic DNA extraction and 16s rRNA sequencing
Bacterial genomic DNA was extracted from sorted samples using the PowerSoil® DNA Isolation Kit (QIAGEN, Dusseldorf, Germany) following the manufacturer’s specifications. Bacterial gDNA extracts were PCR-amplified using the AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Ltd., Nanjing, China). Barcoded primers 514F (GTGYCAGCMGCCGCGGTAA) and 805 R (GGACTACNVGGGTWTCTAAT) [50] were used to amplify the 16S rRNA gene V4 variable region. The PCR cycle conditions were as follows: 94°C for 5 min; followed by 30 cycles at 94°C for 30 s, 52°C for 30 s, and 72°C for 45 s; and a final elongation step at 72°C for 5 min. All PCR amplicons were mixed and sequenced using Illumina paired-end sequencing following the manufacturer’s protocol. The raw sequences were preprocessed and quality controlled using QIIME 2 with default parameters [41,51]; then, they were demultiplexed and clustered into genus-level operational taxonomic units (OTUs) with 97% similarity. OTU generation was based on the USEARCH algorithm [52].
Fusobacterium acid resistance determinations
Seven Fusobacterium strains (Table 1) were employed to establish an acid sensitivity assay to determine their survival ability at low-pH conditions of 1.5 and 3.5 using the two-color bacterial viability assay as previously described in flow cytometry sorting section . The bacterial strains were prepared as previously described, resuspended with the series pH solutions, and incubated in the anaerobic workstation in an atmosphere of 5% CO2, 10% H2, and 85% N2 cultured for 0.5 h at 37°C. The pH of 7.2 was set as the control and the experiment was performed three times.
Membrane fatty acid determination
Total lipids were extracted as previously described [53,54] with some modifications. Briefly, a 5 mL culture aliquot was centrifuged at 10,000 × g for 10 min and washed with ultra-pure distilled water three times. The resulting pellet was resuspended in 7 mL chloroform/methanol (2:1, v/v) and shaken at 150 rpm for 30 min on an Orbital Mini Shaker (VWR International, LLC, Pennsylvania, USA). Next, 1 mL 0.9% NaCl was added to the suspension; then, it was vigorously vortexed and centrifuged at 10,000 × g for 10 min. The upper water phase was removed and the bottom phase was transferred to a new glass screw-cap tube and dried using nitrogen to obtain the crude products. Subsequently, a mixture of 1.5 mL 14% BF3 /methanol and hexane (3 mL) was added to a glass bottle, which was sealed with nitrogen. The mixture was heated in an oil bath at 100°C for 1 h using a heating magnetic stirrer (Thermo Fisher Scientific, Massachusetts, USA) and mixed every 20 min. The mixture was then cooled to room temperature. Fatty acid methyl esters were extracted in hexane after adding 1 mL of ultrapure distilled water. The upper hexane layer was collected and evaporated using an ALPHA 1–4 LD plus freeze dryer (Martin Christ, Osterode am Harz, Germany). The residue was re-dissolved in 100 μL hexane and subsequently subjected to GC – MS analysis.
The samples were analyzed using an Agilent 5977 B BC/MSD Single Quadrupole GC/MS system with a built-in 8890 gas chromatograph and 7693A autosampler (Agilent Technologies, California, USA). Fatty acid methyl esters were separated on a J&W DB-23 (Agilent Technologies, California, USA) fused silica capillary column (60 m × 0.25 mm i.d.) having 0.15-μm-thick film under the following optimized oven temperature program: the initial temperature was set at 50°C and held for 1 min; subsequently, it was ramped to 175°C at a rate of 25ºC/min and held at 175°C for 0 min, followed by ramping to 240°C at a rate of 4ºC/min and held for 5 min. The total run time was 25 min. High-purity helium (≥99.999%) was used as a carrier gas at a flow rate of 0.3 mL/min. The injection volume was 1 μL with a split ratio of 1:20, and the injector temperature was set at 250°C. The mass spectrometer was operated in electron-impact (EI) mode at 70 eV ionization energy, and the spectra were acquired in the m/z range of 50–500 between 3 and 22.25 min at a scan rate of 1.7 scans per second. The quadrupole and ionization source temperatures were set at 150 and 230°C, respectively. The data were analyzed using Agilent MassHunter Qualitative Analysis software, Version B.07.00 (Agilent Technologies, California, USA).
Homologous sequences identification and phylogenetic tree construction
Trans-2-decenoyl-[acyl-carrier-protein] isomerase protein sequence of Streptococcus pneumoniae (strain ATCC BAA-255/R6; UniProtKB/Swiss-Prot: Q8DR19.1) and enoyl-CoA hydratase protein sequence of Streptococcus mutans (strain UA159; GenBank: AVN85541.1) were used as templates for a BLASTP search against the non-redundant protein sequences (nr) database of Fusobacterium nucleatum subsp. nucleatum ATCC 25586 (taxid:190304).
The deduced amino acid sequence of FnFabM was aligned using the MUSCLE algorithm in MEGA software (Version 11.0.13; Pennsylvania State University, Pennsylvania, USA). The evolutionary history was inferred using the Maximum Likelihood method and the JTT matrix-based model [55] with bootstrap values for 100 replicates. The sequences of the enoyl-CoA hydratase/isomerase family proteins of Fusobacterium species applied in this study were retrieved from GenBank, with GenBank accession numbers listed next to the species name. Additionally, two outgroups, namely Helicobacter pylori dehydrogenase/isomerase FabX (GenBank: WJX98257, HpFabX) and Escherichia coli strain K12 dehydratase/isomerase FabA (GenBank: P0A6Q3, EcFabA), were included for comparison; both of which are key unsaturated fatty acid (UFA) biosynthesis enzymes of the elongation stage.
Gene expression analyses
Specific primers (FnfabM Forward primer: TGGACTTGTTCCTGATACTGGTG and FnfabM Reverse primer: TTCTTTTGCTTCTTCTGCACTCAC) were designed using the web server primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast) against the RefSeq Representative Genome Database limited to Fusobacterium nucleatum using the automatic search mode with default primer parameters. The designed primers were used to amplify the samples.
Total bacterial RNA from different pH (1.5, 3.5, and 7.2) acid treatments and different time points (0.5, 1.5, 2.5, 3.5, 6.0, 12, 24, 48 and 72 h) was extracted using TRIzol reagent (Invitrogen; Carlsbad, CA, USA). This was followed by reverse transcription using FastKing gDNA Dispelling RT SuperMix (TIANGEN Biotech (Beijing) Co., Ltd, Beijing, China) to obtain first-strand cDNA. RT-qPCR amplification of cDNA was performed using gene-specific primers and AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Ltd., Nanjing, China) following the manufacturer’s protocol.
For each of the aforementioned treatments, four independent biological replicates were analyzed and the mRNA of the Fusobacterium nucleatum subsp. nucleatum ATCC 25586 DNA-directed RNA polymerase beta chain (rpoB) gene (GenBank: GQ274958.1) was used as a reference gene for the normalization of the expression data (FnrpoB forward primer: TGCAGAAGCAGAAGCTTTCA and FnrpoB Reverse primer: ACTGTTACTTGATCTCCTGGTCT). All the quantification experiments were performed using Applied Biosystems®TM ViiA7DX or ViiA7 Real-Time PCR System (Applied Biosystems; California, USA). The gene expression levels were analyzed using the 2(-Delta Delta C(T)) method [56].
Monounsaturated fatty acid synthase inhibition with cerulenin supplementation with monounsaturated fatty acid
Inhibition of monounsaturated fatty acid synthase with cerulenin and supplementation with monounsaturated fatty acids (MUFAs) was investigated in Fusobacterium nucleatum. F. nucleatum cells were grown in BHI medium until reaching the exponential phase (OD600 ≈0.38). The cells were washed twice with 0.9% NaCl and centrifuged at 10,000 × g for 1 min. Subsequently, they were resuspended in BHI medium at different pH levels (1.5, 3.5, and 7.2) containing 10, 30, and 60 μg/mL of cerulenin or the solvent control DMSO (0.1% of the final volume). Incubation was performed in an anaerobic workstation for 4.5 h as previously described. After incubation, the cell pellet was divided into two parts for RNA and fatty acid extraction. Real-time RT-PCR and GC – MS were used to determine the relative expression of FnfabM and quantify the fatty acids, respectively.
To assess the changes in fatty acid composition and evaluate the bacterial viability in response to varying concentrations of cerulenin and supplementation with MUFA, 10 μg/mL erucic acid (C22:1 n9) was added to the medium, and the cells were incubated for 4.5 h as previously described.
Animal experiments
To investigate the effect of gastric acid in vivo on F. nucleatum, specific pathogen-free (SPF) mice were used as an animal model. Male C57BL/6 mice, aged 7 weeks and weighing 20–24 g, were purchased from Guangdong Medical Laboratory Animal Center and housed under SPF conditions with a fixed 12 light/dark cycle at 25°C, with free access to water and food.
The mice were randomly divided into control and cerulenin-treated groups. In the control group (F. nucleatum + vehicle, n = 12), DMSO was used as the vehicle; the F. nucleatum strain ATCC 25586 was delivered to the mice through oral gavage at a volume of 200 μL containing 2 × 108 CFUs. In the cerulenin treatment group (n = 9), the same volume of bacterial suspension along with cerulenin at a final concentration of 30 μg/mL was administered by gavage.
The mice were sacrificed 8 h after oral gavage and their gastrointestinal contents, including the stomach, jejunum, cecum, and distal colon, were collected for subsequent absolute quantitative PCR analysis. Briefly, bacterial recovery was performed through homogenization and low-speed centrifugation in PBS, followed by high-speed centrifugation to collect bacteria for DNA extraction as previously described to quantify the bacterial load within the gastrointestinal contents. A known concentration of a specific FnnusG fragment of F. nucleatum with a known copy number was used. Serial dilutions of the DNA templates were prepared to cover a range of concentrations. These dilutions were then subjected to qPCR amplification along with the experimental samples using the same primers (FnnusG forward primer: TGAACATGGTAGAGTTAAAGTAATGGTTG and FnnusG Reverse primer: TTGACTTTACTGAGGGAGATTATGTAAAAATC). The resulting qPCR data, including cycle threshold (Ct) values, were used to construct a standard curve by plotting the Ct values against the logarithm of base 10 of the initial DNA template copy number.
Statistical analyses
Statistical analyses were performed using GraphPad Prism (version 9.0.2; GraphPad Software Inc.; San Diego, CA, USA) and SPSS Statistics for Windows (version 20.0; IBM; Armonk, NY, USA). One-way or Two-way ANOVA, paired or unpaired Student’s t test, Mann–Whitney U test, Tukey’s multiple comparison test, and Spearman correlation analysis were used for analyses. Data are expressed as mean ± standard deviation (SD). Sample size calculations were not performed in this study. Statistical significance was determined with a significance threshold set at a P-value <0.05. Statistical significance is denoted as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
The oral flora exhibits varying abilities in resisting acidic environments
To elucidate the survival dynamics of oral bacteria in the oral cavity and gastrointestinal tract, our study initially utilized a mixed consortium of 19 distinct bacterial species. This diverse microbial community was selected to represent a broad spectrum of the oral microbiota and to simulate the natural environmental conditions encountered during the transit through the gastrointestinal tract. The bacterial survival in the simulated salivary fluid and in vitro GI Tract Dynamic pH Model are shown in Figure 2(a). At the simulated stomach stage, the bacterial viability in the acidified artificial gastric fluid treatment rapidly decreased from 72.02%±3.53 to 9.62%±2.37, while in the simulated salivary fluid control group, it decreased slowly from 73.79%±5.98 to 55.86%±15.75 over the 1.5-h study period. In the duodenum reactor, the bacterial viability slightly increased at the first 0.5 h (timepoint, 2.0 h) and then fluctuated within a narrow range before further decreasing to 7.81%±0.84 by the end of the study period. In contrast, the bacterial viability in the simulated salivary fluid control group remained at approximately 68% (Figure 2(a)).
Figure 2. .
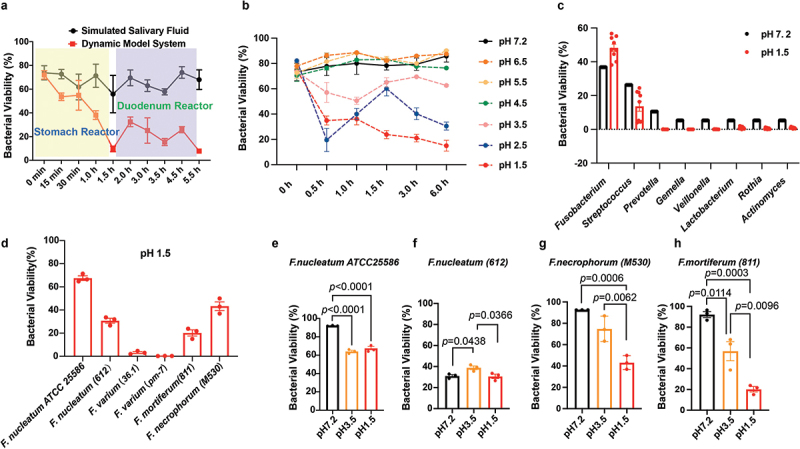
Viability of simulated oral flora in the Dynamic pH Model System and robust viability of Fusobacterium nucleatum, demonstrating superior acid resistance in extreme acid conditions. (a) Bacterial viability determination of the simulating oral flora in the Dynamic in vitro human upper GI tract model system. A total of 19 bacterial suspensions were prepared and mixed together to simulate the oral bacteria communities. Samples (1 ml) were taken from the stomach reactor at 0, 15, 30, 60 and 90 minutes, and from the duodenum reactor at 2, 3, 3.5, 4.5 and 5.5 hours. Bacterial viability was determined using the LIVE/DEAD BacLight bacterial viability kit. (b) Acid resistance test of oral bacterial communities in a series of different pH solutions without enzymes. A total of 19 bacteria suspensions were prepared and mixed together to simulate the oral bacteria communities. The bacterial mixture (1 ml) was centrifuged at 10,000×g for 5 min at 4°C, resuspended with various pH solutions, and incubated in an anaerobic workstation with an atmosphere of 5% CO2, 10% H2 and 85% N2 at 37°C for the corresponding timepoints. Bacterial viability was determined using the LIVE/DEAD BacLight bacterial viability kit. (c) 16S rRNA sequencing of stained bacteria indicated the extraordinarily high viability of Fusobacterium in acid environments. The stained bacteria suspension was transferred into a flow cytometry tube and sorted using a MoFlo XDP cell Sorter (Beckman Coulter), followed by 16S rRNA sequencing. (d) Acid survival tests of six strains of Fusobacterium bacteria members indicated robust viability of F. nucleatum under pH 1.5 condition. F. nucleatum showed the best acid resistance compared with the other tested Fusobacterium bacteria under pH 1.5 condition. Acid survival tests of (e) F. nucleatum ATCC 25586, (f) F. nucleatum (612), (g) F. necrophorum (M530), and (h) F. mortiferum at pH 1.5, pH 3.5, and pH 7.2. The experiment was performed three times. The data are expressed as mean ± standard deviation (SD) or standard error of the mean (SEM). The p values are indicated.
To investigate the effect of different pH values on bacterial viability, a simplified fluid without enzymes was used. The bacterial viability continued to decrease at pH 1.5 from 74.69%±5.4 to 15.01%±4.30 over the 6-h period. The viability of the simulated oral flora also rapidly dropped in the first 0.5 h to 19.50%±9.2 at pH 2.5, similar to that at pH 1.5, but then increased to 60.15%±5.78, approaching the initial stage viability, before dropping again to around 30.52%. The bacterial viability at pH 3.5 exhibited a much gentler decrease compared to that at pH of 1.5 and 2.5. Under the other three conditions, the bacterial viability was similar to that of the control group at pH 7.2 (Figure 2(b)).
In the in vitro GI Tract Dynamic pH Model, bacterial viability at pH 1.5 was lower than that in the simplified fluid, both at the 1.5-h (stomach stage) and 5.5-h (duodenum stage) timepoints, indicating the greater impact of acidity in the in vitro GI Tract Dynamic pH Model. Generally, the lowest viability values were observed at lower pH levels, particularly at pH 1.5 and pH 2.5.
F. nucleatum resists acidic environment at pH 1.5
To determine which bacteria survived in the acidified fluid at pH 1.5 during the 6-h study period, 1 mL samples were collected at 0.5-h intervals. The samples were then stained with PI/CYTO9 and sorted using flow cytometry. After 16S rRNA sequencing and data filtration, the resulting genus-level OTUs were clustered.
The survival rate of the simulated oral flora at 0.5 h was calculated and plotted at the genus level (Figure 2(c)). Compared to the control group treated with simulated salivary fluid only at pH 7.2, Fusobacterium showed the highest survival rate in the acidified fluid, followed by Streptococcus, although no statistically significant difference was observed.
To identify the specific Fusobacterium species that contributed to the high bacterial viability, individual species were tested. As shown in Figure 2(d), the effect of pH on bacterial viability varied among the species. F. nucleatum ATCC 25586 exhibited the highest viability at pH 1.5 (70.53%±7.00), and showed no significant difference (P = 0.2506) in viability at pH 3.5 (Figure 2(e)). F. nucleatum (612) displayed similarly high viability at pH 1.5 and pH 7.2, with the highest viability observed at pH 3.5 (Figure 2(f)). F. varium (36.1, pm-7) exhibited the weakest acid resistance at pH 1.5 compared to the other investigated Fusobacterium species (Figure 2(d)). F. mortiferum (811) and F. necrophorum (M530) demonstrated comparable acid resistance at pH 1.5, with viabilities of 20.20%±4.60 and 43.26%±6.49, respectively (Figures 2(d,g,h)); the observed pattern was similar to that of the F. nucleatum ATCC 25586 and 612 strains (Figures 2(d,f)).
F. nucleatum exhibits high levels of erucic acid (C22:1(n9)) in the cell membranes at pH 1.5
Cell membranes are the primary targets of environmental stress because they directly interact with the external environment. Cell viability under acidic stress conditions depends on the state of the cell membranes. Owing to its exceptional viability in acidic environments having pH 1.5 and its emerging clinical relevance, further research needs to investigate F. nucleatum ATCC 25586.
The bacterial growth at pH 1.5 significantly increased (p = 0.0017) the relative fatty acid content of MUFAs and decreased (p = 0.0072) the proportion of saturated fatty acids (SFAs) in F. nucleatum ATCC 25586 (Figures 3(a,b)).
Figure 3. .
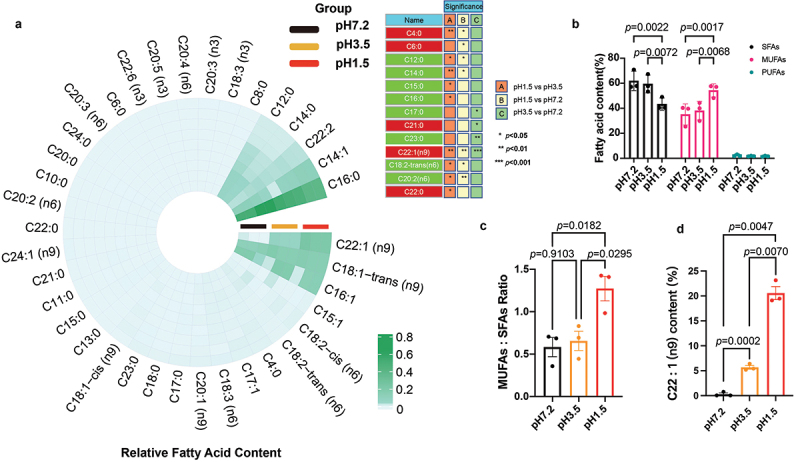
The response of Fusobacterium nucleatum to acidic environmental stress by upregulating the content of monounsaturated fatty acids, particularly C22:1. (a) Circular heatmap of fatty acid composition in Fusobacterium nucleatum ATCC 25586. The circular heatmap was generated using https://www.bioinformatics.com.cn (accessed on July 10th, 2023), an online platform for data analysis and visualization; the red color represents the upregulation, while the green color represents downregulation. (b) Fatty acid composition of Fusobacterium nucleatum under pH 1.5 and pH 3.5 acidic conditions. The total composition of saturated fatty acids (SFAs), monounsaturated fatty acids (MUFAs), and polyunsaturated fatty acids (PUFAs) were calculated, and compared to the fatty acid content at pH 7.2, which served as the control. (c) MUFAs/SFAs ratio of Fusobacterium nucleatum was upregulated at pH 1.5. The MUFAs/SFAs ratio was calculated by dividing the content of monounsaturated fatty acids by the content of saturated fatty acids. (d) Significant upregulation of the monounsaturated fatty acid erucic acid (C22:1) at pH 1.5. The experiments were performed three times. The data are expressed as mean ± standard deviation (SD). The p values are indicated.
Membrane fluidity is a critical factor that influences various membrane functions such as biochemical reactions, transport systems, and protein secretion. The ratio of MUFAs to SFAs is a key determinant of membrane fluidity; an increased ratio indicates higher membrane fluidity, whereas a decreased ratio indicates reduced membrane fluidity. Figure 3(c) demonstrates the changes in the MUFAs: SFAs ratio of low-pH-adapted cells compared with that of cells grown in neutral conditions. The ratio slightly increased at pH 3.5 (p = 0.9103) and significantly (p = 0.0182) increased when adapted to environments with pH 1.5, suggesting enhanced membrane fluidity.
Palmitic acid (C16:0) was the most abundant fatty acid identified at pH 1.5 (Figure 3(a)), where it accounted for 41.2%±4.83 of the total fatty acid composition. Erucic acid C22:1 (n9) was the second most prevalent fatty acid, constituting 20.57%±2.24 of the sample. Elaidic acid (C18:1-trans (n9)), the third most prevalent fatty acid, accounted for 14.17%±1.15 of the total fatty acid composition. Other notable fatty acids included palmitoleic acid (C16:1), tetradecenoic acid (C14:1), and docosadienoic acid (C22:2) which accounted for 13.18%±0.17, 9.91%±1.31, and 1.76%±0.19 of the total fatty acid composition, respectively. The remaining fatty acids were present in relatively small proportions in the range of 0.004–0.329%.
At pH 3.5 (Figure 3(a)), palmitic acid (C16:0) was the most abundant fatty acid, constituting 55.10%±6.61 of the total fatty acid composition. Palmitoleic acid (C16:1), elaidic acid (C18:1-trans (n9)), and C14:1 accounted for 17.65%±0.6, 15.28%±2.03, and 5.78%±2.13 of the total fatty acid composition, respectively. C22:1 (n9) represented 5.68%±0.64 of the total fatty acid composition, while the remaining fatty acids had smaller proportions (lauric acid (C12:0), 2.18% ±0.27; C22:2, 2.05%±0.24; and myristic acid (C14:0), 1.60%±0.18).
Similarly, at pH 7.2 (Figure 3(a)), C16:0 was the dominant fatty acid, comprising 54.42%±6.83 of the total fatty acid composition. C16:1, C18:1-trans (n9), C14:1, C12:0, C14:0, and C22:2 accounted for 17.38%±0.10, 11.56%±1.38, 10.27%±1.36, 4.22%±0.53, 2.90%±0.38, and 1.88%±0.24 of the total fatty acid composition, respectively.
Palmitic acid (C16:0) was consistently the most abundant fatty acid at all pH levels. The relative proportions of other fatty acids varied at different pH levels, indicating pH-dependent changes in fatty acid composition. The proportion of palmitic acid decreased with increasing the acidity of the environment from 54.42%±6.83 to 41.21%±4.83, suggesting a higher abundance of palmitic acid in neutral compared to extremely acidic environments; however, the change was not statistically significant. Interestingly, the saturated fatty acid myristic acid (C14:0) showed a significant (p = 0.0232) decrease at pH 1.5, indicating the potential impact of acidic conditions on its abundance in F. nucleatum ATCC 25586. In contrast, the relative content of the MUFA erucic acid (C22:1 n9) showed a significant (p = 0.0047) increase up to 70 folds at pH 1.5 (Figures 3(a,d)).
MUFA generation in F. nucleatum is associated with FnFabM and phylogenetic tree analysis revealed the origin of the FnfabM gene
The enzyme responsible for generating monounsaturated membrane fatty acids in F. nucleatum is unknown. However, a putative protein annotated as an enoyl-CoA hydratase-related protein, which displayed a sequence identity of 30.92% with an expected e-value of 2e−37, was identified through homology searches based on FabM sequences. The enoyl-CoA hydratase gene (FN0271) encodes a 264 amino acid protein belonging to the enoyl-CoA hydratase/isomerase family. This protein family acts as enoyl-CoA hydratases and exhibits isomerase activity. In this study, this gene was designated as the putative FnfabM.
Analysis of the submitted sequences of key enzymes involved in the elongation stage of UFA synthesis resulted in grouping the Fusobacterium spp. into two main clusters within the phylogenetic tree. The phylogenetic tree with the highest log likelihood (−5733.44) was constructed using the Maximum Likelihood method (Figure 4). Phylogenetic tree construction revealed the evolutionary relationships among the analyzed enoyl-CoA hydratase/isomerase family proteins. The tree exhibited several distinct clusters representing different species and strains. One cluster was represented by Helicobacter pylori FabX (GenBank: WJX98257, HpFabX), categorized as a dehydrogenase/isomerase, whereas the other cluster was represented by Escherichia coli strain K12 FabA (GenBank: P0A6Q3, EcFabA), categorized as a dehydratase/isomerase.
Figure 4. .
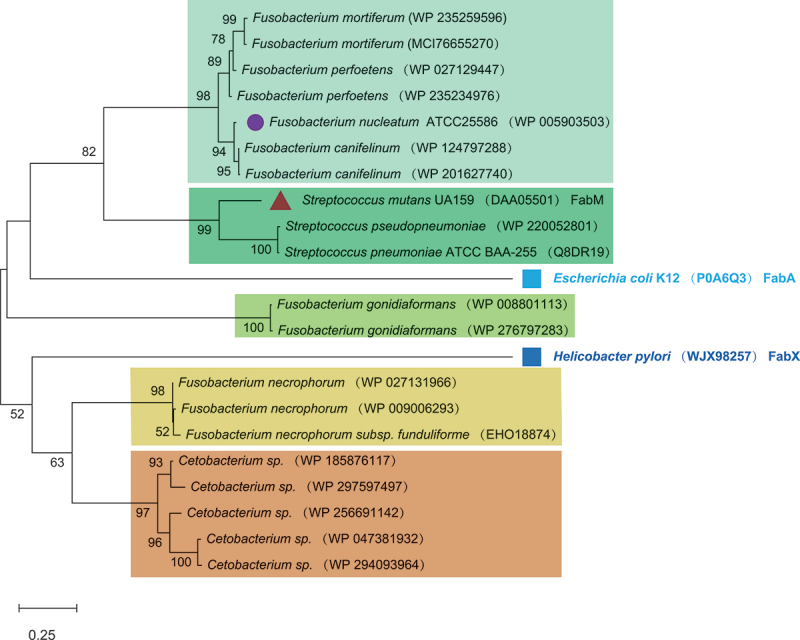
Phylogenetic tree analysis reveals the origin of the FnfabM gene. Phylogenetic tree analysis: the phylogenetic tree was constructed using sequences of the enoyl-CoA hydratase/isomerase family protein downloaded from GenBank, with the accession numbers listed next to the species names. For comparison, two outgroups (solid squares) were included: Helicobacter pylori Dehydrogenase/isomerase FabX (GenBank: WJX98257, HpFabX) and Escherichia coli strain K12 Dehydratase/isomerase FabA (GenBank: P0A6Q3, EcFabA), both representing the key enzymes of the elongation stage of the unsaturated fatty acid biosynthesis. Streptococcus mutans UA159 StrmFabM and Fusobacterium nucleatum FnFabM are marked with a solid circle and triangle, respectively. The scale bar at the bottom indicates 0.25 differences per residue. The numbers indicate the frequencies with which the tree topology was replicated after 100 bootstrap iterations.
In the FabA cluster, F. mortiferum (GenBank accession numbers WP_235259596 and MCI_7665527) and F. perfoetens (GenBank accession numbers WP_027129447 and WP_235234976) formed one subgroup; F. nucleatum (GenBank accession numbers WP_005903503) and F. canifelinum (GenBank accession numbers WP_124797288 and WP_201627740) constituted another subgroup. Notably, Streptococcus mutans UA159 (GenBank accession numbers: DAA05501, FabM), Streptococcus pseudopneumoniae (GenBank accession number: WP_220052801), and Streptococcus pneumoniae ATCC_BAA-255 (GenBank accession number: Q8DR19) branched off into separate clusters. Additionally, Escherichia coli strain K12 FabA was observed as a solitary, independent sequence within this cluster. Another interesting observation was that the two sequences from F. gonidiaformans (GenBank: WP_008801113 and WP_276797283) were grouped together. Within the FabX cluster, F. necrophorum (GenBank accession numbers: WP_027131966, WP_009006293, and EHO18874) clustered together. This cluster was further linked to a group of sequences from Cetobacterium sp. (GenBank accession numbers: WP_185876117, WP_297597497, WP_256691142, WP_047381932, and WP_294093964). However, Helicobacter pylori (GenBank accession no. WJX98257; FabX) branched out separately from this group.
FnfabM gene expression governs C22:1(n9) erucic acid synthesis in F. nucleatum
The expression pattern of FnfabM was investigated under different pH conditions at nine time points (Figure 5(a)). At pH 1.5, FnfabM gene expression increased rapidly, reaching its highest level at 1.5 h (37.42 ± 7.62), compared to an already elevated level at 0.5 h (22.7 ± 7.054). After the peak, expression rapidly decreased, becoming equivalent to pH 7.2 levels after 6 h (p = 0.0462). At pH 3.5, the initial upregulation of FnfabM was less pronounced (p = 0.1188) compared to pH 1.5, with peak expression occurring later at 2.5 h. Despite this delay, the overall expression trend at pH 3.5 mirrored the pattern observed at pH 1.5, albeit with lower magnitude. The FnfabM gene showed rapid upregulation in response to acidic conditions. At 3.5 h, expression levels were significantly higher at both pH 1.5 and pH 3.5 compared to neutral conditions (pH 7.2) (Figure 5(b)).
Figure 5. .
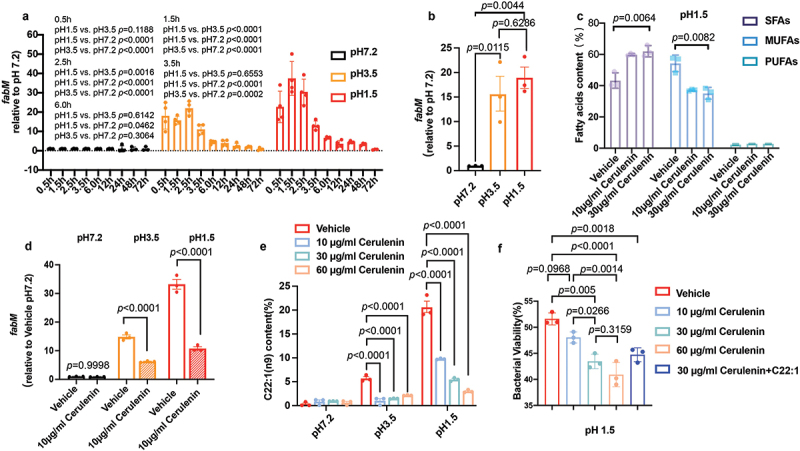
Investigation of the role of the FnfabM gene in the synthesis of monounsaturated fatty acids (MUFAs) in Fusobacterium nucleatum. (a) Spatiotemporal expression profile of FnfabM. The histogram displays the expression levels of the FnfabM gene in Fusobacterium nucleatum under acid stress conditions at different timepoints. Four independent biological replicates were analyzed. To normalize the expression data, the Fusobacterium nucleatum subsp. nucleatum ATCC 25586 DNA-directed RNA polymerase beta chain (rpoB) gene (GenBank: GQ274958.1) was used as a reference gene. (b) The expression of FnfabM was significantly upregulated when incubated at pH 1.5 for 3.5 h to simulate the gastric emptying time. (c) Unsaturated fatty acids synthase inhibition by cerulenin. Cerulenin, an inhibitor obtained from the fungus Cephalosporium caerulens specifically targets and inhibits the FabB and FabF enzyme β-ketoacyl-acp synthase of E. coli, which is involved in the elongation of fatty acids during their synthesis. (d) The FnfabM gene was significantly inhibited by 10 μg/ml cerulenin at pH 1.5 and pH 3.5, but not at pH 7.2. (e) Alterations in erucic acid C22:1(n9) content in Fusobacterium nucleatum with different concentrations of cerulenin for 4.5 h at different pH conditions. Changes in the composition of erucic acid (C22:1 n9) were observed in the presence of 10 μg/ml cerulenin in a dose-dependent manner. (f) Bacterial viability was significantly inactivated by cerulenin in a dose-dependent manner. Erucic acid (C22:1 n9) supplementation (10 μg/ml) restored Fusobacterium nucleatum acid resistance at pH 1.5 even in the presence of cerulenin, the fatty acid synthase inhibitor. The experiments were performed three times. The data are expressed as mean ± standard deviation (SD). The p values are indicated.
The difficulty in obtaining potent FabM inhibitors was highlighted in several studies [57,58]. Cerulenin, a natural compound from the fungus Cephalosporium caerulens, was discovered in 1960 to inhibit fatty acid synthesis [59]. It is particularly known for its ability to specifically target and inhibit both enzymes FabB and FabF β-ketoacyl-ACP synthase of E. coli, which are involved in the elongation of fatty acids during their synthesis [60]. In this study, cerulenin was used for the exploitation of fatty acid synthesis by Fusobacterium nucleatum subsp. nucleatum ATCC 25586.
Increasing cerulenin concentration at pH 1.5 resulted in a significant decrease (p = 0.0082) in the MUFAs content and substantial increase (p = 0.0064) in the SFAs content (Figure 5(c)). The investigation of FnfabM expression serves as a logical continuation, as it provides insights into the potential regulatory mechanisms and interplay between FnfabM and the observed variations in fatty acid composition under different pH conditions and cerulenin concentrations. Interestingly, 10 μg/mL of cerulenin led to the sufficient inhibition of the FnfabM transcription in the observed pH gradients (Figure 5(d)).
GC – MS was used to determine the content of fatty acids treated with the inhibitor cerulenin at pH 1.5. The content of the highly UFA C22:1 (n9), which showed the highest levels at pH 1.5, decreased more than four-fold in the presence of the cerulenin inhibitor in a concentration-dependent manner (Figure 5(e)).
Inhibition of FnfabM gene expression resulted in the decreased colonization efficiency of F. nucleatum in the stomach and jejunum of mice
A high UFAs/SFAs ratio enhances the integrity of the bacterial cell membranes and reduces their permeability to acidic environments [61]. Cerulenin significantly increased the SFAs content (p = 0.0064) and decreased the MUFAs content (p = 0.0082) at pH 1.5 (Figure 5(c)). It remains unclear whether the suppression of UFAs synthesis or FnfabM gene enhances bacterial sensitivity to strong acids. Therefore, in vitro and in vivo experiments were performed to determine the sensitivity of these cells to acids after suppression. Cerulenin at a concentration of 30 μg/mL was sufficient to decrease the viability of F. nucleatum subsp. nucleatum ATCC 25586 at pH 1.5 in a dose-dependent manner (Figure 5(f)). Notably, when supplemented with exogenous C22:1 (n9), the bacterial viability was restored to the level similar to that under cerulenin inhibition treatment (Figure 5(f)).
In the in vivo study, significant differences were observed between the control and cerulenin treatment groups in terms of bacterial load within the gastrointestinal contents of different intestinal segments (Figure 6(a)). Notably, the F. nucleatum bacterial load was significantly lower in the cerulenin treatment group compared to that in the control group in the stomach and jejunum of mice, p = 0.0158 and p = 0.0123, respectively (Figures 6(b, c)). To verify whether the expression of the corresponding FnfabM gene in these intestinal segments was suppressed, RT-PCR was performed. The corresponding FnfabM gene expression in the same intestinal segments was also significantly (stomach p = 0.0007, jejunum p = 0.0464) suppressed (Figure 6(d)), raising intriguing questions regarding the potential impact of cerulenin on MUFA synthesis and metabolism of F. nucleatum in the presence of gastric acid.
Figure 6. .
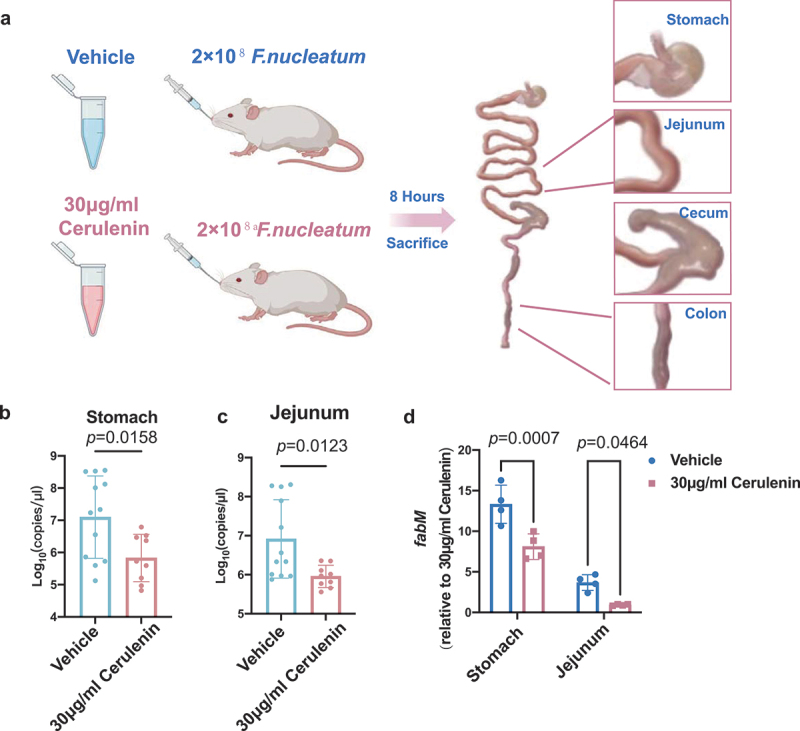
FnfabM gene inhibition using cerulenin reduced the bacterial load in the stomach and jejunum in mice. (a) Schematic diagram of the animal experiments set-up. Quantification of the bacterial load in the contents of (b) stomach and (c) jejunum using absolute quantitative PCR 8 h post gavage. (d) FnfabM gene expression levels of F. nucleatum in the contents of stomach and jejunum 8 h post gavage. The dots represent the numbers of mice (vehicle group, n = 12 and cerulenin treatment group, n = 9). The data are expressed as mean ± standard deviation (SD). The p values are indicated.
Discussion
The oral-gut axis is emerging as an important pathway contributing to the development of systemic conditions independent of blood circulation. In this study, we introduced a dynamic pH simulation model of the human upper gastrointestinal tract used for probiotic screening and investigated its impact on different oral bacterial species. We investigated four phyla, namely Bacteroidetes, Firmicutes, Actinobacteria, and Fusobacteria, as well as eight genera of oral bacteria, based on their composition in oral microorganisms [62]. Streptococcus, a member of the yellow complex associated with healthy pockets, and Fusobacterium, a member of the orange complex, have been identified as transitional populations between healthy and severe periodontal disease, demonstrating high acid resistance. The intricate structure and functionality of microbial communities extend beyond a mere aggregation of their constituents, as interspecies interactions can profoundly influence the resilience and adaptability of the consortium to environmental perturbations. Streptococcus, the predominant genus in whole saliva and healthy adult dental plaque [62], plays a crucial role in the assembly of oral microbiota. It contributes to the immune function in the saliva, biofilm formation, and colonization of dental plaque, thereby impacting the development of gingivitis and dental caries through carbohydrate metabolism and acid production. Notably, among the Streptococcus genus, the S. mutans stands out for its significant contribution to dental caries due to its high acidogenic and aciduric properties [63]. Paradoxically, S. sanguinis and S. gordonii generate ammonia through the arginine deiminase system (ADS), buffering acids in dental plaque [63]. S. salivarius, have been noted for their probiotic properties, which include the suppression of pathogen proliferation and the preservation of oral microbial homeostasis [64]. It is noteworthy to incorporate recent advancements in our understanding of the S. anginosus species, as elucidated by Kaili Fu et al [65]. This study has delineated the potential oncogenic role of S. anginosus in gastric tumorigenesis, underscoring its remarkable resilience to acidic conditions within the gastric mucosa, which facilitates its colonization and subsequent promotion of gastric inflammation and atrophy-key precursors to neoplastic transformation.
Fusobacteria, produce butyric acid and other acids during fermentation, which can influence the microbiome’s overall adaptability. These microbes may engage in symbiotic relationships that enhance the community’s resilience, particularly in acidic environments, by exchanging metabolic byproducts that support mutual survival. Microscopic imagery has captured the symbiotic adhesion of oral bacteria, including Corynebacterium, Fusobacterium, Streptococcus, Porphyromonas, and Haemophilus/Aggregatibacter, illustrating their formation of distinctive corncob clusters within dental plaque structures [66]. To date, more than 12 human-associated Fusobacterium species have been adequately described [67]. F. nucleatum and F. necrophorum are the most frequently isolated species from humans and animals, respectively [68], and are notably prevalent in oral and neck cancers [69]. In the oral cavity, F. nucleatum is particularly well-studied due to its highly adhesive surface properties, which allow to act as a bridge organism, facilitating the diversification of dental plaque by linking primary and secondary colonizers [66]. The relevance of F. nucleatum extends beyond the oral cavity, with its role increasingly recognized in various conditions, including adverse pregnancy outcomes, cardiovascular disease, rheumatoid arthritis, and colorectal cancers [66]. Our previous study [70] has indicated that different Fusobacterium species exhibit distinct correlation patterns with host diseases. For instance, F. varium is enriched in colorectal cancer (CRC), F. mortiferum is associated with host metabolism, and F. nucleatum may have the ability to translocate to distant organs. Additionally, we have observed regional variability in the distribution of Fusobacterium species, with the F. mortiferum lineage being the most prevalent and abundant in the gut, and it is negatively correlated with F. varium and F. ulcerans. These findings underscore the importance of understanding the mechanisms that may enable acid-resistant strains to translocate to the gastrointestinal tract and thrive in diverse ecological niches, even though F. mortiferum and F. varium are more commonly found in the intestines [68]. Given the increasing clinical significance of Fusobacterium species, particularly F. nucleatum, our focus has been directed towards elucidating how this species, known for its extensive study and clinical relevance, achieves its remarkable acid resistance.
Being of increasing clinical importance and the most extensively studied species within the genus, F. nucleatum displayed the highest acid resistance level at pH 1.5. We observed altered fatty acid composition in F. nucleatum, characterized by increased levels of MUFAs, which enhanced acid resistance in vitro and in vivo. In a recent study, it was reported that the Fna C2 clade of F. nucleatum exhibits enhanced acid tolerance due to its distinctive metabolic capabilities and glutamate-dependent acid resistance system, thereby gaining a competitive edge in the acidic tumor microenvironment of colorectal cancer [71]. Incorporating our evidence, it is further demonstrated that F. nucleatum can transition from its oral niche to the gastrointestinal and tumor microenvironments through various acid resistance mechanisms.
Bacteria adapt to environmental changes by modifying their fatty acid composition. This adaptation is crucial for surviving in acidic conditions, where maintaining membrane integrity and fluidity is essential. The ratio of unsaturated fatty acids (UFAs) to saturated fatty acids (SFAs) plays a key role in this process [72,73]. Interestingly, F. nucleatum synthesizes UFAs despite lacking the typical UFA synthesis pathway [74]. This ability is likely due to the presence of FabM, a novel protein first identified in S. pneumoniae and S. mutans. FabM isomerizes trans-unsaturated bonds to cis-isomers, enabling MUFA synthesis and survival at low pH [75,76]. FabM homologues have primarily been found in Streptococcal and Staphylococcal species, with F. nucleatum being a notable exception as a Gram-negative bacterium [76], which is consistent with the phylogenetic tree analysis result in here. This finding supports the theory that Fusobacterium spp. are essentially Gram-positive bacteria that have acquired a Gram-negative ‘cloak’ to evade immune responses and enhance adhesion [77]. The genetic diversity within microbial communities may provide bacteria with a broader range of adaptive strategies, including those for acidic stress tolerance. This diversity highlights the complex and sophisticated mechanisms bacteria employ to thrive in challenging environments.
The rapid upregulation of FnfabM in acidic conditions suggests a quick metabolic adjustment as the bacterium senses pH changes. The temporal divergence in expression patterns between pH 1.5 and pH 3.5, along with sustained high expression levels, points to a complex regulatory mechanism. This may indicate different adaptive strategies for extreme (pH 1.5) versus strong (pH 3.5) acid stress. A key component of this survival strategy is the bacterium’s two-component systems, CarRS and ModRS, which enhance interspecies interactions and provide defenses against oxidative stress, respectively [78,79]. These systems, coupled with the proton motive force (PMF) and the global stress response governed by the extracytoplasmic function sigma factor (ECF) sigma E (σE) and its encoding gene RpoE, are instrumental in F. nucleatum’s resilience to a spectrum of environmental challenges, particularly those posed by acidic conditions [80].
F. nucleatum’s remarkable adaptability across diverse environments is underpinned by a suite of sophisticated mechanisms that extend beyond FabM-mediated fatty acid adjustments. The sensor histidine kinase ArlS is necessary for S. aureus to activate ArlR in response to nutrient availability [81] and the response regulator ArlR from F. nucleatum had just been characterized [82]. This intricate regulatory network likely underpins the differential gene expression of FnfabM under the acidic conditions of pH 1.5 and pH 3.5, highlighting the bacterium’s adaptability in the face of environmental extremes.
Our investigation into the FnfabM gene, an Enoyl-CoA hydratase and isomerase, has shed light on its pivotal role in the bacterium’s acid adaptation. We found that FnfabM is significantly upregulated under acidic conditions, and its expression is specifically curbed by cerulenin in these conditions, underscoring a nuanced regulatory network at play.
Our study revealed significant differences in fatty acid composition changes among Fusobacterium strains under acidic conditions. In F. nucleatum, a complex adaptive network modulates fatty acid composition, elevating levels of UFAs, particularly C22:1(n-9). These alterations reinforce membrane integrity and attenuate acid permeability, enabling the bacterium to navigate the harsh acidic milieu of the stomach and intestine effectively. Interestingly, the F. necrophorum (530) strain, which possesses a FabM homolog (Figure 4), exhibited a notable increase in MUFAs under acid stress. However, this increase was not specific to C22:1 fatty acid but likely resulted from the accumulation of various fatty acids. In contrast, F. mortiferum (811), despite also having a FabM homolog, did not show a similar significant elevation in MUFAs when incubated at pH 1.5. The F. varium (pm-7) strain, which lacks a FabM homolog, also did not demonstrate an increase in MUFAs under acidic conditions. The absence of FabM in F. varium may be associated with its poorer acid survival capability, as demonstrated in Figure 2(d). Surprisingly, the F. nucleatum (612) strain did not display the same pattern of fatty acid changes as the F. nucleatum ATCC25586 strain, possibly due to strain-specific genetic variations (Supplemental Figures 1(a–h)).
In our in vivo animal study, we observed a lower F. nucleatum load in the cerulenin-treated group than that in the control group in the stomach and jejunum. This suggests that cerulenin disrupts key metabolic pathways in F. nucleatum when exposed to the acidic environments of the stomach and small intestine. Acid adaptation in bacteria can confer resistance to various stress conditions, and the acid-producing F. nucleatum coexists with the same acid-producing Streptococcus in the oral environment; the acid tolerance of F. nucleatum may aid in withstanding the subsequent stress conditions encountered in the intestine [83,84].
The adaptive mechanism triggered by the exposure to an acidic environment, which involves an increase in the UFA composition of membrane phospholipids, is crucial for maintaining membrane integrity [65], as evidenced by a high UFAs: SFAs ratio observed in F. nucleatum at pH 1.5 in our study. This adaptation, akin to the response to cold stress, enhances membrane fluidity and is associated with increased bacterial survival [85]. While UFAs exhibit weakly proinflammatory or neutral properties, SFAs are notably proinflammatory [86], suggesting a subtle yet critical role in modulating host responses. Long-chain MUFAs have been studied for their potential effects [87]; however, more studies are needed to understand their impact. Contrastingly, the host response to microbial communities is significantly modulated by the microbiota, which has profound implications for bacterial survival and pathogenicity within acidic environments. Interestingly, while S. anginosus infection at the precancerous stage has been noted to cause an elevation in gastric pH [65], our unpublished data intriguingly reveal a comparable elevation in pH induced by F. nucleatum, suggesting a common yet enigmatic mechanism by which these bacteria modulate the gastric environment.
Acknowledging the limitations of this work is necessary and provides a foundation for future research directions. While our study has focused on selected oral bacteria, we recognize the rich spatiotemporal dynamics of the oral polymicrobial community, which likely plays a critical role in bacterial adaptation to acidic conditions and survival. Firstly, we concede that our current investigation did not encompass the full complexity of the oral microbiome, which may lead to an incomplete understanding of microbial survival strategies in acidic environments. This limitation could potentially affect the generalizability of our findings to the broader oral microbial community. Future studies will leverage the oral microbiome from the human saliva pool, which is known to contain a diverse array of microorganisms. We plan to use co-culture experiments and metagenomic analyses to investigate how microbe–microbe interactions could potentially enhance collective acid tolerance and influence the expression of genes involved in acid adaptation, such as FnfabM. Secondly, regarding the biochemical mechanisms underpinning membrane adaptation, we have not yet experimentally validated the role of the enoyl-CoA hydratase in generating MUFA within the context of the polymicrobial community. Our preliminary attempts to knock out this enzyme in F. nucleatum strain 25586 were met with challenges due to inherent genetic defenses and limitations in gene editing techniques for this strain. This limitation in our ability to manipulate F. nucleatum genetically has restricted our capacity to fully elucidate the in vivo role of the enoyl-CoA hydratase in acid adaptation within the complex oral environment. In light of this, we intend to pursue recombinant prokaryotic expression systems to assess enzyme activity and plan to conduct knock-out experiments that will take into account the broader microbial interactions within the oral community. By integrating these approaches, including co-culture experiments, metagenomic analyses, and recombinant enzyme studies, we aim to provide a more comprehensive understanding of how oral microbes interact and adapt to acidic stress. This holistic approach is essential for elucidating their roles in oral health and disease, potentially leading to new strategies for managing acid-related oral conditions.
Conclusions
Our findings suggest that F. nucleatum adapts to the hostile gastric acid environment by upregulating MUFAs, primarily C22:1(n9). In the intestinal milieu, F. nucleatum exhibits distinct characteristics marked by the emergence of C22:1(n9). This adaptation may lead to the colonization of F. nucleatum in the intestine; thus, playing a role in dampening proinflammatory responses and influencing macrophage polarization. Understanding these mechanisms could offer potential avenues for preventive and therapeutic interventions targeting acid resistance mechanisms.
Supplementary Material
Acknowledgments
We would like to thank J. Xu (Single-Cell Center, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences) for providing the bacterial strains. We thank Dr. Prabhakar Mujagond for isolating the Fusobacterium. This study was designed and carried out at the Microbiome Medicine Center of Zhu Jiang Hospital of Southern Medical University. We thank the Laboratory Medicine and Microbiome Medicine Center at the Zhu Jiang Hospital of Southern Medical University and the Oral Microbiome Medicine Center at the Shenzhen Stomatology Hospital (Pingshan) of Southern Medical University. We acknowledge using BioRender (https://www.biorender.com) to provide creative illustration materials and platforms. We also acknowledge using the online platform (https://www.bioinformatics.com.cn) for the data analysis and visualization.
Funding Statement
This study was supported by the National Key Research and Development Program of China under Grant number [2022YFA0806400 HWZ].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions:
X. C. L. and H. W. Z. designed the study. X. C. L., S. P. Z., and H. F. S. conducted the experiments. X. C. L. and Z. L. analyzed and interpreted the data. H. W. Z. and B. L. W. oversaw project development. X. C. L., H. W. Z., and Z. L. prepared the manuscript for publication. X. C. L., Y. Z., D. Q. C., Z. L., and H. W. Z. engaged in insightful discussions and contributed to the overall discourse. All authors have read the journal’s authorship agreement, and the manuscript has been reviewed and approved by all the named authors.
Data availability statement
The 16s rRNA raw data that support the findings of this study are openly available in the NCBI SRA metadata under accession number PRJNA1050633. The data generated in this study via experiments, including the bacterial viability assays, inhibitor and animal experiments are available from the corresponding author upon reasonable request.
Ethics approval
The animal experiments in the present study were approved by the Ethics Committee for Animal Care and Research of Zhujiang Hospital of Southern Medical University (Approval ID: LAEC-2021-216).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/20002297.2025.2453964
References
- [1].Aas JA, Paster BJ, Stokes LN, et al. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43(11):5721–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Olsen I, Yamazaki K.. Can oral bacteria affect the microbiome of the gut? J Oral Microbiol. 2019;11(1):1586422. doi: 10.1080/20002297.2019.1586422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Xian P, Xuedong Z, Xin X, et al. The oral microbiome bank of China. Int J Oral Sci. 2018;10(2):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu XX, Jiao B, Liao XX, et al. Analysis of salivary microbiome in patients with Alzheimer’s disease. J Alzheimer’s Disease. 2019;72(2):633–640. [DOI] [PubMed] [Google Scholar]
- [5].Carelli M, Maguolo A, Zusi C, et al. Oral microbiota in children and adolescents with type 1 diabetes mellitus: novel insights into the pathogenesis of dental and periodontal disease. Microorganisms. 2023;11(3):668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Qin H, Li G, Xu X, et al. The role of oral microbiome in periodontitis under diabetes mellitus. J Oral Microbiol. 2022;14(1):2078031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu XR, Xu Q, Xiao J, et al. Role of oral microbiota in atherosclerosis. Clinica (Rome) Acta. 2020;506:191–195. [DOI] [PubMed] [Google Scholar]
- [8].Seerangaiyan K, Jüch F, Winkel EG. Tongue coating: its characteristics and role in intra-oral halitosis and general health-a review. J Breath Res. 2018;12(3):34001. [DOI] [PubMed] [Google Scholar]
- [9].Nagy KN, Sonkodi I, Szöke I, et al. The microflora associated with human oral carcinomas. Oral Oncol. 1998;34(4):304–308. [PubMed] [Google Scholar]
- [10].Michaud DS. Role of bacterial infections in pancreatic cancer. Carcinogenesis. 2013;34(10):2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].McCoy AN, Araujo-Perez F, Azcarate-Peril A, et al. Fusobacterium is associated with colorectal adenomas. PLOS ONE. 2013;8(1):e53653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Marchesi JR, Dutilh BE, Hall N, et al. Towards the human colorectal cancer microbiome. PLOS ONE. 2011;6(5):e20447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Suh JS, Kim S, Boström KI, et al. Periodontitis-induced systemic inflammation exacerbates atherosclerosis partly via endothelial–mesenchymal transition in mice. Int J Oral Sci. 2019;11(3):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Slocum C, Coats S, Hua N, et al. Distinct lipid a moieties contribute to pathogen-induced site-specific vascular inflammation. PLOS Pathog. 2014;10:e1004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pan W, Wang Q, Chen Q. The cytokine network involved in the host immune response to periodontitis. Int J Oral Sci. 2019;11(3):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Reyes L. Porphyromonas gingivalis. Trends Microbiol. 2021;29(4):376–377. [DOI] [PubMed] [Google Scholar]
- [17].Mei F, Xie M, Huang X, et al. Porphyromonas gingivalis and its systemic impact. Curr Status Pathog. 2020;9(11):944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kamer AR, Pushalkar S, Gulivindala D, et al. Periodontal dysbiosis associates with reduced CSF Aβ42 in cognitively normal elderly. Alzheimer’s & Dementia: Diagnosis, Assess Disease Monit. 2021;13(1):e12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol And Immunol. 2002;17(2):113–118. doi: 10.1046/j.0902-0055.2001.00100.x [DOI] [PubMed] [Google Scholar]
- [21].Chhibber-Goel J, Singhal V, Bhowmik D, et al. Linkages between oral commensal bacteria and atherosclerotic plaques in coronary artery disease patients. NPJ Biofilms Microbiomes. 2016;2(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Madianos PN, Bobetsis YA, Offenbacher S. Adverse pregnancy outcomes (APO s) and periodontal disease: pathogenic mechanisms. J Clin Periodontol. 2013;40:S170–80. [DOI] [PubMed] [Google Scholar]
- [23].Bobetsis YA, Graziani F, Gürsoy M, et al. Periodontal disease and adverse pregnancy outcomes. Periodontology 2000. 2020;83(1):154–174. [DOI] [PubMed] [Google Scholar]
- [24].Fardini Y, Wang X, Témoin S, et al. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol. 2011;82(6):1468–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Huang Z, Chen J, Li B, et al. Faecal microbiota transplantation from metabolically compromised human donors accelerates osteoarthritis in mice. Ann Rheum Dis. 2020;79(5):646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hong M, Li Z, Liu H, et al. Fusobacterium nucleatum aggravates rheumatoid arthritis through FadA-containing outer membrane vesicles. Cell Host & Microbe. 2023;31(5):798–810. doi: 10.1016/j.chom.2023.03.018 [DOI] [PubMed] [Google Scholar]
- [27].Gaffen SL, Moutsopoulos NM. Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci Immunol. 2020;5(43):eaau4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fine N, Chadwick JW, Sun C, et al. Periodontal inflammation primes the systemic innate immune response. J Dent Res. 2021;100(3):318–325. [DOI] [PubMed] [Google Scholar]
- [29].Elmaghrawy K, Hussey S, Moran GP. The oral microbiome in pediatric IBD: a source of pathobionts or biomarkers? Front Pediatr. 2021;8:620254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jackson MA, Goodrich JK, Maxan ME, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut. 2016;65(5):749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Filardo S, Scalese G, Virili C, et al. The potential role of hypochlorhydria in the development of duodenal dysbiosis: a preliminary report. Front Cell Infect Microbiol. 2022:12 :854904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li B, Ge Y, Cheng L, et al. Oral bacteria colonize and compete with gut microbiota in gnotobiotic mice. Int J Oral Sci. 2019;11(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bao J, Li L, Zhang Y, et al. Periodontitis may induce gut microbiota dysbiosis via salivary microbiota. Int J Oral Sci. 2022;14(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bernard-Raichon L, Venzon M, Klein J, et al. Gut microbiome dysbiosis in antibiotic-treated COVID-19 patients is associated with microbial translocation and bacteremia. Nat Commun. 2022;13(1):5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cowardin CA, Syed S, Iqbal N, et al. Environmental enteric dysfunction: gut and microbiota adaptation in pregnancy and infancy. Nat Rev Gastroenterol & Hepatol. 2023;20(4):223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ryu EP, Davenport ER. Host genetic determinants of the microbiome across animals: from Caenorhabditis elegans to cattle. Annu Rev Anim Biosci. 2022;10:203–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guo W, Zhou X, Li X, et al. Depletion of gut microbiota impairs gut barrier function and antiviral immune defense in the liver. Front Immunol. 2021;12:636803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nighot M, Liao PL, Morris N, et al. Long-term use of proton pump inhibitors disrupts intestinal tight junction barrier and exaggerates experimental colitis. J Crohn’s Colitis. 2023;17(4):565–579. doi: 10.1093/ecco-jcc/jjac168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fujimori S. Gastric acid level of humans must decrease in the future. World J Gastroenterol. 2020;26(43):6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Martinsen TC, Fossmark R, Waldum HL. The phylogeny and biological function of gastric juice—microbiological consequences of removing gastric acid. Int J Mol Sci. 2019;20(23):6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].He Y, Wu W, Zheng HM, et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat Med. 2018;24(10):1532–1535. [DOI] [PubMed] [Google Scholar]
- [42].Uchino Y, Ken-Ichiro S. A simple preparation of liquid media for the cultivation of strict anaerobes. J Pet Environ Biotechnol S. 2011;3(2).S3–001 [Google Scholar]
- [43].Mainville I, Arcand Y, Farnworth ER. A dynamic model that simulates the human upper gastrointestinal tract for the study of probiotics. Int J Food Microbiol. 2005;99(3):287–296. [DOI] [PubMed] [Google Scholar]
- [44].Carpenter GH. The secretion, components, and properties of saliva. Annu Rev Food Sci Technol. 2013;4(1):267–276. [DOI] [PubMed] [Google Scholar]
- [45].Berrada N, Lemeland JF, Laroche G, et al. Bifidobacterium from fermented milks: survival during gastric transit. J Dairy Sci. 1991;74(2):409–413. [DOI] [PubMed] [Google Scholar]
- [46].Smith JL. The role of gastric acid in preventing foodborne disease and how bacteria overcome acid conditions. J Food Prot. 2003;66(7):1292–1303. [DOI] [PubMed] [Google Scholar]
- [47].Minekus M, Marteau P, Havenaar R, et al. A multicompartmental dynamic computer-controlled model simulating the stomach and small intestine. Altern To Lab Anim. 1995;23(2):197–209. [Google Scholar]
- [48].Hall JE, Hall ME.Secretory Functions of the Alimentary Tract. Guyton and Hall Textbook of Medical Physiology ,14 th Editiion (Philadelphia: Elsever; ). 2020: 807–822. [Google Scholar]
- [49].Nyambe L, Mkandawire SA. Composition, in vitro digestibility, and sensory evaluation of extruded whole grain sorghum breakfast cereals. Lebensm Wiss Und-Tech Food Sciamp; Technol. 2015;62(1):662–667. [Google Scholar]
- [50].Walters W, Hyde ER, Berg-Lyons D, et al. Improved bacterial 16S rRNA gene (V4 and V4–5) and fungal internal transcribed spacer Marker gene primers for microbial community surveys. mSystems. 2016;1(1). doi: 10.1128/mSystems.00009-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Caporaso JG, Kuczynski J, Stombaugh J et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336 doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. [DOI] [PubMed] [Google Scholar]
- [53].Araujo P, Nguyen TT, Frøyland L, et al. Evaluation of a rapid method for the quantitative analysis of fatty acids in various matrices. J Chromatogr A. 2008;1212(1–2):106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang XJ, Huang LL, Su H, et al. Characterizing plasma phospholipid fatty acid profiles of polycystic ovary syndrome patients with and without insulin resistance using GC–MS and chemometrics approach. J Pharm Biomed Anal. 2014;95:85–92. [DOI] [PubMed] [Google Scholar]
- [55].Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Bioinformatics. 1992;8(3):275–282. [DOI] [PubMed] [Google Scholar]
- [56].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- [57].Zhang L, Liu W, Hu T, et al. Structural basis for catalytic and inhibitory mechanisms of β-hydroxyacyl-acyl carrier protein dehydratase (FabZ). J Biol Chem. 2008;283(9):5370–5379. [DOI] [PubMed] [Google Scholar]
- [58].Sharma SK, Kapoor M, Ramya TNC, et al. Identification, characterization, and inhibition of Plasmodium falciparum β-hydroxyacyl-acyl carrier protein dehydratase (FabZ). J Biol Chem. 2003;278(46):45661–45671. [DOI] [PubMed] [Google Scholar]
- [59].Bibens L, Becker JP, Dassonville-Klimpt A, Sonnet, P. A review of fatty acid biosynthesis enzyme inhibitors as promising antimicrobial drugs. Pharmaceuticals (Basel). 2023;16(3):425. doi: 10.3390/ph16030425. PMID: 36986522; PMCID: PMC10054515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Price AC, Choi KH, Heath RJ, et al. Inhibition of β-ketoacyl-acyl carrier protein synthases by thiolactomycin and cerulenin: structure and mechanism. J Biol Chem. 2001;276(9):6551–6559. [DOI] [PubMed] [Google Scholar]
- [61].Eckhardt TH, Skotnicka D, Kok J, et al. Transcriptional regulation of fatty acid biosynthesis in Lactococcus lactis. J Bacteriol. 2013;195(5):1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Palmer RJ Jr. Composition and development of oral bacterial communities. Periodontology 2000. 2014;64(1):20–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bowen WH, Burne RA, Wu H, et al. Oral biofilms: pathogens, matrix, and polymicrobial interactions in microenvironments. Trends Microbiol. 2018;26(3):229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Burton JP, Wescombe PA, Moore CJ, et al. Safety assessment of the oral cavity probiotic streptococcus salivarius K12. Appl Environ Microbiol. 2006;72(4):3050–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fu K, Cheung AHK, Wong CC, et al. Streptococcus anginosus promotes gastric inflammation, atrophy, and tumorigenesis in mice. Cell. 2024;187(4):882–896. [DOI] [PubMed] [Google Scholar]
- [66].Welch JLM, Rossetti BJ, Rieken CW, et al. Biogeography of a human oral microbiome at the micron scale. Proceedings of the National Academy of Sciences of the United States of America; 2016;(6):E791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ludwig W, Viver T, Westram R, et al. Release LTP_12_2020, featuring a new ARB alignment and improved 16S rRNA tree for prokaryotic type strains. Syst Appl Microbiol. 2021;44(4):126218. [DOI] [PubMed] [Google Scholar]
- [68].Hofstad T. The genus fusobacterium BT - the prokaryotes: volume 7: proteobacteria: delta, epsilon subclass. In: Dworkin M, Falkow S, Rosenberg E,Schleifer KH, Stackebrandt E, editors. New York, NY: Springer New York; 2006. p. 1016–1027. [Google Scholar]
- [69].Bronzato JD, Bomfim RA, Edwards DH, et al. Detection of fusobacterium in oral and head and neck cancer samples: a systematic review and meta-analysis. Arch Oral Biol. 2020;112:104669. [DOI] [PubMed] [Google Scholar]
- [70].He Y, Mujagond P, Tang W, et al. Non-nucleatum fusobacterium species are dominant in the Southern Chinese population with distinctive correlations to host diseases compared with F. nucleatum. Gut. 2021;70(4):810–812. [DOI] [PubMed] [Google Scholar]
- [71].Zepeda-Rivera M, Minot SS, Bouzek H, et al. A distinct fusobacterium nucleatum clade dominates the colorectal cancer niche. Nature. 2024;628(8007):424–432. doi: 10.1038/s41586-024-07182-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sohlenkamp C. Membrane Homeostasis in Bacteria upon pH Challenge. In: Geiger, O Biogenesis of Fatty Acids, Lipids and Membranes. Handbook of Hydrocarbon and Lipid Microbiology.Cham,Springer International Publishing. 2017:1–13 doi: 10.1007/978-3-319-43676-0_57-1. [DOI] [Google Scholar]
- [73].Álvarez-Ordóñez A, Fernández A, López M, et al. Relationship between membrane fatty acid composition and heat resistance of acid and cold stressed salmonella senftenberg CECT 4384. Food Microbiol. 2009;26(3):347–353. [DOI] [PubMed] [Google Scholar]
- [74].Kapatral V, Anderson I, Ivanova N, et al. Genome sequence and analysis of the oral bacterium fusobacterium nucleatum strain ATCC 25586. J Bacteriol. 2002;184(7):2005–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Marrakchi H, Choi KH, Rock CO. A new mechanism for anaerobic unsaturated fatty acid formation inStreptococcus pneumoniae. J Biol Chem. 2002;277(47):44809–44816. [DOI] [PubMed] [Google Scholar]
- [76].Fozo EM, Quivey RG Jr. The fabM gene product of streptococcus mutans is responsible for the synthesis of monounsaturated fatty acids and is necessary for survival at low pH. J Bacteriol. 2004;186(13):4152–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Robinson A, Wilde J, Allen-Vercoe E. Fusobacteria: physiology, form, and function Floch H, Martin. In: Colorectal neoplasia and the colorectal microbiome. United States: Elsevier; 2020. p. 95–134. [Google Scholar]
- [78].Ponath F, Zhu Y, Cosi V, et al. Expanding the genetic toolkit helps dissect a global stress response in the early-branching species fusobacterium nucleatum. Proceedings of the National Academy of Sciences; 2022;(40):e2201460119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Falk P, Yan Z, Jörg V. Transcriptome fine-mapping in Fusobacterium nucleatum reveals FoxJ, a new σE-dependent small RNA with unusual mRNA activation activity. MBio. 2024. Mar 4;15(4):e03536–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Krulwich TA, Sachs G, Padan E. Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol. 2011. May;9(5):330–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Párraga Solórzano PK, Shupe AC, Kehl-Fie TE. The sensor histidine kinase ArlS is necessary for staphylococcus aureus to activate ArlR in response to nutrient availability. J Bacteriol. 2021;203(24):e0042221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Fan R, Li Z, Shi X, et al. Expression, purification, and characterization of the recombinant, two-component, response regulator ArlR from Fusobacterium nucleatum. Appl Biochem Biotechnol. 2022;194(5):2093–2107. [DOI] [PubMed] [Google Scholar]
- [83].Bearson S, Bearson B, Foster JW. Acid stress responses in enterobacteria. FEMS Microbiol Lett. 1997;147(2):173–180. [DOI] [PubMed] [Google Scholar]
- [84].Procházková N, Falony G, Dragsted LO, et al. Advancing human gut microbiota research by considering gut transit time. Gut. 2023;72(1):180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Yoshida K, Hashimoto M, Hori R, et al. Bacterial long-chain polyunsaturated fatty acids: their biosynthetic genes, functions, and practical use. Mar Drugs. 2016;14(5):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ohue‐Kitano R, Yasuoka Y, Goto T, et al. α‐Linolenic acid‐derived metabolites from gut lactic acid bacteria induce differentiation of anti‐inflammatory M2 macrophages through G protein‐coupled receptor 40. The FASEB J. 2018;32(1):304–318. [DOI] [PubMed] [Google Scholar]
- [87].Tsutsumi R, Yamasaki Y, Takeo J, et al. Long-chain monounsaturated fatty acids improve endothelial function with altering microbial flora. Transl Res. 2021;237:16–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 16s rRNA raw data that support the findings of this study are openly available in the NCBI SRA metadata under accession number PRJNA1050633. The data generated in this study via experiments, including the bacterial viability assays, inhibitor and animal experiments are available from the corresponding author upon reasonable request.