

Abstract
Recent studies reported hundreds of genes linked to chronic lymphocytic leukemia (CLL). However, many of these candidate genes were lack of replication and results were not always consistent. Here, we proposed a computational workflow to curate and evaluate CLL-related genes. The method integrates large-scale literature knowledge data, gene expression data, and related pathways/network information for quantitative marker evaluation. Pathway Enrichment, Sub-Network Enrichment, and Gene-Gene Interaction analysis were conducted to study the pathogenic profile of the candidate genes, with four metrics proposed and validated for each gene. By using our approach, a scalable CLL genetic database was developed including CLL-related genes, pathways, diseases and information of supporting references. The CLL case/control classification supported the effectiveness of the four proposed metrics, which successfully identified nine well-studied CLL genes (i.e., TNF, BCL2, TP53, VEGFA, P2RX7, AKT1, SYK, IL4, and MDM2) and highlighted two newly reported CLL genes (i.e., PDGFRA and CSF1R). The computational biology approach and the CLL database developed in this study provide a valuable resource that may facilitate the understanding of the genetic profile of CLL.
Keywords: : chronic lymphocytic leukemia, genetic network analysis, literature data mining, pathway enrichment analysis, sub-network enrichment analysis
1. Introduction
Being the most common type of leukemia in adults, chronic lymphocytic leukemia (CLL) affects more men than women (Shanshal and Haddad, 2012). The onset age of CLL are mostly over 50, few occur in children (Montserrat et al., 1991; Mauro et al., 1999; Demir et al., 2014). Although the cause of CLL remains unclear, it has been hypothesized that a combination of both environmental and genetic factors play important roles in the development of CLL, and multiple genes of small effect contribute to the risk (Goldin et al., 2010; Puiggros et al., 2014).
In recent years, hundreds of genetic variations have been related to CLL. For instance, TNF, IL4, TP53, VEGFA, P2RX7, and IL10 are suggested in many studies as important markers for the pathogenic development of CLL (Mainou-Fowler et al., 2001). Some other genes (e.g., BCL2 [NCT02427451] and CD40 [NCT00224354]) have been studied in clinical trials. However, approximately half of these CLL-gene linkages were reported once with no further replication. Moreover, due to the variation in data collection and processing approaches, results from different studies were not always consistent. Meanwhile, there are dozens of new CLL risk genes being reported every year, posing an increased need for further validation of these candidate genes to CLL. While biological experiments were effective toward this validation task, they could be very costly. To address this issue, we propose a computational biology approach for a systematic evaluation of these CLL candidate genes.
In recent years, Pathway Studio ResNet relation data have been widely used to study modeled relationships between proteins, genes, complexes, cells, tissues, and diseases (http://pathwaystudio.gousinfo.com/Mendeley.html). In this study, we integrated large-scale CLL-related ResNet literature knowledge data, independent gene expression data, and related pathway/network information to study the functional profile of a large gene pool that has been reported being linked to CLL. The purpose of the study is to provide an easy-update computational evaluation workflow, through which a CLL genetic database (CLL_GD) could be generated to present a weighted landscape view of the genetic basis underlying the pathogenic development of CLL. Our results support the hypothesis that CLL candidate genes were functionally linked to each other, forming a large genetic network to regulate the pathogenic development of CLL through multiple pathways.
2. Methods
Figure 1 presents the diagram of the proposed computational gene marker evaluation system, with detailed description in the following subsections. Using our approach, a genetic database (CLL_GD) was developed and deposited into an open source “Bioinformatics Database” online available at http://database.gousinfo.com, including 753 genes (with metric scores), 125 pathways, and 159 diseases that are linked to CLL. Also included in CLL_GD are information of 3000+ supporting references for CLL-gene relationships, including the titles and relevant sentences where the relations were identified. The CLL_GD database is scalable and will be updated monthly or upon request using our approach.
FIG. 1. .

Diagram for the integrative computational marker evaluation approach for CLL. CLL, chronic lymphocytic leukemia.
2.1. ResNet literature knowledge data
ResNet relation data (CLL-Gene) were acquired from the Pathway Studio ResNet® Mammalian database updated November 2016. The ResNet Mammalian database are a group of real-time updated literature knowledge databases, including curated signaling, cellular processes and metabolic pathways, ontologies and annotations, and molecular interactions and functional relationships (http://pathwaystudio.gousinfo.com/ResNetDatabase.html). Modeled relation data are extracted from the 41M+ references covering entire PubMed abstracts and Elsevier and third party full text journals. The ResNet database employs an automated natural language processing-based information extraction system, MedScan, with precision of over 91% (Daraselia et al., 2004). Each relationship data within the database is supported with one or more references. By far, Pathway Studio ResNet Databases is the largest database among known competitors in the field (Lorenzi et al., 2014).
2.2. Enrichment and gene–gene interaction analysis
Pathway enrichment analysis (PEA) and sub-network enrichment analysis (SNEA) (http://pathwaystudio.gousinfo.com/SNEA.pdf) was conducted using Pathway Studio to identify genetic pathways and diseases potentially linked to CLL (Sivachenko et al., 2007). Furthermore, a pathway based gene–gene interaction (GGI) analysis was conducted to generate weighted edges/linkage between genes. The weight of an edge is the number of pathways where both nodes were included.
2.3. Metrics analysis
For the gene network built through the aforementioned steps, four metrics were proposed for each node/gene, including two literature-based metric scores (reference score [RScore] and age score [AScore]), and two enrichment-based metric scores (PScore and significance score [SScore]). The logic is that, a gene is likely linked to CLL if it satisfies one or more of the following conditions: the gene has been frequently observed in independent studies to be linked to CLL (high RScore), plays roles within multiple pathways associated with CLL (high PScore), and demonstrates strong functional linkage to many of other genes that were associated with CLL (high SSCore). Additionally, an AScore was proposed to present the history of each CLL-gene relation. The detailed definitions of the proposed metrics are described as follows.
2.3.1. Two literature metrics
The RScore of a gene is defined as the reference number underlying a gene-disease relationship, as shown in Equation (1).
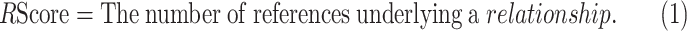 |
The AScore of a gene is defined as the earliest publication age of a gene-disease relationship, as shown in Equation (2).
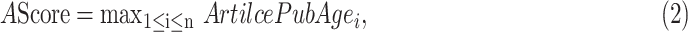 |
where n is the total number of references supporting a gene-disease relation, and
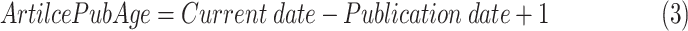 |
2.3.2. Two enrichment metrics
We define a network significance score (SScore) of a node as Freeman's formalized node degree centrality (Freeman, 2012), as defined in Equation (4).
 |
where n is the total number of nodes, and j represents all other nodes within the network; x is the adjacency matrix, in which the cell xj is 1 if the jth node is connected with the current node, other with is 0.
Given a disease is associated with a set of genetic pathways  , we define the PScore for the gene as Equation (5).
, we define the PScore for the gene as Equation (5).
 |
2.4. Validation using independent gene expression data
We hypothesized that significant CLL-related genes should contribute to distinguishing CLL patients from healthy controls. To validate the effectiveness of the selected genes and the proposed metrics, we performed a Euclidean distance-based multivariate classification (Wang et al., 2015) on two independent gene expression data sets (NCBI GEO: GSE50006 and GSE8835), followed by a leave-one-out (LOO) cross validation, using the overall gene set and the subsets selected by different scores as tentative markers. In each run of LOO, gene expression data of one subject is used for testing and the rest for training. A permutation of 5000 runs was then conducted to test the hypothesis that a randomly selected gene set in same size can reach equal or higher classification accuracy (classification ratio [CR]).
The expression profile of GSE50006 was acquired from 279 subjects, including samples from 247 CLL tumors and 32 sorted CD19pos B cells from healthy donors (NCBI GEO: GSE50006), with 691 genes overlapped with the 753 CLL gene-pool identified by ResNet database. For data set GSE8835, expression profile were acquired from CD4 T cells and CD8 T cells of peripheral blood mononuclear cells, including 42 blood samples from 42 CLL patients and 24 healthy individuals. The GSE8835 data set contains 625 genes overlapped with the 735 AD gene-pool tested in this study.
3. Results
3.1. Target CLL genes for evaluation
CLL-Gene literature knowledge data analysis identified 753 CLL candidate genes, supported by 3156 scientific articles (see CLL_GD→Related Genes and CLL_GD→Ref for Disease-Gene Relation). Of the 753 genes, 408 (54.18%) have been reported with one reference (RScore = 1), 113 (15.01%) with 2, 64 (8.50%) with 3, and 103 (13.68%) with more than 5, as shown in Supplementary Figure S1a. Publication date statistics of the 3078 supporting references are presented in Supplementary Figure S1b, with novel genes reported in each year (Supplementary Fig. S1c).
3.2. Enrichment analysis results
PEA showed that, 605 out of 753 genes got significantly enriched within 125 CLL candidate pathways/gene sets (p-values <1e-10, q = 0.001 for false discovery rate (FDR); CLL_GD→Related Pathways). Many of these pathways were previously implicated with CLL, including, 9 pathways/groups related to cell apoptosis (232 unique genes) (Campàs et al., 2006; Billard, 2014), 11 to cell growth and proliferation (261 unique genes) (Messmer et al., 2005; Herishanu et al., 2011), 9 to protein kinase (155 unique genes) (Davids and Brown, 2013), 6 to protein phosphorylation (127 unique genes) (Paterson et al., 2012; Hojjat-Farsangi et al., 2013), 8 to transcription factors (205 unique genes) (Pepper et al., 2009), 5 to drug effects (158 unique genes) (Rosen et al., 1983), and 4 pathways/groups related to immune system (218 unique genes) (Friedrichs et al., 2008). Due to lack of space, we only present the top 10 pathways enriched in Table 1 (p-value ≤2.5e-34, including 440 out of 753 genes).
Table 1.
Top 10 Molecular Function Pathways/Groups Enriched by 440 Genes Reported
| Pathway/gene set name | GO ID | No. of entities | Overlap | p-Value | p-Value (before FDR) |
|---|---|---|---|---|---|
| External side of plasma membrane | 0009897 | 288 | 99 | 5.32E-73 | 5.45E-77 |
| Cell surface | 0009929 | 645 | 120 | 3.35E-56 | 4.58E-60 |
| Positive regulation of cell proliferation | 0008284 | 568 | 107 | 6.16E-49 | 1.05E-52 |
| Response to drug | 0017035 | 509 | 98 | 2.17E-45 | 4.45E-49 |
| Immune response | 0006955 | 468 | 94 | 4.21E-45 | 1.01E-48 |
| Inflammatory response | 0006954 | 404 | 87 | 4.30E-44 | 1.18E-47 |
| Apoptotic process | 0008632 | 790 | 118 | 1.12E-43 | 3.46E-47 |
| Negative regulation of apoptotic process | 0006916 | 650 | 100 | 1.51E-37 | 5.67E-41 |
| Innate immune response | 0002226 | 792 | 108 | 6.75E-36 | 2.77E-39 |
| Response to lipopolysaccharide | 0032496 | 252 | 62 | 2.46E-34 | 1.18E-37 |
For each gene set, the p-value was calculated using Fisher-Exact test against the hypothesis that a randomly selected gene group of same size (753) can generate a same or higher overlap with the corresponding gene set (p = 0.001 for FDR correction).
FDR, false discovery rate.
A SNEA was also performed to identify the pathogenic significance of the reported genes to other disorders that are potentially related to CLL. There are 159 significantly enriched disease seeded sub-networks (p-value <4.5e-80; q = 0.001 for FDR; 717 out of 753 genes are included; see CLL_GD→Related Diseases). Not surprisingly, besides other leukemia subtypes (e.g., acute myeloid leukemia, acute lymphoblastic leukemia, etc.) and cancers (e.g., multiple myeloma, melanoma, hepatocellular carcinoma, nonsmall cell lung cancer, breast cancer), genes linked to CLL tend to play roles within many immune-related diseases (e.g., rheumatoid arthritis, autoimmune disease).
3.3. GGI results
Figure 2 presents the genetic network for CLL, which was built through GGI analysis. The nodes of the network are 605 out of 753 genes that were enriched within the 125 CLL target pathways. There were 72,538 edges within the network, the weight of which are the numbers of pathways shared by the corresponding pair of nodes. The average node strength (sum of the number of genes directly connected) of the network was 120, and the node strength for the 148 unconnected genes was signed with 0. More statistics of the network (Fig. 2) are provided in CLL_GD→ Network Statistics.
FIG. 2. .

Gene–gene interaction network for CLL. The network contains 605 out of 753 CLL target genes that enriched within the 125 CLL target pathways. The weight of an edge between two nodes is the number of pathways shared by both nodes. The larger the size of a node, the larger the number of CLL candidate pathways including the gene (high PScore); the brighter the color, the larger number of CLL candidate genes associated with gene (high SScore). One hundred forty-eight out of 753 genes were not included in the network as they were not enriched within the top 125 CLL candidate pathways.
Along with GGI, SScore, and PScore were calculated for each gene (CLL_GD→ Related Genes). The value of a PScore represents how many CLL candidate pathways involve the gene, and an SScore represents how many other genes within the network (Fig. 2) were functionally linked to the gene.
3.4. Validation results
Classification and LOO cross validation were conducted on two independent public RNA expression dataset (NCBI GEO: GSE50006 and GSE8835), followed by a permutation test of 5000 runs. For the LOO cross validation, the 753 genes were first ranked according to different metric scores in descending order, then the top n (n = 1, 2 …) genes were used as input variables for classification and LOO cross validation. Table 2 and Figure 3 presents the results, with the maximum CRs marked at the position of corresponding number of genes.
Table 2.
Permutation Test on top Genes Corresponding to Highest Classification Ratios
| Data sets | Items | RScore | AScore | PScore | SScore | All genes |
|---|---|---|---|---|---|---|
| GSE50006 (case/control:247/32) | MaxCRs | 98.92 | 98.92 | 98.57 | 98.57 | 98.57 |
| #Genes | 186 | 218 | 619 | 619 | 691 | |
| p-Value | 0.0038 | 0.0048 | 0.0676 | 0.0724 | 0.0738 | |
| GSE8835 (case/control:42/24) | MaxCRs | 84.85 | 86.37 | 80.30 | 80.30 | 80.30 |
| #Genes | 15 | 77 | 36 | 126 | 625 | |
| p-Value | 0.002 | 0.0006 | 0.0018 | 0.005 | 0.001 |
AScore, age score; CRs, classification ratios; PScore; RScore, reference score; SScore, significance score.
FIG. 3. .

Validation of different metrics through a leave-one-out cross validation. (a) Results from GSE50006. (b) Results from GSE8835. Mean of CRs by randomly selected genes are displayed in a dash-gray line (legend: Random). The maximum CRs by different metrics are presented at the corresponding positions. CRs, classification ratios.
From Figure 3 we see that the top genes selected by different scores (in descending order) can lead to the highest classification accuracies, which are significantly higher than the average CRs of randomly selected gene set in same size (see the “Random” line in Fig. 3), while adding more genes with lower score may not necessarily lead to improved CRs.
3.5. Cross metrics analysis
Results from CLL case/control classification (Table 2 and Fig. 3) demonstrate the effectiveness of the proposed metrics. Therefore, it is worthy to study the overlaps among these top genes by different scores. Cross metrics analysis of the top 8% (63 genes, corresponding to the number of genes reported this years, 2016) of 753 genes selected using different scores showed that (see Veen diagram at Supplementary Fig. S2), there was a strong overlap between PScore group and SScore group (48/63). Among these 48 genes, 9 were also identified to be within RScore group, including TNF, BCL2, TP53, VEGFA, P2RX7, AKT1, SYK, IL4, and MDM2, with RScore = 26 ± 17 references, PScore = 29 ± 8 pathways, SScore = 458 ± 35 connected genes. Network analysis using Pathway Studio showed that, these 10 genes also demonstrated strong correlation with the top disorders that are linked to CLL (Fig. 4, highlighted in red). The genes related to these diseases present significant overlap with these genes linked to CLL (see CLL_GD→ Related Diseases). On the other hand, two genes (i.e., PDGFRA and CSF1R) were identified to be the overlap of AScore, PScore, and SScore groups (Fig. 4, highlighted in yellow), which also linked to several other diseases (e.g., acute myeloid leukemia, hepatocellular carcinoma, and rheumatoid arthritis) that are genetically linked to CLL.
FIG. 4. .

CLL genes selected by cross metrics analysis and their relation with other diseases. The 9-gene overlap of RScore, PScore, and SScore are highlighted in green; The 2-gene overlap of AScore, PScore, and SScore groups are highlighted in yellow. The network was built using the “network building” module of Pathway Studio. AScore, age score; PScore, pathway involvement score; RScore, reference score; SScore, significance score.
4. Discussion
In this study, we integrated large-scale literature knowledge data, gene expression data and related pathways, and disease-seeded networks to evaluate 753 CLL target genes. Four metric scores have been proposed and validated. A scalable genetic database, CLL_GD, was developed, which is available at “Bioinformatics Database” (http://database.gousinfo.com).
PEA results showed that most genes within the network (605 out of 753) were significantly enriched (FDR corrected p-value <1e-10) in the pathways previously implicated with CLL, including pathways/groups related to drug effects, cell apoptosis, cell growth and proliferation, protein phosphorylation, protein kinase, and transcription factors (Rosen et al., 1983; Messmer et al., 2005; Campàs et al., 2006; Friedrichs et al., 2008; Pepper et al., 2009; Herishanu et al., 2011; Paterson et al., 2012; Davids and Brown, 2013; Hojjat-Farsangi et al., 2013; Billard, 2014). In addition, SNEA results demonstrated that over 95% (717) of the 753 CLL-genes were as well identified as causal genes for other disorders that were linked to CLL (CLL_GD→Related Diseases). These results support the hypothesis that CLL genes were functionally linked to each other to play roles within multiple pathways influencing CLL.
Four metrics were proposed to measure the significance of each CLL target gene from different aspects: (1) publication frequency (RScore), (2) novelties (AScore), (3) Number of associated CLL candidate pathways (PScore), and (4) Network centrality (SScore). The effectiveness of our four proposed metrics were supported by the CLL case/control classification study using two independent gene expression data sets (GSE50006 and GSE8835). Results of the LOO cross validation and permutation process showed that, the top genes by the each metric score can lead to significantly higher CR than using randomly selected gene sets (Table 2 and Fig. 3). While using target gene set as a whole (691 and 625 out of 753 for GSE50006 and GSE8835, respectively) do not necessarily gain better classification accuracy. These results suggest the advantages of using our network metrics for further analysis of the candidate CLL genes. Notably, for each score, the number of top genes corresponded to the maximum CRs for the data sets were different, which may reflect the unique variation of different subjects' genome in case of CLL (Lu et al., 2014).
Cross metrics analysis showed that nine genes were overlapped within RScore, SScore, and PScore groups (Fig. 4, highlighted in green). These genes were frequently identified by different studies to be linked to CLL (RScore = 26 ± 17 references), play roles with in multiple CLL candidate pathways (PScore = 29 ± 8 pathways), and demonstrate strong network centrality (SScore = 458 ± 35 direct gene connections). Therefore, our results suggest that they are among the top CLL risk genes that likely pose biological significance with the disease. As a matter of fact, these genes were also identified to play roles within many other disorders that were linked to CLL (Fig. 4), demonstrating the effectiveness of the proposed metric scores in the identification of top genes for CLL.
Additionally, two newly reported genes (AScore  ), PDGFRA and CSF1R (Fig. 4, highlighted in yellow) were also highlighted from cross metrics analysis. These two genes have not been frequently replicated in their association with CLL (RScore = 1 reference, see CLL_GD→ Related Genes), and present less relationships with other CLL-related disorders. However, they demonstrate high SScore (439 and 431 connected genes, respectively) and PScore (31 and 29 pathways), suggesting them worthy of further study. As a matter of fact, PDGFRA rearrangement has been identified to play roles in other CLL-related diseases, including hyper-eosinophilia, T-lymphoblastic lymphoma, myeloproliferative neoplasm, and precursor B-cell acute lymphoblastic leukemia (Huang et al., 2011). In addition, studies showed that αPDGFR, a subtype of platelet-derived growth factor (PDGF) receptors encoded by PDGFRA, is linked to the increased levels of intracellular reactive oxygen species (ROS) (Bäumer et al., 2008), which contributes to the death of CLL cells (Ma et al., 2010). This may partially explain the rearrangement of αPDGFR in case of CLL. On the other hand, soluble CSF1R has been shown to inhibit CSF-1-mediated ROS production (Rieger et al., 2015) and activates nitric oxide synthesis (Kettenmann et al., 2011). Different from ROS, nitric oxide is an antiapoptotic molecule for CLL cells (Quiney et al., 2004). These observations support the conclusion from a recent study that inhibition of CSF1R could be a therapeutic direction toward CLL treatment (Galletti et al., 2016).
), PDGFRA and CSF1R (Fig. 4, highlighted in yellow) were also highlighted from cross metrics analysis. These two genes have not been frequently replicated in their association with CLL (RScore = 1 reference, see CLL_GD→ Related Genes), and present less relationships with other CLL-related disorders. However, they demonstrate high SScore (439 and 431 connected genes, respectively) and PScore (31 and 29 pathways), suggesting them worthy of further study. As a matter of fact, PDGFRA rearrangement has been identified to play roles in other CLL-related diseases, including hyper-eosinophilia, T-lymphoblastic lymphoma, myeloproliferative neoplasm, and precursor B-cell acute lymphoblastic leukemia (Huang et al., 2011). In addition, studies showed that αPDGFR, a subtype of platelet-derived growth factor (PDGF) receptors encoded by PDGFRA, is linked to the increased levels of intracellular reactive oxygen species (ROS) (Bäumer et al., 2008), which contributes to the death of CLL cells (Ma et al., 2010). This may partially explain the rearrangement of αPDGFR in case of CLL. On the other hand, soluble CSF1R has been shown to inhibit CSF-1-mediated ROS production (Rieger et al., 2015) and activates nitric oxide synthesis (Kettenmann et al., 2011). Different from ROS, nitric oxide is an antiapoptotic molecule for CLL cells (Quiney et al., 2004). These observations support the conclusion from a recent study that inhibition of CSF1R could be a therapeutic direction toward CLL treatment (Galletti et al., 2016).
To note, although in this study we focused on the evaluation of 753 known CLL candidate genes acquired from ResNet database, which already received literature support for their association with CLL (CLL_GD→Ref for Disease-Gene Relation), the proposed PScore and SScore can be applied to any given genes and therefore could be used for evaluation and discovery of novel target genes for CLL. Moreover, the genetic database built through our approach, namely CLL_GD, is scalable and can be automatically updated using the computational workflow proposed in this study. Any novel CLL-gene relationships can be added to update the database. Moreover, further network analysis with more experiment data may extract additional meaningful features that can be added into our proposed system to gain improved evaluation of existing and/or novel CLL genes.
To our knowledge, this is the first study integrating large-scale literature knowledge data, experiment data, and related pathway/network data for a systematical evaluation of CLL candidate genes. The computational biology approach of this study provides a comprehensive weighted genetic network and genetic database for CLL, which may help in the evaluation and prioritization of CLL candidate genes for further study in the field.
Acknowledgments
This study is partly supported by Shanghai Three-Year Plan of the Key Subjects Construction in Public Health-Infectious Diseases and Pathogenic Microorganism (15GWZK0102).
Author Disclosure Statement
The author H.C. is with Elsevier Inc., the company that owns the software Pathway Studio used in this study.
Supplementary Material
References
- Bäumer A.T., Ten Freyhaus H., Sauer H., et al. 2008. Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for alpha-platelet-derived growth factor receptor-induced production of reactive oxygen species. J. Biol. Chem. 283, 7864–7876. [DOI] [PubMed] [Google Scholar]
- Billard C. 2014. Apoptosis inducers in chronic lymphocytic leukemia. Oncotarget 5, 309–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campàs C., Cosialls A.M., Barragán M., et al. 2006. Bcl-2 inhibitors induce apoptosis in chronic lymphocytic leukemia cells. Exp. Hematol. 34, 1663–1669. [DOI] [PubMed] [Google Scholar]
- Daraselia N., Yuryev A., Egorov S., et al. 2004. Extracting human protein interactions from MEDLINE using a full-sentence parser. Bioinformatics 20, 604–611. [DOI] [PubMed] [Google Scholar]
- Davids M.S., and Brown J.R. 2013. Phosphoinositide 3′-kinase inhibition in chronic lymphocytic leukemia. Hematol. Oncol. Clin. North. Am. 27, 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir H.A., Bayhan T., Üner A., et al. 2014. Chronic lymphocytic leukemia in a child: A challenging diagnosis in pediatric oncology practice. Pediatr. Blood Cancer 61, 933–935. [DOI] [PubMed] [Google Scholar]
- Freeman L.C. 2012. Centrality in social networks conceptual clarification. Soc. Networks 1, 215–239. [Google Scholar]
- Friedrichs B., Siegel S., Kloess M., et al. 2008. Humoral immune responses against the immature laminin receptor protein show prognostic significance in patients with chronic lymphocytic leukemia. J. Immunol. 180, 6374–6384. [DOI] [PubMed] [Google Scholar]
- Galletti G., Caligaris-Cappio F., and Bertilaccio M.T. 2016. B cells and macrophages pursue a common path toward the development and progression of chronic lymphocytic leukemia. Leukemia 30, 2293–2301. [DOI] [PubMed] [Google Scholar]
- Goldin L.R., Slager S.L., and Caporaso N.E. 2010. Familial chronic lymphocytic leukemia. Curr. Opin. Hematol. 17, 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herishanu Y., Pérez-Galán P., Liu D., et al. 2011. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 117, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojjat-Farsangi M., Khan A.S., Daneshmanesh A.H., et al. 2013. The tyrosine kinase receptor ROR1 is constitutively phosphorylated in chronic lymphocytic leukemia (CLL) cells. PLoS One 8, e78339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Snyder D.S., Chu P., et al. 2011. PDGFRA rearrangement leading to hyper-eosinophilia, T-lymphoblastic lymphoma, myeloproliferative neoplasm and precursor B-cell acute lymphoblastic leukemia. Leukemia 25, 371–375. [DOI] [PubMed] [Google Scholar]
- Kettenmann H., Hanisch U.K., Noda M., et al. 2011. Physiology of microglia. Physiol. Rev. 91, 461–553. [DOI] [PubMed] [Google Scholar]
- Lorenzi P.L., Claerhout S., Mills G.B., et al. 2014. A curated census of autophagy-modulating proteins and small molecules: Candidate targets for cancer therapy. Autophagy 10, 1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.F., Goldstein D.B., Angrist M., et al. 2014. Personalized medicine and human genetic diversity. Cold Spring Harb. Perspect. Med. 4, a008581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Wang Y., Lo A.S., et al. 2010. Cell density plays a critical role in ex vivo expansion of T cells for adoptive immunotherapy. J. Biomed. Biotechnol. 2010, 386545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainou-Fowler T., Proctor S.J., Miller S., et al. 2001. Expression and production of interleukin 4 in B-cell chronic lymphocytic leukaemia. Leuk. Lymphoma 42, 689–698. [DOI] [PubMed] [Google Scholar]
- Mauro F.R., Foa R., Giannarelli D., et al. 1999. Clinical characteristics and outcome of young chronic lymphocytic leukemia patients: A single institution study of 204 cases. Blood 94, 448–454. [PubMed] [Google Scholar]
- Messmer B.T., Messmer D., Allen S.L., et al. 2005. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Invest. 115, 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montserrat E., Gomis F., Vallespi T., et al. 1991. Presenting features and prognosis of chronic lymphocytic leukemia in younger adults. Blood 78, 1545. [PubMed] [Google Scholar]
- Paterson A., Mockridge C.I., Adams J.E., et al. 2012. Mechanisms and clinical significance of BIM phosphorylation in chronic lymphocytic leukemia. Blood 119, 1726–1736. [DOI] [PubMed] [Google Scholar]
- Pepper C., Hewamana S., Brennan P., et al. 2009. NF-kappaB as a prognostic marker and therapeutic target in chronic lymphocytic leukemia. Future Oncol. 5, 1027–1037. [DOI] [PubMed] [Google Scholar]
- Puiggros A., Blanco G., and Espinet B. 2014. Genetic abnormalities in chronic lymphocytic leukemia: Where we are and where we go. Biomed Res Int. 2014, 435983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiney C., Dauzonne D., Kern C., et al. 2004. Flavones and polyphenols inhibit the NO pathway during apoptosis of leukemia B-cells. Leuk Res. 28, 851–861. [DOI] [PubMed] [Google Scholar]
- Rieger A.M., Havixbeck J.J., Belosevic M., et al. 2015. Teleost soluble CSF-1R modulates cytokine profiles at an inflammatory site, and inhibits neutrophil chemotaxis, phagocytosis, and bacterial killing. Dev. Comp. Immunol. 49, 259–266. [DOI] [PubMed] [Google Scholar]
- Rosen S.T., Maciorowski Z., Wittlin F., et al. 1983. Estrogen receptor analysis in chronic lymphocytic leukemia. Blood 62, 996–999. [PubMed] [Google Scholar]
- Shanshal M., and Haddad R.Y. 2012. Chronic lymphocytic leukemia. Dis. Mon. 58, 153–167. [DOI] [PubMed] [Google Scholar]
- Sivachenko A.Y., Yuryev A., Daraselia N., et al. 2007. Molecular networks in microarray analysis. J. Bioinform. Comput. Biol. 5, 429–456. [DOI] [PubMed] [Google Scholar]
- Wang J., Cao H., Liao Y., et al. 2015. Three dysconnectivity patterns in treatment-resistant schizophrenia patients and their unaffected siblings. Neuroimage Clin. 8, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.