

Summary
Phenotypic screening of existing drugs is a good strategy to discover new drugs. Herein, 33 psychotherapeutic drugs in our drug library were screened by phenotypic screening and penfluridol (PFD) was found to exhibit excellent anti-endometrial cancer (EC) activity both in vitro and in vivo. Furthermore, the molecular target of PFD was identified as septin7, a tumor suppressor in EC. In septin7-deficient EC cells and xenograft mouse models, PFD exhibited weaker anti-cancer properties, indicating that septin7 was essential for the tumor inhibitory activity. Notably, PFD could induce cell apoptosis by regulating the septin7-Orai/IP3R-Ca2+-PIK3CA pathway. In addition, PFD attenuates the interaction of septin7-tubulin, thereby inhibiting microtubule polymerization. In summary, this study revealed a target and mechanistic insights into EC therapeutic strategies and identified a potential candidate agent for the treatment of EC.
Subject areas: Cell biology, Functional aspects of cell biology, Cancer
Graphical abstract
Highlights
-
•
Penfluridol exhibits excellent anti-EC activities both in vitro and in vivo
-
•
Chemical proteomics identifies septin7 as a potential target of penfluridol
-
•
Penfluridol exhibits anti-EC activities by septin7-Orai/IP3R-Ca2+-PIK3CA axis
-
•
Penfluridol attenuates septin7-tubulin interaction
Cell biology; Functional aspects of cell biology; Cancer
Introduction
Endometrial cancer (EC), often referred to as uterine corpus cancer, is the most commonly diagnosed gynecological cancer in developed countries.1,2 The American Cancer Society predicted diagnoses of ∼66,500 new EC cases and 12,900 EC-related deaths in the United States in 2023.3 The lifetime risk of EC is ∼3%, with a median age at diagnosis of 61 years.4 Currently, the mainstay of treatment for EC is surgery, supplemented chemotherapy, and radiotherapy. The standard first-line drug for therapy is a combination of carboplatin and paclitaxel, with a progression-free survival of 13 months.5 However, the side effects of chemotherapy are typically severe, the tolerability of which is of concern as two-thirds of the patients demonstrated a grade 3–4 treatment-related toxicity.6 Hormone therapy had fewer side effects than chemotherapy, which is suitable for young patients who need to preserve reproductive function in the early stage and with advanced, recurrent, or inoperable EC.7 But it faces the challenge of inducing drug resistance and is ineffective for the treatment of type II EC. Due to the hectic pace of life observed in recent years, the age of onset of EC has lowered, about 5%–14% of EC patients younger than 40 years old,8 which coupled with the increasingly common phenomenon of marriage and conceiving at a late age, has led to an increase in the demand for conservative treatment of EC. Additionally, patients with advanced and recurrent EC account for 10%–20% of the total EC cases,9 among which the 5-year survival rate of patients with advanced EC is only 30%–40% and that of patients with recurrent EC is 42%–65%.10 Moreover, the response of these patients to continuous chemotherapy was only 20%, and the side effects seriously affected their quality of life. Therefore, for young patients with a requirement for the preservation of reproductive function, as well as those with advanced, recurrent, or inoperable EC, there is an urgent requirement for the development of conservative treatment drugs with a good response and fewer side effects to improve the survival rate and quality of life of such patients.
Repurposing existing drugs is a good strategy to discover new candidate drugs given their shorter duration of development, low-cost, high efficiency, and reduced risk of failure.11 There are usually two approaches to repurposing existing drugs, target screening and phenotypic screening, and the latter is superior to discover candidate drugs for complex diseases with multi pathogenic pathways participation, such as tumor and neurodegenerative diseases, because single target drugs based on target screening are easily compensated by other pathways in complex diseases. In a previous study, through the phenotypic screening of 20 clinically approved tricyclic antipsychotic drugs available in our drug library, chlorpromazine (CPZ) and perphenazine (PPZ) were identified to demonstrate a good anti-EC activity by inhibiting the phosphorylation of PI3K/AKT.12,13 Since both of them belong to phenothiazine structural type, to further expand the diversity in the structural types of anti-EC agents, 33 non-tricyclic psychotherapeutic drugs including antipsychotics and antidepressants available in our drug library were screened. Of these, penfluridol (PFD) was found to exhibit excellent anti-EC activity where the IC50 values for ISK and KLE cell proliferation were 3.48 μM and 3.14 μM, respectively.
PFD is a long-acting antipsychotic drug targeting D2 dopamine receptor (DRD2) and calcium channel approved by the FDA in the 1960s, which belongs to the diphenylbutylpiperidine antipsychotics. Recently, PFD has been shown to exhibit good anti-cancer activities in melanoma, breast cancer, colon cancer, esophageal cancer, etc.14,15,16,17 For example, PFD suppressed esophageal cancer by targeting PFKL to activate AMPK/FOXO3a/BIM signaling pathway.16 PFD could inhibit the migration and invasion of breast cancer cells by down-regulating the expression of integrin β4, α6, α5, and, β1 in a dose-dependent manner.17 Although PFD inhibited the growth of a variety of tumors,12,13,14,15,16,17 the mechanism underlying its activity and the identification of its target in EC have not yet been investigated.
Activity-based protein profiling (ABPP) is a chemical proteomic technique that applies activity-based probes derived from bioactive compounds to identify the potential target proteins in biological systems. It has been widely applied in drug discovery, especially to identify targets of small therapeutic molecules based on phenotypic screening, and has proved to be an efficient method to identify the underlying cellular targets that are responsible for the manifestation of the phenotypic effects.18 By using ABPP coupled with mass spectrometry-based proteomics, septin7 was discovered as a potential direct target for the inhibition of proliferation and induction of apoptosis in EC cells by PFD. Septin7 is an important member of the septin family of proteins in turn belonging to the GTPase superfamily; is recognized as the fourth component of the cytoskeleton.19 Septins are involved in various cellular processes, including exocytosis, leukemogenesis, carcinogenesis, and neurodegeneration,20 and demonstrated roles in a variety of biological activities and physiological functions, such as the development of cancer, cell proliferation, and cytokinesis. Present studies on the role of septin7 in cancer have been rare, with only a few reports describing its functions in glioma,21,22,23,24 thyroid papillary carcinoma,25 hepatocellular carcinoma (HCC),26 and breast cancer.27 Septin7 functions as a tumor suppressor in glioma and thyroid papillary carcinoma. The expression of septin7 in brain tumors was much lower than that in normal tissues, and its overexpression inhibits the growth of glioma cells.21,22,23,24 However, septin7 acts as an oncogene in HCC and breast cancer. MiR-127 suppressed the expression of septin7 and inhibited the growth of HCC cells.26 Compared to normal cells, the levels of septin2 and 7 were significantly enhanced in breast cancer cell lines; the down-regulation of their expression could inhibit the proliferation, migration, and invasion of breast cancer cells.27 These antithetical effects of septin7 in different types of cancer may be related to its subcellular localization and post-translational modification.20
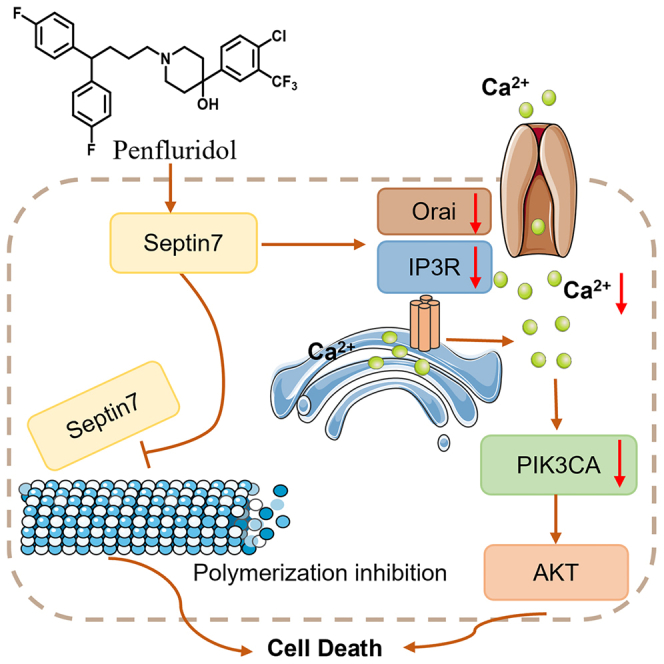
In this study, 33 psychotherapeutic drugs available in our drug library were screened and PFD exhibited excellent anti-EC activity in vitro and in vivo. Based on ABPP coupled with proteomics, the molecular target of PFD was revealed as septin7, which plays different roles in different tumors. However, the physiologic function of septin7 in EC is unclear and there is no small molecule ligands reported for septin7. Further, the role of septin7 in EC was explored either by its knockdown or overexpression in both cells and xenograft mice, and a target for PFD was identified through a series of functional assays. Moreover, RNA-seq analysis and western blotting revealed that PFD inhibited the growth of EC cells by regulating septin7-Orai/IP3R-Ca2+-PIK3CA pathway.
Results
Discovery of antipsychotic drug PFD exhibiting excellent anti-EC activities in vitro and in vivo
To discover the anti-EC agents with new structural types, the phenotypic screening of 33 non-tricyclic psychotherapeutic drugs available in the drug library constituted by our group was conducted. Through the use of a cell counting kit-8 (CCK-8) assay, the inhibitory rates were obtained to evaluate the effects of small molecule compounds at 40 μM in ISK cells (Figure 1A; Table S1). Among them, PFD demonstrated the best inhibition rate of 100.3% at 40 μM. To verify the anti-EC property of PFD, the IC50 values of PFD in ISK and KLE cells at 24 h, 48 h, and 72 h were measured. The results obtained demonstrated that PFD impaired EC cells (ISK and KLE cells) proliferation in a dose- and time-dependent manner with IC50 values of 5.34 μM and 5.38 μM at 24 h, 3.48 μM and 3.14 μM at 48 h, 2.77 μM and 2.88 μM at 72 h, respectively (Figures 1B; Table S2). In addition, PFD significantly impaired the migration and colony-forming abilities of EC cells (Figures 1C and 1D). Annexin V-FITC/PI staining of PFD-treated EC cells demonstrated that PFD could enhance their apoptosis in a dose-dependent manner (Figure 1E). Thus, the tumor inhibitory activity of PFD could be attributed to the induction of apoptosis.
Figure 1.

PFD significantly inhibits the proliferation and growth of EC cells in vitro and in vivo
(A) Proliferation inhibition of 33 non-tricyclic psychotherapeutic drugs at 40 μM in ISK cells.
(B) Structure of Penfluridol (PFD) and its IC50 values at 48 h in ISK and KLE cells.
(C) The migration ability of EC cells was impaired by PFD. Data are presented as mean ± SD (n = 3) and comparisons were made using one-way ANOVA. ∗∗∗p < 0.001.
(D) The colony-forming ability of EC cells was impaired by PFD. Data are presented as mean ± SD (n = 3) and comparisons were made using one-way ANOVA. ∗∗p < 0.01, ∗∗∗p < 0.001.
(E) PFD could enhance EC cells apoptosis. Data are presented as mean ± SD (n = 3) and comparisons were made using one-way ANOVA. ∗∗p < 0.01, ∗∗∗p < 0.001.
(F) Body weight of mice from each group during the whole observation period. Data are presented as mean ± SEM (n = 5) and comparisons were made using one-way ANOVA. ∗∗p < 0.01.
(G) The images of tumors from mice at 14 days after initiation of treatment.
(H and I) Tumor weight and tumor volume of mice from each group. Data are presented as mean ± SEM (n = 5) and comparisons were made using one-way ANOVA. ∗∗∗p < 0.001.
Next, nude mouse xenograft models were established to determine the effects of PFD in vivo. KLE cells were subcutaneously injected into the right armpits of nude mice to obtain the tumor xenograft model mice, the tumor tissue of which was in turn subcutaneously implanted into the right armpits of new nude mice to acquire a sufficient number of xenograft model mice. The model mice were then treated with PFD (2 or 5 mg/kg), cisplatin (DDP, 2 mg/kg), and vehicle (normal saline solution) through intraperitoneal injection for 14 consecutive days and the body weight of each mouse in each group was monitored daily. All the PFD-treated groups revealed a significant attenuation in tumor volume and weight after 14 days when compared with the vehicle group, but the body weight did not change significantly (Figures 1F–1I). PFD exhibited a satisfactorily good activity of tumor growth inhibition at 2 mg/kg with tumor volume growth inhibition (TGIV) of 78.2% and tumor weight growth inhibition (TGIW) of 70.5%. The efficacy was comparable to that of 2 mg/kg cisplatin, the first-line drug used for the systemic treatment of EC, but cisplatin demonstrated obvious side effects of loss in body weight. Moreover, PFD is a butylbenzene-based antipsychotic with long-acting effects and is absorbed through the gastrointestinal tract after oral administration with peak blood concentrations reached within 24–72 h. In mice, its oral LD50 is 87 mg/kg,28 with the primary toxic effects being myocardial damage, interference with intracardiac conduction, severe arrhythmia, and chest discomfort. In the mouse efficacy study, 2 mg/kg and 5 mg/kg PFD significantly inhibited the growth of EC tumors, indicating a good safety profile. Further, safety was evaluated by HE staining of the main organs of the mice and blood-based biochemical tests. The tissue sections of the main organs such as the heart, liver, spleen, and kidneys of mice treated with 2 or 5 mg/kg PFD showed no conspicuous pathological changes (Figure S1). In addition, there were no differences in the levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (CREA), and urea between the treated and the vehicle mice, indicating that PFD did not impair the proper functioning of the liver and kidneys (Table S6). In summary, all of the results obtained in this study revealed that the antipsychotic drug PFD was a potential anti-EC agent with good safety and efficacy.
ABPP identified septin7 as a potential target of PFD in EC cells
To explore the molecular target of PFD affecting the EC progression, ABPP experiment was performed in EC cell lysate29 (Figure 2A). First, a PFD activity-based probe BP containing a diazirine photo-cross-linking group and a ‘clickable’ handle consisting of an alkynyl group was designed and synthesized (Supporting Information). Then the anti-proliferative activity of BP was tested in ISK cells, and the results indicated that BP maintained the activity of PFD (BP: IC50 = 7.12 ± 0.58 μM vs. PFD: IC50 = 3.47 ± 0.02 μM, Figure 2B). The overall scheme of the ABPP experiment was shown in Figure 2A. Briefly, cell lysates were incubated with different concentrations of BP and the competition group consisting of 10×PFD and BP, then reacted with TAMRA-N3 (Tetramethylrhodamine-N3) under a CuAAC-mediated click reaction and separated by SDS-PAGE. Subsequent in-gel fluorescence scanning demonstrated that the BP probe could label different cellular targets and the labeled proteins at 35–60 kDa were dose-dependent (Figure 2C), suggesting that they might be probe-targeted proteins instead of those with abundant non-specific labeling. To enrich the potential cellular targets of PFD, pull-down experiment was conducted. Samples were conjugated with biotin-N3 instead of TAMRA-N3, then enriched by streptavidin miniprep columns and separated using SDS-PAGE. Silver staining revealed that proteins with an MW of ∼50 kDa were enriched in the BP-treated group, but decreased in the PFD competitive group (Figure 2D). LC-MS/MS analysis revealed a lot of possible binding proteins (Table S4), from which potential targets were filtered by comparing the differences in the proteins identified in probe samples but reduced in the competition group and absent in the control (DMSO group). Among the top ten proteins (Table S5), septin7, as a fourth class of cytoskeletal protein, plays an important role in many cellular physiological functions, such as cytokinesis, cell polarity formation, intracellular material transport, apoptosis, cell migration, and DNA repair18 and is closely related to the progression of various malignant tumors such as glioma,21 liver cancer26 and breast cancer.27 Moreover, researches indicated septin7-Akt-NF-kB pathway influenced the glioma progress30,31 and septin7 acted as an upstream regulator of Akt, which is closely associated with our previously validated PIK3CA-AKT pathway. In addition, the expression of SEPT7 was significantly reduced in EC according to the TCGA database (Figure 2E). These findings suggest that septin7 could be a possible target for molecules that influence EC progression. Thus, septin7 was prioritized for further analysis as a prospective molecular target of PFD.
Figure 2.

Target identification by probe BP and validation using different assays
(A) Outline of the ABPP strategy for the target identification of small molecule probes in the cell lysates. After probes binding to their protein targets, a click reaction was performed to ligate a fluorophore or enrichment tag, such as biotin, to the probe. Enriched proteins were subsequently analyzed using SDS-PAGE or mass spectrometry.
(B) Structures of ABPP probes BP and their anti-proliferation on ISK cells according to CCK-8 assay (n = 3).
(C) BP dose-dependent labeling proteins (n = 3). Left: In gel fluorescence (FL). Right: Coomassie brilliant blue staining (CBB). M: marker.
(D) Silver staining of probes BP with or without competitor PFD (10×). Probe labeled proteins were labeled with biotin and enriched by streptavidin columns, separated by SDS-PAGE (n = 3). M: marker.
(E) The expression of SEPT7 in EC is significantly reduced. UALCAN (https://ualcan.path.uab.edu/) was employed to analyze the expressions of SEPT7 in EC.
(F) Labeling of recombinant septin7 with different concentrations of BP. Left: In gel fluorescence (FL). Right: Coomassie brilliant blue staining (CBB). Data are representative of at least three independent experiments. M: marker.
(G and H) Cellular thermal shift binding assay of PFD with septin7 in ISK cells. Data are representative of at least three independent experiments. Data are presented as mean ± SD (n = 3) and comparisons were made using t test. ∗∗p < 0.01, ∗∗∗p < 0.001.
(I) MST analysis of the binding affinity between PFD and human recombinant septin7 protein (WT and mutant). The measured Kd value has been shown.
(J) Computational docking between PFD and septin7. PFD forms conventional H-bond with septin7 at Glu58, Ser59′ and Ser168. The “′” indicates that the residue is located in the second strand in the dimer.
(K) The result of superposition of molecular dynamics (MD) simulation stable structure and the crystal structure of septin7. The crystal structure is shown in grayish-white, and the MD stable structure is shown in in light green.
(L) The 3D binding mode of septin7 with PFD from MD simulation. PFD forms conventional H-bond with septin7 at Glu58.
Validation of PFD binding to septin7
The target of PFD was verified by different experiments. In the in-gel fluorescence experiments, BP was successfully labeled with recombinant septin7 in a dose-dependent manner and PFD could compete its binding septin7, indicating that BP and PFD possessed excellent sensitivity toward septin7 (Figure 2F). Thermal shift assay indicated that PFD decreased the thermal stability of septin7 in cell lysates in a temperature-dependent manner compared with the DMSO treated control (Figures 2G and 2H), suggesting the possibility of direct binding between septin7 and PFD. Furthermore, the microscale thermophoresis (MST) assay was conducted to measure the binding capacity of PFD with septin7 with an obtained Kd value of 0.98 μM (Figure 2I). The human septin family is composed of 13 subtypes, which are categorized into four main groups based on sequence similarity and functional properties: septin2 group (septin1, septin2, septin4, septin5), septin 3 group (septin3, septin9, septin12), septin 6 group (septin6, septin8, septin10, septin11, septin14) and septin 7 group (septin7).19 Therefore, the binding of PFD to representative septins of other three group (septin2, septin3 and septin6) were also tested. PFD did not bind to septin3 (Kd > 1000μM), and showed weaker binding affinity for septin2 and septin6 (Kd values were 54.75 μM and 43.09 μM, respectively) than that of septin7, indicating that PFD has a selectivity for binding septin7 (Figure S2). Molecular docking indicated that PFD bound to septin7 through H-bond interaction with the Glu58, Ser59′ [the ‘′’ indicates that the residue was located in the second strand in the dimer], and Ser168 (Figure 2J). In addition, molecular dynamics (MD) simulation also suggested H-bonds formation of PFD with septin7 at Glu58 (Figures 2K and 2L; Figure S3). PFD showed extremely weak binding affinity with mutant septin7 (altering Glu58, Ser59 and Ser168 to Ala), whose Kd > 1000 μM (Figure 2I). These results indicated that PFD could bind to its potential anti-EC molecular target septin7.
PFD suppressed the Orai/IP3R-Ca2+-PIK3CA-AKT signaling pathway in EC cells
To explore the anti-EC mechanism of PFD, transcriptome analysis using RNA-seq was performed. A total of 3344 differentially expressed genes (DEGs) including 1027 up-regulated and 2317 down-regulated genes were identified in EC cells treated with 5 μM PFD compared to DMSO (adjusted p < 0.05). Then Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed to explore the functional roles of the DEGs, revealing that 20 signaling pathways were significantly affected by PFD (adjusted p < 0.01) with the top ranking functional cluster being the TNF signaling pathway (adjusted p = 7.6 × 10−5, Figure 3A). Heatmap analysis of 33 genes associated with the TNF signaling pathway (Table S3) revealed that PFD significantly down-regulated the expression of PIK3CA (Figure 3B), the mutations and amplifications in which were commonly identified in patients with EC metastases and a poor prognosis.32,33 PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha), encoding the catalytic subunit p110α of the PI3K,34 was identified to be the significant gene with the second highest frequency of mutations after PTEN in primary EC lesions.35 Subsequently, qRT-PCR was conducted to detect the expression levels of total PI3K and PIK3CA. The levels of both significantly decreased in EC cells treated with 5 μM PFD with that of PIK3CA relatively lower than that of total PI3K (Figure 3C), indicating that PFD may selectively influence PIK3CA. In addition, western blot analysis revealed that PFD could down-regulate the expression of PIK3CA and its downstream components, AKT phosphorylation, and up-regulated the pro-apoptotic protein cleaved caspase-3 in EC cells (Figures 3D and S4A–S4D). Researches suggested that intracellular Ca2+ is an important upstream regulator of PI3K/AKT signaling pathway.36 Orai, the classical calcium channel located in the cell membrane, and IP3R located in the endoplasmic reticulum (ER) could affect the intracellular Ca2+.37 Thus, Orai and IP3R may be essential for PIK3CA exerting its biological functions. It has been reported that septin7 could regulate the expression of Orai protein.38 A possible mechanism hypothesis was proposed that PFD may exhibit its anti-EC activities by targeting septin7 to down-regulate Orai, IP3R, intracellular Ca2+ and PIK3CA. Thus, whether PFD could influence the expression of Orai and IP3R was investigated. As shown in Figures 3E, S4E, and S4F, PFD reduced the expression of Orai and IP3R in EC cells. In addition, PFD significantly decreased intracellular Ca2+(Figure 3F). These experimental evidences suggested that PFD decreased PIK3CA via Orai/IP3R and Ca2+ suppression and then induced EC cells apoptosis. In summary, PFD could inhibit the Orai/IP3R-Ca2+-PIK3CA-AKT pathway in EC cells.
Figure 3.

The effects of PFD on Orai/IP3R-Ca2+-PIK3CA-AKT pathway in EC cells were determined by transcriptomics analysis, qRT-PCR and western blot
(A) KEGG enrichment analysis of DEGs altered by 5 μM PFD. All pathways significantly enriched in the DEGs (adjusted p < 0.01) were included.
(B) A heatmap depicting DEGs altered by PFD revealed a significant correlation with the PIK3CA.
(C) Relative mRNA expression of PIK3CA (n = 4) and PI3K (n = 4). Data are presented as mean ± SD (n = 4) and comparisons were made using t-test. ∗∗∗p < 0.001.
(D) Protein expression of PIK3CA, p-AKT, AKT, caspase-3 and cleaved caspase-3 in ISK and KLE cells and quantitative statistics is also presented in Figures S4A–S4D. (n = 3).
(E) Protein expression of Orai and IP3R in ISK and KLE cells and quantitative statistics is also presented in Figures S4E and S4F. (n = 3).
(F) PFD significantly reduced the intracellular Ca2+ in ISK and KLE cells (n = 3). Data are presented as mean ± SD (n = 3) and comparisons were made using one-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
PFD attenuates the interaction of septin7-tubulin, resulting in microtubule polymerization and cell-cycle arrest at the G1 phase
Furthermore, as binding proteins of microtubules, septins play an important role in their assembly, monomer, and multimer dynamics, and post-translational modification.18 For example, the knockdown of SEPT7 in HeLa cells led to an increase in the acetylation of the microtubules thus enhancing their stability.39 To explore the possibility of the interaction of septin7 with tubulin and whether PFD affected their interaction, EC cells were co-transfected with a gene encoding Flag-septin7 and Myc-tubulin (β-tubulin, after which all are denoted by tubulin). The two proteins were co-immunoprecipitated (Co-IP) from the transfected cells in reciprocal experiments and PFD reduced the amount of protein produced by Co-IP, suggesting the interaction of septin7 with tubulin that could be inhibited by PFD (Figures 4A and 4B; Figures S7A and S7B). In addition, to further explore the effects of PFD on microtubules, immunofluorescence experiments were conducted in EC cells treated with DMSO and varying concentrations of PFD for 24 h, and the microtubule polymerization agonist paclitaxel (PTX), and the microtubule polymerization inhibitor colchicine (COL) as positive controls. PFD inhibited the polymerization of microtubules in EC cells in a dose-dependent manner, as did COL but unlike PTX (Figures 4C and 4D), suggests that PFD is an inhibitor of microtubule polymerization. More importantly, flow cytometry revealed that PFD induced the arrest of the cell cycle at the G1 phase (Figure 4E), further illustrating the effects of PFD on microtubules. Given that PFD did not alter septin7 expression (Figure S5) and septins form oligomeric complexes composed of different septin subgroup members and could assemble into higher-order structures involved in various biological processes, the influence of PFD on septin7 oligomerization were examined. Results in Figure S6 revealed that PFD promotes the oligomerization of septin7, thereby enhancing its activation. In short, PFD could affect the interaction of septin7-tubulin, inhibit the microtubule polymerization and promote septin7 oligomerization, thereby arresting the EC cell cycle in the G1 phase and inhibiting the EC cell growth.
Figure 4.

PFD attenuates the interaction between septin7 and tubulin, resulting in microtubule polymerization and cell-cycle arrest at the G1 phase
(A and B) Tubulin or septin7 was immunoprecipitated and then revealed by Western blotting with an antibody against septin7 or tubulin. The input of tubulin and septin7 was shown at the bottom of each panel and quantitative statistics is also presented in Figures S7A and S7B. Data are representative of at least three independent experiments.
(C and D) Immunofluorescence images of EC cells treated with DMSO, different concentrations of PFD, paclitaxel (PTX) and colchicine (COL) for 24 h. The nuclei and microtubules have been labeled with DAPI, tubulin antibody. Data are representative of at least three independent experiments. Scale bar: 10 μm.
(E) The effects of PFD on the cell cycle of ISK and KLE cells by flow cytometry analysis. Data are presented as mean ± SD (n = 3) and comparisons were using one-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Septin7 functions as a tumor suppressor in EC
To investigate the biological functions of septin7 in EC, septin7 overexpressing or knockdown ISK and KLE cell lines were generated (Figures 5A and 5B; Figures S8A and S8B). Septin7 overexpression significantly suppressed the proliferation of EC cells while septin7 knockdown promoted their proliferation (Figures 5C and 5D). Similarly, septin7 overexpression significantly inhibited the colony formation of EC cells, while septin7 knockdown achieved the opposite effects (Figures 5E and 5F). In addition, in contrary to septin7 knockdown, septin7 overexpression down-regulated the Orai, IP3R, Ca2+ and PIK3CA levels, inhibited AKT phosphorylation, and increased the cleaved caspase-3 levels, indicating that septin7 inhibited the Orai/IP3R-Ca2+-PIK3CA-AKT pathway (Figures 5G–5I; Figures S9A–S9L), which was consistent with the role of PFD. These results suggested that septin7 functions as a tumor suppressor in EC.
Figure 5.

Septin7 functions as a tumor suppressor that inhibits the Orai/IP3R-Ca2+-PIK3CA-AKT pathway in EC
(A and B) Septin7 protein level with septin7 overexpression (A) or septin7 knockdown (B) lentivirus plasmid infection in ISK or KLE cells was analyzed by western blot.
(C) CCK-8 assays showing the effect of overexpression of septin7 in ISK and KLE cells proliferation (OD450). Data are presented as mean ± SD (n = 3) and comparisons were made using t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(D) CCK-8 assays showing the effect of knockdown of septin7 in ISK and KLE cells proliferation (OD450). Data are presented as mean ± SD (n = 3) and comparisons were made using t test. ∗∗p < 0.01, ∗∗∗p < 0.001.
(E) Cell colony formation assays showing the effect of overexpression or knockdown of septin7 in ISK proliferation. Data are presented as mean ± SD (n = 3) and comparisons were made using t test. ∗∗p < 0.01, ∗∗∗p < 0.001.
(F) Cell colony formation assays showing the effect of overexpression or knockdown of septin7 in KLE proliferation. Data are presented as mean ± SD (n = 3) and comparisons were made using t test. ∗∗p < 0.01, ∗∗∗p < 0.001.
(G and H) The protein expression of Orai, IP3R, PIK3CA, p-AKT was decreased, and cleaved caspase-3 was increased after treatment with septin7 overexpression or knockdown in ISK and KLE cells. And the quantitative statistics is also presented in Figures S9A–S9L. Data are representative of at least three independent experiments.
(I) Intracellular Ca2+ measurements were performed to determine the levels of intracellular Ca2+ in septin7 overexpression ISK and KLE cells. Data are presented as mean ± SD (n = 4) and comparisons were made using t-test. n.s, not significant; ∗p < 0.05, ∗∗p < 0.01.
PFD exhibited its activity through septin7 in EC cells and xenograft mice
The role of septin7 in the PFD’s anti-EC activity was investigated in septin7 knockdown or overexpression of EC cells. It seems that septin7 overexpression augmented the anti-EC potential of PFD, compared to the vector control (Figure 6A). PFD inhibited the proliferation of EC cells (sh-CON), whereas sh-septin7 weakened this effect on cell viability, indicating the necessity of septin7 for PFD’s anti-EC activity (Figure 6B). However, cisplatin (DDP) significantly inhibited cell proliferation in septin7 knockdown or overexpression of EC cells (Figures S10A and S10B). Cell cycle analysis consistently exhibited a disappeared suppression of the G1 phase after treatment with PFD in sh-septin7 (Figures 6C and 6D). The ability of PFD to inhibit microtubule polymerization was also investigated in septin7 knockdown EC cells. As shown in Figure 6E, septin7 knockdown promoted microtubule polymerization (sh-CON vs. sh-Septin7). PFD inhibited microtubule polymerization (sh-CON), but this effect was reduced when septin7 was knocked down (sh-septin7). What is more, PFD suppressed Orai, IP3R, and PIK3CA expression in EC cells, while this effect of PFD was diminished in septin7 mutant EC cells (Figure 6F; Figures S11A–S11F). Additionally, PFD inhibited their expression in control cells (sh-CON), while this inhibition was weakened in sh-Septin7 cells (Figure 6G; Figures S11G–S11L). Similarly, the intracellular Ca2+ determination assay showed that a disappeared suppression of the intracellular Ca2+ after PFD treatment with sh-septin7 (Figure 6H). These results above indicated the regulations of cell cycle, microtubule polymerization, Orai/IP3R/PIK3CA expression and intracellular Ca2+ by PFD were depended on septin7 in EC cells. The role of septin7 in EC tumorigenesis and whether the anti-tumor activity of PFD depended on it in vivo were also investigated. Septin7-deficient KLE cells were subcutaneously injected into the right armpits of nude mice. As shown in Figures 6I–6L, the septin7-deficient mice showed increased tumor volume and tumor weight compared with the vector control sh-CON, and the effects of PFD in the septin7-deficient mice were not significantly reduced (sh-septin7 vs. sh-septin7 + PFD). Collectively, these data validated that PFD’s tumor progression suppression in EC depending on septin7.
Figure 6.

PFD exhibited its activity through septin7 in EC cells and xenograft mice
(A) Cells proliferation assay to determine the effect of PFD (4 μM) on proliferation in septin7 overexpression ISK and KLE cells. Data are presented as mean ± SD (n = 3) and comparisons were made using t test. ∗p < 0.05, ∗∗p < 0.01; ∗∗∗p < 0.001.
(B) Cells proliferation assay to determine the effect of PFD (4 μM) on proliferation in septin7 knockdown ISK and KLE cells. Data are presented as mean ± SD (n = 3) and comparisons were made using t test. n.s, no significance; ∗p < 0.05, ∗∗p < 0.01.
(C and D) Cell cycle assay to determine the effect of PFD (4 μM) on cell cycle in septin7 knockdown ISK and KLE cells. Data are presented as mean ± SD (n = 3) and comparisons were made using t test. n.s, no significance; ∗p < 0.05, ∗∗∗p < 0.001.
(E) The effect of PFD on microtubule polymerization in septin7 knockdown or overexpression EC cells. Scale bar: 15 μm.
(F) The protein expression of Orai, IP3R and PIK3CA after PFD treatment in septin7 mutant ISK and KLE cells. And the quantitative statistics is also presented in Figures S11A–S11F. Data are representative of at least three independent experiments.
(G) The protein expression of Orai, IP3R and PIK3CA after PFD treatment in septin7 knockdown ISK and KLE cells. And the quantitative statistics is also presented in Figures S11G–S11L. Data are representative of at least three independent experiments.
(H) Intracellular Ca2+ measurements to determine the effect of PFD (4 μM) on intracellular Ca2+ in septin7 knockdown ISK and KLE cells. Data are presented as mean ± SD (n = 3) and comparisons were made using t-test. n.s, no significance; ∗p < 0.05, ∗∗p < 0.01.
(I) Relative tumor growth curves showing that the anti-tumor effects of PFD (5 mg/kg) were decreased with the septin7 knockdown; n = 8. Data are presented as mean ± SEM (n = 8) and comparisons were made using t-test. n.s, no significance; ∗p < 0.05, ∗∗∗p < 0.001.
(J) The images of tumors from mice at 14 days after initiation of treatment.
(K and L) Tumor weight and relative tumor volume of mice from each group. Data are presented as mean ± SEM (n = 8) and comparisons were made using t-test. n.s, no significance; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
Endometrial cancer is the most commonly diagnosed gynecological cancer in developed countries. There is an urgent need to develop conservative treatment drugs with good therapeutic effect and few adverse effects to improve the survival rate and quality of life of patients with a requirement for the preservation of reproductive function, as well as those with advanced, recurrent, or inoperable EC. Drug repositioning is an effective strategy for discovery of innovative drugs. It has been shown that antipsychotics had great potential for anti-cancer.15 Phenotype-based drug discovery is a traditional method for new drug screening, which directly observed the efficacy of drugs on pathological models or humans without relying on the target.18 In this study, we found PFD had significant effects in vitro and in vivo through phenotypic screening. Further, molecular target of PFD was identified as septin7 by ABPP techniques. Moreover, two pathways of PFD were revealed to be involved in its anti-EC activity. One pathway was that PFD could induce cell apoptosis by regulating septin7-Orai/IP3R-Ca2+-PIK3CA pathway. Another pathway was that PFD could impede the interaction of septin7 with tubulin, thereby inhibiting the polymerization of microtubules and arresting the cell cycle in G1 phase.
Septins were first identified in 1971 in budding yeast (Saccharomyces cerevisiae) and were proven to be essential for the regulation of the cell division cycle in yeast.40 As a fourth class of cytoskeletal proteins, they play an important role in several cellular physiological functions, such as cytokinesis, DNA repair, apoptosis, etc. Septin7 is an important member of the septins family, which belongs to the GTPase superfamily, and is a GTP-binding protein widely expressed in cells.19 However, its role in human tumors including EC remains to be elucidated, as no small molecular ligand for septin7 has been reported so far. In this study, we identified PFD as a small molecule ligand of septin7 based on ABPP techniques and a series of data including MST, thermal shift assay, and pure protein in-gel fluorescence assay supported the direct binding of PFD to septin7. In addition, through molecular docking and molecular dynamics simulation, we found that PFD bound to septin7 through H-bond interaction with the Glu58, Ser59′, and Ser168. This was confirmed by the extremely weak affinity (Kd > 1000 μM) of PFD to mutant septin7. More importantly, a series of functional data obtained demonstrated that septin7 overexpression suppressed the growth of EC cells by inhibiting the Orai/IP3R-Ca2+-PIK3CA-AKT pathway. The anti-cancer bioactivity of PFD was reduced or disappeared in septin7-deficient EC cells and xenograft mice, suggesting PFD’s anti-EC activity depending on septin7. These findings provide initial evidence suggesting that septin7 is a tumor suppressor in EC and may serve as a potential therapeutic target.
PIK3CA, located on chromosome 3q26.32 encodes the catalytic subunit p110α of PI3K, which plays an important role in the PI3K/AKT pathway and regulates important cellular functions such as cell proliferation, metabolism, angiogenesis, and apoptosis.41 It is an oncogene and is involved in the progression of many tumors, including EC. Its abnormal activation and amplified expression play a critical role in the initiation and progression of cancerous tumors.33,42,43 Unlike most PIK3CA mutations that occur in the regions encoding the helical and kinase domains, those in EC are distributed throughout the entire gene.1 As an upstream regulator of PIK3CA, Ca2+ plays an important role in regulating the biological function of PIK3CA.36 This study showed that PFD could inhibit EC cell growth by regulating the Orai/IP3R-Ca2+-PIK3CA-AKT pathway. These findings not only unraveled the mechanism of PFD action but also identified it as an anti-cancer agent regulating the Orai/IP3R-Ca2+-PIK3CA-AKT axis. Septins are an important class of microtubule-binding proteins,43 play an important role in microtubule assembly, monomer, and multimer dynamics, and also exhibited type-specific differences in their effects. For example, impeding the expression of SEPT7 in HeLa cells increased the stability of microtubules,39 while a knockdown of the SEPT9 in human breast epithelial cells led to a significant reduction in the levels of polymerized microtubules.44,45 In this study, exogenous Co-IP showed that septin7 directly interacted with tubulin and that PFD could inhibit this interaction. More importantly, PFD could inhibit microtubule polymerization and arrest the cell cycle in the G1 phase, thereby acting as an anti-EC agent. These findings provide insight into the regulation of tubulin homeostasis by septin7. Moreover, since septin7 has been suggested as neuroblastoma therapeutic target,46 it will be very interesting to test whether PFD inhibits neuroblastoma. Additionally, research has demonstrated that PFD exerts anti-esophageal cancer effects by targeting PFKL. In breast cancer, PFD inhibits tumor growth by suppressing the integrin pathway, and so on.14,15,16,17 These findings suggest that PFD possesses broad-spectrum antitumor properties. In summary, our study demonstrates that PFD targeting septin7 to inhibit Orai/IP3R-Ca2+-PIK3CA-AKT signaling pathway and microtubule polymerization opens a promising avenue for alleviation of EC and possibly even neuroblastoma. What’s more, in the era of personalized medicine, the introduction of molecular/genomic analysis will help to formulate the most appropriate treatment regimen. On the basis of the outcomes of the Cancer Genome Atlas, and the ProMisE (Proactive Molecular Risk Classifier for EC), tumors are divided into four subgroups according to the presence of polymerase epsilon (POLE) exonuclease domain mutations (EDMs), and protein 53 (p53) immunohistochemistry and mismatch repair (MMR) proteins, creating four different subgroups: POLEmut, p53 wild type (low copy number–CNL-or nonspecific molecular profile-NSMP), p53 null/missense mutations (high copy number) and mismatch repair deficient (MMRd). In advanced or metastatic cases, molecular signatures provide critical information for determining the most appropriate treatment. For instance, patients with hypermutation (POLE) and hypermutation (MSI-H) signatures may respond well to immune checkpoint inhibitors, while dostarlimab monotherapy is beneficial for patients with MMRd/MSI-H endometrial cancer.47,48 In summary, this molecular classification optimizes the clinical management of EC by defining distinct risk classes. It is anticipated that molecular classification will become the standard approach for the treatment of gynecological tumors, particularly endometrial cancer, thereby avoiding unnecessary treatment for patients with favorable molecular profiles. In this study, we found that PFD can inhibit the growth of type II EC tumors (where p53 mutations exceed 90%). Therefore, PFD may be a personalized treatment option for patients with p53mutations.
Limitations of the study
Septin7 plays important roles in many cellular biological functions and is closely related to various human diseases, such as malignant tumors, Alzheimer’s disease, schizophrenia and male infertility, indicating it is a potential therapeutic target in human disease, but there are still lack of molecular agents targeting septin7 at present.
In this study, PFD, a drug clinically used to treat schizophrenia, was found to inhibit EC tumor growth in vitro and in vivo by phenotypic screening. Furthermore, by ABPP technology coupled with proteomics, septin7 was identified as the direct target required for the anti-EC activity of PFD. Mechanistically, the direct binding of PFD to septin7 not only suppressed the EC progression by inhibiting the Orai/IP3R-Ca2+-PIK3CA signaling pathway, but also influenced septin7-tubulin interaction in turn affecting the microtubule assembly. This provides a preclinical rationale for the development of PFD as a potential drug for the treatment of EC. However, KLE cells are an innate progestin-resistant cell line. In the future, we need to construct acquired progestin-resistant EC cells and further verify the efficacy of PFD in vitro and in vivo.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Fei Mao (maofei@ecust.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Data: RNA-Seq data have been deposited at GEO: GSE236228 and are publicly available as of the data of publication. Accession number is listed in the Key resources table.
-
•
Code: This paper does not report original code.
-
•
Other items: Additional information in this paper is available from the lead contact upon request.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (22477028, 22077033, 32121005), National Key R&D Program of China (2021YFA0804904), Shanghai Frontier Science Center of Optogenetic Techniques for Cell Metabolism (2021 Sci & Tech 03–28), Innovative Research Team of High-level Local Universities in Shanghai (SHSMU-ZDCX20212702), Chinese Special Fund for State Key Laboratory of Bioreactor Engineering (2060204), Program of Shanghai Academic Research Leader (22XD1403500), Central Guidance on Local Science and Technology Development Fund of Shanghai Province (YDZX20223100003006).
Author contributions
F.M., Y.D.W., and J.L. designed the study. L.Y.S., H.W.W., and X.H.L. wrote the manuscript. X.H.L. synthesized the probes. H.W.W. analyzed RNA-seq data. L.Y.S. and H.W.W. performed ABPP and functional validation of Septin7. X.S. performed the in vivo experiments. F.M. and J.L. modified the manuscript. W.N., J.L.W., X.K.L., L.J.L., B.B.L., and X.W.L. contributed to interpretation of data. All co-authors contributed comments.
Declaration of interests
F.M., Y.D.W., H.W.W., X.S., X.H.L., L.J.L., and J.L. are inventors on patent WO2023035200.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| septin7 | Abcam | Cat#ab17522; RRID: AB_2876836 |
| PIK3CA | Cell Signaling Technology | Cat#4249S; RRID: AB_2165248 |
| Orai | ABclonal | Cat#A7412; RRID: AB_2767942 |
| IP3R | Abways | Cat# Q14643; RRID: AB_3086675 |
| AKT | Cell Signaling Technology | Cat#4691S; RRID: AB_915783 |
| Caspase-3 | Cell Signaling Technology | Cat#14420T; RRID: AB_2798429 |
| cleaved-caspase-3 | Cell Signaling Technology | Cat#9664; RRID: AB_2070042 |
| Tubulin | Proteintech | Cat#66301-1-Ig; RRID: AB_11042766 |
| β-actin | Yeasen | Cat#30103ES; RRID: AB_2923152 |
| Secondary antibodies HRP-linked anti-rabbit IgG antibody | Cell Signaling Technology | Cat#7074; RRID: AB_2099233 |
| HRP-linked anti-mouse IgG antibody | Cell Signaling Technology | Cat#7076S; RRID: AB_330924 |
| Goat anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 555 | Thermo Fisher Scientific | Cat#A32727; RRID: AB_2633276 |
| FLAG Tag Antibody (magnetic bead conjugate) | Cell Signaling Technology | Cat#82103S; RRID:AB_3073821 |
| Myc-Tag Antibody (magnetic bead conjugate) | Cell Signaling Technology | Cat#91856S; RRID: AB_3073821 |
| control IgG (magnetic bead conjugate) | Cell Signaling Technology | Cat#8726S; RRID: AB_10828938 |
| Chemicals, peptides, and recombinant proteins | ||
| Polybrene | Sigma-Aldrich | Cat# 107689 |
| Puromycin | Thermo Fisher Scientific | Cat# A1113803 |
| RIPA lysate | Yeasen | Cat#20114ES60 |
| PMSF | Yeasen | Cat#20104ES03 |
| septin7 protein | Abcam | Cat#ab158072 |
| cocktail protease inhibitors | Abcam | Cat#ab158072 |
| DAPI | Coolabor | Cat# SL1086 |
| CCK-8 | Beyotime | Cat# C1002 |
| Crystal Violet Staining Solution | TargetMol | Cat#C0005 |
| TRIzol reagent | Beyotime | Cat# C1021 |
| Critical commercial assays | ||
| BCA Protein Quantification Kit | Yeasen | Cat#20201ES76 |
| Annexin V-FITC Apoptosis Detection Kit | Elabscience | Cat#E-CK-A211 |
| Cell Cycle and Apoptosis Analysis Kit | Beyotime | Cat#C1052 |
| Total RNA Kit II | Omega | Cat#R6934-01 |
| Hifair II 1st Strand cDNA Synthesis SuperMix | Yeasen | Cat# 11123ES60 |
| Hieff qPCR SYBR® Green Master Mix | Yeasen | Cat#11201ES08 |
| Capturem Streptavidin Miniprep Columns Kit | Takara | Cat#635733 |
| Fast Silver Stain Kit | Beyotime | Cat#P0017S |
| Fluo-4 a.m. (Calcium ion fluorescent probe, 2mM) | Beyotime | Cat#S1060 |
| Deposited data | ||
| RNA-Seq data | This paper | GSE236228 |
| Experimental models: Cell lines | ||
| ISK | ATCC | HTB-113 |
| KLE | ATCC | CRL-1622 |
| Experimental models: Organisms/strains | ||
| Mouse: BALB/c nude | Shanghai SLAC Laboratory Animal Co., Ltd | https://www.bioon.com.cn/company/index/c42d1349852 |
| Oligonucleotides | ||
| shRNA targeting sequences (see Table S7) | This paper | N/A |
| Primers for RT-qPCR (see Table S8) | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism version 8.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | ImageJ software | https://imagej.net/ij/download.html |
| FlowJo v10.8.1 | FlowJo software | https://www.flowjo.com/solutions/flowjo |
| Real-time PCR | Bio-Rad | CFX384 |
| CytExpert2.4 | Beckman Coulter Life Sciences | https://www.beckman.ae/flowcytometry/instruments/cytoflex/software |
Experimental model and study participant details
Animals
5-6-week-old female BALB/c nude mice were procured from a commercial supplier (Shanghai SLAC Laboratory Animal Co., Ltd). Animals were housed in individually ventilated cages and kept under controlled environmental conditions (12 h-light-dark cycle, 23 ± 1°C with 30–50% relative humidity). Mice were provided with standard rodent chow and ultra-pure water. All mice were healthy during the experiment. In the study of xenograft tumors in animals, when the average tumor volume reached 80–150 mm3, the nude mice were randomly divided into different groups. All the animal experiments were approved by the committee on the ethics of animal experiments of shanghai jiao tong university, China.
Cell lines
The human endometrial cancer cell line ISK and KLE (obtained from Shanghai Fuheng Biotechnology Co., Ltd, China, ATCC: HTB-113, CRL-1622, respectively) was grown in complete DMEM/F12 medium (DMEM/F12 supplemented with 10% fetal serum). The ISK cell was derived from a 39-year-old woman with differentiated endometrial adenocarcinoma (source: ATCC). The KLE is a cell line that was isolated from the endometrium of a White, 64-day-old, female patient with adenocarcinoma (source: ATCC). All cell lines were profiled, authenticated, and Mycoplasma-tested in the Cell Line Core Facility at Shanghai·Biowing Biotechnology Co. LTD in 2023.
Method details
Cell culture
ISK and KLE cell lines were cultured in DMEM/F12 (Biosharp). All media were supplemented with 10% FBS (Bio-Channel), 1% penicillin-streptomycin (Yeasen). Cells were cultured at 37°C in an incubator containing 5% CO2.
Cell proliferation assay
Cells were seeded in 96-well culture plates at a confluence of 70%–80% per well, which equated to approximately 5000 cells per well. Next day, the cells were treated with 200 μL of fresh culture medium containing varying concentrations of test compounds. After 48 h of incubation, the culture medium was removed, and 100 μL of serum-free medium containing 10% CCK-8 (TargetMol) was added to each well, followed by incubation in the incubator for 1–2 h. Then, the absorbance at 450 nm was measured in a microplate reader (Synergy H1, Biotek). The resultant data were analyzed using GraphPad Prism, and the IC50 values of drugs was calculated using nonlinear regression (curve fitting) to automatically fit the dose-response curve.
Colony formation assay
The cells were digested and inoculated into 6-well plates with a density of 1000–2000 cells per well. After overnight culture, 2 mL medium containing different concentrations of drug was added. Cultures were grown for 12 days and the medium was changed every 2 days. After the cell clones are visible in the 6-well plates, rinsed with PBS, and then fixed with methanol at −20°C then stained with 0.1% Crystal Violet Staining Solution (Beyotime) for 30 min, and finally counted for analysis with gel imager (4600SF, Tanon). The ImageJ software counted manually and counted the number of clones formed in each well.
Cell apoptosis assay
Cells were seeded into 24-well plates at a density of 4×104 cells per well, incubated overnight in an incubator to allow adherence, and then various concentrations of drugs were added. After 48 h, cell apoptosis was detected by flow cytometry using an annexin V-FITC apoptosis detection kit (Elabscience) according to the manufacturer’s instructions. Apoptosis was analyzed using CytExpert 2.0.
Cell migration assay
The cells were digested and added to the upper chambers at a density of about 1×105 cells per well. 600 μL of medium (containing 20% FBS) was added below the chamber, and 150 μL of serum-free medium containing the corresponding concentration of drug was added to the upper chamber. After 24 h, the migrated cells were counted. Five fields were randomly selected under the microscope and photographed (200 times). ImageJ software was used to manually count the number of cells in each field.
Cell cycle assay
Cells were grown to the 70%–80% confluency in 6-well plates and synchronized by culturing in serum-free medium for 12 h, following by 24 h treatment of different concentrations of drugs. Subsequently, cells were trypsinized and fixed in ice-cold 70% ethanol for overnight at 4°C. Cells were then centrifugated and the pellet was resuspended in PBS, stained using cell cycle staining solution according to the protocol provided by the cell cycle and apoptosis analysis kit (Beyotime). Cell cycle analysis was performed using a flow cytometer (CytoFLEX LX, Beckman Coulter, CA, USA). The analysis of the obtained data was carried out using FlowJo.
Transcriptome analysis by RNA-seq
When adherent cells reached a density of 30%–40%, drugs were added and incubated. After 48 h, the cells were rinsed with PBS and then digested with trypsin, and the cell precipitate obtained by centrifugation was washed twice with PBS and transferred to a 1.5 mL centrifuge tube. Cells were lysed by adding 1 mL TRIzol reagent (Beyotime) to each tube, snap-frozen in liquid nitrogen, and then transferred to −80°C for storage. More detailed protocols were assisted by Shanghai Majorbio Bio-Pharm Technology Co., Ltd. (China).
RT-qPCR
Cells were grown to 80%–90% confluency in 6-well plates as described above. Different concentrations of PFD in fresh medium (the total volume was 1.5 mL, DMSO <0.01%) were added to the wells and incubated for 48 h. Total RNA was extracted using a total RNA kit II (Omega). Reverse transcription was performed using Hifair II 1st Strand cDNA Synthesis SuperMix (Yeasen) according to the manufacturer’s instructions. Quantitative PCR was performed using Hieff qPCR SYBR Green Master Mix (Yeasen) on a quantitative PCR system (CFX96 Touch, Bio-Rad). qPCR Primers used to perform qPCR are listed below:
for GAPDH, Forward 5′-GCAAGTTCAACGGCACAG-3′,
Reverse 5′-CTCAACAGTATAAAGAGC-3′;
for PI3K, Forword 5′-TGCTATGCCTGCTCTGTAGTGGT-3′,
Reverse 5′-GTGTGACATTGAGGGAGTCGTTG-3′;
for PIK3CA, Forward 5′-AGGATGCCCAACTTGATGCTGATG-3′,
Reverse 5′-CCGTTCATATAGGGTGTCGCTGT-3’.
Western blot analysis
When cells were grown to 70%–90% confluence density, Cells treated with drugs were lysed in 1× SDS loading buffer. Cell lysates were separated by SDS–PAGE and transferred to nitrocellulose membrane (Bio-Rad). After blocked with 5% fat-free dry milk in Tris-buffered saline Tween 20, the membranes incubated with the corresponding primary antibodies. Primary antibodies against septin7 (Abcam), Orai (ABclonal), IP3R (Abways), PIK3CA (Cell Signaling Technology), AKT (Cell Signaling Technology), p-AKT (Cell Signaling Technology), caspase-3 (Cell Signaling Technology), cleaved-caspase-3 (Cell Signaling Technology), Tubulin (Proteintech), β-actin (Yeasen), GAPDH (Proteintech). Secondary antibodies HRP-linked anti-rabbit IgG antibody (Cell Signaling Technology) and HRP-linked anti-mouse IgG antibody (Cell Signaling Technology) were further used. Finally, proteins were visualized with the ECL System from Tanon (4600SF).
In-gel fluorescence analysis
ISK cells were washed with 1×PBS, rapidly lysed by adding weak RIPA lysate (Yeasen) containing 1 mM PMSF (Yeasen). The precipitate was discarded by centrifugation at 13,000 rpm for 15 min at 4°C. The cell lysate supernatant was collected for quantification by BCA Protein Quantification Kit (Yeasen). Different concentrations of BP and competition group (7 BP μM + PFD 70 μM) was added to the same amount of lysate and further incubated for 2 h at room temperature and photocross-linking was then performed. Subsequently, a freshly premixed click chemistry reaction cocktail was added to the protein solution, so that the molar ratio of probe, TAMRA-N3, TBTA, TCEP, CuSO4 was 1:2:5:50:50. After 2 h of incubation, the proteins were directly mixed with loading buffer (5×) at a volume ratio of 4:1 and denatured for 5 min at 95°C in a metal bath. 20 μg of protein was sampled per lane on SDS-PAGE and subsequently visualized by in-gel fluorescence scanning (Typhoon FLA 9500).
For recombinant protein labeling, different concentrations of probes were incubated with purified septin7 protein at different final concentrations in PBS buffer for 1 h at 37°C. The subsequent labeling process is similar to that described above for the total proteome of cells. Purified septin7 protein was purchased from Abcam.
Pull down/LC-MS/MS
To identify the potential targets of BP, pull-down experiments were carried out, accompanied by western blot and LC-MS/MS analysis. ISK cells were grown in 10 cm dishes to 90% confluency and lysed. Then the protein solution in the lysate was incubated with different concentrations of BP and photocross-linking was performed. Subsequently, a freshly premixed click chemistry reaction cocktail was added to the protein solution, so that the molar ratio of probe, biotin-N3, TBTA, TCEP and CuSO4 was 1:2:5:50:50. After 2 h of incubation. Pull down experiments were performed using the Capturem Streptavidin Miniprep Columns Kit (Takara) according to the standardized procedure in the manufacturer’s instructions. The columns were washed three times with PBS to remove unbound proteins, the streptavidin-bound proteins were eluted with a buffer containing 0.1 mol/L glycine (pH 2.5), neutralized in a buffer containing 1 mol/L Tris (pH 8.5), separated by SDS-PAGE, and visualized by silver staining. The specific process of silver staining was according to the instructions of Fast Silver Stain Kit (Beyotime). After silver staining, the enriched protein bands were excised and analyzed by LC-MS/MS by Luming Biological Testing Company.
Molecular docking
The septin7 dimer structure (PDB ID: 7M6J) was pocked by sitemap in Schrodinger software, and the highest ranked pocket was located on the dimer interface. The Induced Fit Docking module in Glide was used to perform molecular docking on the PFD to obtain the complex structure.
Molecular dynamics simulation
The Amber molecular dynamics simulation system was constructed, the protein force field was ff14SB, and the small molecule force field was gaff. The final simulation system contained 85632 atoms with A size of 77 × 88 × 152 Å3. After the construction of the simulation system, Amber22 was used for molecular dynamics simulation, and the total time was 80 ns. The simulation temperature was 303.15 k, the pressure was 1.0 atm, and the integration step was 2 fs. The PME method was used for the long-range electrostatic potential, and the 9 Å cutoff distance was used for the short-range van der Waals force.
Thermal shift assay
ISK cells were grown to 80%–90% confluency in 6-well plates as described above and lysed in RIPA lysis buffer supplemented with 1 mmol/L PMSF. Cell lysates were collected and centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant protein solution was taken and divided equally into two tubes to which DMSO and 7 μM PFD were added. After 2 h of reaction at room temperature, each group was divided into 12 tubes and heated for 5 min at 40°C, 41.4°C, 43.3°C, 46.3°C, 49.8°C, 53.3°C, 56.4°C, 60.5°C, 64.0°C, 66.8°C, 68.8°C, 70°C, respectively, using PCR instrument CETSA program. Proteins were mixed with loading buffer (5×) at a volume ratio of 4:1, denatured in a metal bath at 95°C for 5 min, and the proteins were subjected to western blot analysis.
Microscale thermophoresis assay
To evaluate the binding affinity of PFD and septin7 (WT and mutant), septin2, septin3 and septin6, an MST assay was conducted by Monolith NT. Automated (NanoTemper Technologies). The recombinant protein was labeled with the His-Tag Labeling Kit RED-tris-NTA 2nd Generation (MO-l018, Nano Temper) according to the manufacturer’s instructions. The final labeled recombinant protein concentration was 50 nM, which was mixed with different concentrations of PFD by multiple pipetting. All samples were diluted in 1×PBST and contained the same amount of DMSO. MST analysis was performed using the Monolith NT. Automated (Nano Temper Technologies), and Kd was measured in MO. Control software.
Measurements of intracellular Ca2+
The intracellular calcium ion levels of EC cells were measured with the diluted Fluo-4 a.m. ester stock solution (Beyotime, Cat#S1060) (10 μM with HBSS). After adhesion, the culture medium was removed, and the cells were rinsed with HBSS for three times. The Fluo-4 a.m. ester solution and 5 μM PFD was added (80 μL per well) and incubated with EC cells for 30 min at 37°C. Then, after removing the Fluo-4 a.m. ester solution, the cells were rinsed by HBSS for three times and 100 μL of 10mM CaCl2 was added. the fluorescence intensity was measured with microplate reader (Synergy H1, Biotek) at the excitation wavelength of 488 nm.
Cell transfection
Genomeditech (Shanghai, China) assisted the construction of recombinant adenovirus vector systems of tubulin over-expression, septin7 knockdown and over-expression. ISK or KLE cells were inoculated with 1×105 cells in 12-well plates. After adherence, cells were transfected by adding the recombinant adenovirus to the culture at a multiplicity of infection (MOI) for 20 h. On the fifth day, cells were screened with appropriate antibiotics. Then the cells were maintained by DMEM/F-12 + 10% FBS +1% penicillin-streptomycin +0.5 μg/mL puromycin or 30 μg/mL blasticidin complete culture medium. The recombinant adenovirus containing septin7 short hairpin RNA (shRNAs) or negative control shRNA (sh-CON) was packaged using PGMLV-SB3 RNAi vector. The shRNAs for septin7 were 5′-GCCTGTTATCGACTACATTGA-3′(shRNA-1),
5′-GCTGTGGTAGGTAGTAATACT-3′(shRNA-2),
5′-GGGAAGCTCAACAACGTATTT-3′(shRNA-3),
5′-GCAGCTGACTAAGAGCCCTCT-3′ (shRNA-4).
The shRNA for negative control was 5′- TTCTCCGAACGTGTCACGT-3′(sh-CON). Septin7 over-expression was packaged with PGMLV-CMV-MCS-3×Flag-PGK-Puro vector. Septin7 primer: Forward: 5′-CGCGAATTCGAAGTATACCTCG-3′, Reverse:5′-GTCATGGTCTTTGTAGTCGGATCC -3′. β-tubulin over-expression was packaged with PGMLV-CMV-MCS-EF1-ZsGreen1-T2A-Blasticidin vector. β-tubulin primer: Forward: 5′- GCGAATTCGAAGTATACCTCGAGGCCACCATGCGTG-3′, Reverse:5′-CGATCGCAGATCCTTGGATCCTCACAGATCCTCTTCAGAGATGAGTTTCTGCTCG-3′. Zoonbio Biotechnology Co Ltd assisted the construction of Septin7 mutant plasmids. Septin7 mutant plasmids were transfected into cells with PEI (Proteintech, Cat#PR40001) were transfected into cells according to the manufacturer’s instructions.
Co-immunoprecipitation
Co-immunoprecipitation (Co-IP) were performed as previously described.49 Flag-septin7 and Myc-β-tubulin were successfully transfected into ISK cells according to the above cell transfection methods. When cells had grown to a confluence density of 70–80%, the fresh medium containing 4 μM PFD was added. After 48h, Cells were lysed in RIPA lysis buffer supplemented with 1 mmol/L PMSF and centrifuged at 12,000 rpm for 15 min in 4°C. 60 μL of the supernatant was removed as input. The remaining half of the lysate was mixed with 5 μL of FLAG Tag Antibody (magnetic bead conjugate) (Cell Signaling Technology) or Myc-Tag Antibody (magnetic bead conjugate) (Cell Signaling Technology, Cat#91856S), and the other half was mixed with 5 μL of control IgG (magnetic bead conjugate) (Cell Signaling Technology). After overnight incubation, the precipitates were washed five times with lysis lysate and subjected to western blot analysis.
Immunofluorescence
The immunofluorescence was performed as described previously.31 Cells were fixed with 4% paraformaldehyde for 30 min and was blocked and permeable with the PBS solution containing 1% BSA and 0.3% Triton X-100 at 37°C for 1 h. The cells were incubated with the primary antibody against β-tubulin (Proteintech) overnight at 4°C, and then the Goat anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 (Thermo Fisher Scientific) was added incubation for 1.5 h at room temperature. After DAPI counterstaining, the images were collected Ti-S microscope (Nikon).
Mouse tumor xenograft studies
Female nude mice (6–8 weeks old) were purchased from Shanghai Slac Laboratory Animal CO. LTD. KLE cells (5 × 107/mice) were subcutaneously injected into nude mice. The nude mice were killed when the tumor grew to 800–1000 mm3, and then the tumor tissue was cut into evenly sized tumor tissue blocks, which were surgically transplanted into the right subcutaneous axilla of new 6–8 weeks old nude mice (the mice were anesthetized with isoflurane). For septin7-deficient KLE xenograft models, septin7-deficient KLE cells (5 × 107/mice) were subcutaneously injected into nude mice. When the average tumor volume reached 80–150 mm3, the nude mice were randomly divided into 5 groups. Each group of mice were intraperitoneally injected with vehicle (normal saline), 2 mg/kg cisplatin (DDP), 2 mg/kg PFD and 5 mg/kg PFD every day and weighed the nude mice the next day. After 14 days of continuous administration, the nude mice were sacrificed for cervical dislocation. The tumors were removed and photographed; its volume was calculated with the formula V = 1/2 (length × width2). Then, major organs such as heart, liver, spleen and kidney of nude mice were taken for HE staining to observe pathological changes caused by toxicity. Whole blood serum was collected to detect blood biochemical indexes to evaluate hepatorenal toxicity after drugs administration.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism software, version 8.4 (GraphPad, San Diego, CA, USA). In animal experiments, data are presented as mean ± standard error of mean (SEM), and other data are presented as mean ± standard deviation (SD). In animal experiments, n represents the number of animals, and in other experiments, n represents the number of repeated experiments. For the cell migration assay, colony formation assay, cell apoptosis assay, Ca2+ measurements, and cell cycle assay (Figures 1C–1E, 3F, and 4E), statistical significance was determined using a one-way ANOVA. Mouse tumor xenograft studies (Figures 1F, 1H, and 1I), statistical significance was also determined using a one-way ANOVA. For Cellular thermal shift binding assay (Figure 2H), relative mRNA expression of PIK3CA and PI3K (Figure 3C), statistical significance was assessed using t-test. The effect of overexpression or knockdown of septin7 in ISK and KLE cells, including CCK-8 assays (Figures 5C and 5D), cell colony formation assay (Figures 5E and 5F), Intracellular Ca2+ measurements (Figure 5I), and the effect of PFD on proliferation and cell cycle in septin7 overexpression or knockdown ISK and KLE cells (Figures 6A–6D), statistical significance was assessed using t-test. The effect of PFD on Intracellular Ca2+ in septin7 knockdown ISK and KLE cells (Figure 6H), statistical significance was also assessed using t-test. The anti-tumor effects of PFD with the septin7 knockdown (Figures 6I, 6K, and 6L), statistical significance was assessed using t-test. p < 0.05 was used as the test standard, ns, no significance, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Published: December 19, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111640.
Contributor Information
Jian Li, Email: jianli@ecust.edu.cn.
Yudong Wang, Email: wangyudong@shsmu.edu.cn.
Fei Mao, Email: maofei@ecust.edu.cn.
Supplemental information
References
- 1.Miller K.D., Nogueira L., Devasia T., Mariotto A.B., Yabroff K.R., Jemal A., Kramer J., Siegel R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022;72:409–436. doi: 10.3322/caac.21731. [DOI] [PubMed] [Google Scholar]
- 2.SGO Clinical Practice Endometrial Cancer Working Group. Burke W.M., Orr J., Leitao M., Salom E., Gehrig P., Olawaiye A.B., Brewer M., Boruta D., Herzog T.J. Endometrial cancer: a review and current management strategies: part II. Gynecol. Oncol. 2014;134:393–402. doi: 10.1016/j.ygyno.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R.L., Miller K.D., Wagle N.S., Jemal A. Cancer statistics, 2023. CA Cancer J. Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 4.Crosbie E.J., Kitson S.J., McAlpine J.N., Mukhopadhyay A., Powell M.E., Singh N. Endometrial cancer. Lancet. 2022;399:1412–1428. doi: 10.1016/S0140-6736(22)00323-3. [DOI] [PubMed] [Google Scholar]
- 5.Miller D.S., Filiaci V.L., Mannel R.S., Cohn D.E., Matsumoto T., Tewari K.S., DiSilvestro P., Pearl M.L., Argenta P.A., Powell M.A., et al. Carboplatin and Paclitaxel for Advanced Endometrial Cancer: Final Overall Survival and Adverse Event Analysis of a Phase III Trial (NRG Oncology/GOG0209) J. Clin. Oncol. 2020;38:3841–3850. doi: 10.1200/JCO.20.01076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makker V., Colombo N., Casado Herráez A., Santin A.D., Colomba E., Miller D.S., Fujiwara K., Pignata S., Baron-Hay S., Ray-Coquard I., et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N. Engl. J. Med. 2022;386:437–448. doi: 10.1056/NEJMoa2108330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacLean J.A., 2nd, Hayashi K. Progesterone Actions and Resistance in Gynecological Disorders. Cells. 2022;11 doi: 10.3390/cells11040647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duska L.R., Garrett A., Rueda B.R., Haas J., Chang Y., Fuller A.F. Endometrial cancer in women 40 years old or younger. Gynecol. Oncol. 2001;83:388–393. doi: 10.1006/gyno.2001.6434. [DOI] [PubMed] [Google Scholar]
- 9.Creasman W.T., Odicino F., Maisonneuve P., Beller U., Benedet J.L., Heintz A.P., Ngan H.Y., Pecorelli S. Carcinoma of the corpus uteri. Int. J. Gynaecol. Obstet. 2003;83:79–118. doi: 10.1016/s0020-7292(03)90116-0. [DOI] [PubMed] [Google Scholar]
- 10.Deleon M.C., Ammakkanavar N.R., Matei D. Adjuvant therapy for endometrial cancer. J. Gynecol. Oncol. 2014;25:136–147. doi: 10.3802/jgo.2014.25.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vlachos N., Lampros M., Voulgaris S., Alexiou G.A. Repurposing Antipsychotics for Cancer Treatment. Biomedicines. 2021;9 doi: 10.3390/biomedicines9121785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y., Cui Y., Sun X., Wu H., Ou M., Tang Y., Ni S., Li X., Zhu J., Mao F., et al. Repurposing of antipsychotics perphenazine for the treatment of endometrial cancer. Bioorg. Med. Chem. Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127239. [DOI] [PubMed] [Google Scholar]
- 13.Cui Y., Wu H., Yang L., Huang T., Li J., Gong X., Li L., Sun X., Mao F., Wang Y. Chlorpromazine Sensitizes Progestin-Resistant Endometrial Cancer Cells to MPA by Upregulating PRB. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.665832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu L., Liu Y.Y., Li Z.X., Zhao Q., Wang X., Yu Y., Wang Y.Y., Wang Y.Q., Luo F. Anti-tumor effects of penfluridol through dysregulation of cholesterol homeostasis. Asian Pac. J. Cancer Prev. APJCP. 2014;15:489–494. doi: 10.7314/apjcp.2014.15.1.489. [DOI] [PubMed] [Google Scholar]
- 15.Shaw V., Srivastava S., Srivastava S.K. Repurposing antipsychotics of the diphenylbutylpiperidine class for cancer therapy. Semin. Cancer Biol. 2021;68:75–83. doi: 10.1016/j.semcancer.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng C., Yu X., Liang Y., Zhu Y., He Y., Liao L., Wang D., Yang Y., Yin X., Li A., et al. Targeting PFKL with penfluridol inhibits glycolysis and suppresses esophageal cancer tumorigenesis in an AMPK/FOXO3a/BIM-dependent manner. Acta Pharm. Sin. B. 2022;12:1271–1287. doi: 10.1016/j.apsb.2021.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranjan A., Gupta P., Srivastava S.K. Penfluridol: An Antipsychotic Agent Suppresses Metastatic Tumor Growth in Triple-Negative Breast Cancer by Inhibiting Integrin Signaling Axis. Cancer Res. 2016;76:877–890. doi: 10.1158/0008-5472.CAN-15-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu J., Li X., Ding K., Li Z. Applications of Activity-Based Protein Profiling (ABPP) and Bioimaging in Drug Discovery. Chem. Asian J. 2020;15:34–41. doi: 10.1002/asia.201901500. [DOI] [PubMed] [Google Scholar]
- 19.Hall P.A., Russell S.E.H. The pathobiology of the septin gene family. J. Pathol. 2004;204:489–505. doi: 10.1002/path.1654. [DOI] [PubMed] [Google Scholar]
- 20.Wang X., Fei F., Qu J., Li C., Li Y., Zhang S. The role of septin 7 in physiology and pathological disease: A systematic review of current status. J. Cell Mol. Med. 2018;22:3298–3307. doi: 10.1111/jcmm.13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia Z.F., Huang Q., Kang C.S., Yang W.D., Wang G.X., Yu S.Z., Jiang H., Pu P.Y. Overexpression of septin 7 suppresses glioma cell growth. J. Neuro Oncol. 2010;98:329–340. doi: 10.1007/s11060-009-0092-1. [DOI] [PubMed] [Google Scholar]
- 22.Jia Z., Wang K., Wang G., Zhang A., Pu P. MiR-30a-5p antisense oligonucleotide suppresses glioma cell growth by targeting SEPT7. PLoS One. 2013;8 doi: 10.1371/journal.pone.0055008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Jiang H., Hua D., Zhang J., Lan Q., Huang Q., Yoon J.G., Han X., Li L., Foltz G., Zheng S., Lin B. MicroRNA-127-3p promotes glioblastoma cell migration and invasion by targeting the tumor-suppressor gene SEPT7. Oncol. Rep. 2014;31:2261–2269. doi: 10.3892/or.2014.3055. [DOI] [PubMed] [Google Scholar]
- 24.Hou M., Liu X., Cao J., Chen B. SEPT7 overexpression inhibits glioma cell migration by targeting the actin cytoskeleton pathway. Oncol. Rep. 2016;35:2003–2010. doi: 10.3892/or.2016.4609. [DOI] [PubMed] [Google Scholar]
- 25.Igci Y.Z., Erkilic S., Arslan A. Septin 7 immunoexpression in papillary thyroid carcinoma: a preliminary study. Pathol. Res. Pract. 2014;210:426–431. doi: 10.1016/j.prp.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Zhou J., Lu S., Yang S., Chen H., Shi H., Miao M., Jiao B. MicroRNA-127 post-transcriptionally downregulates Sept7 and suppresses cell growth in hepatocellular carcinoma cells. Cell. Physiol. Biochem. 2014;33:1537–1546. doi: 10.1159/000358717. [DOI] [PubMed] [Google Scholar]
- 27.Zhang N., Liu L., Fan N., Zhang Q., Wang W., Zheng M., Ma L., Li Y., Shi L. The requirement of SEPT2 and SEPT7 for migration and invasion in human breast cancer via MEK/ERK activation. Oncotarget. 2016;7:61587–61600. doi: 10.18632/oncotarget.11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janssen P.A., Niemegeers C.J., Schellekens K.H., Lenaerts F.M., Verbruggen F.J., Van Nueten J.M., Schaper W.K. The pharmacology of penfluridol (R 16341) a new potent and orally long-acting neuroleptic drug. Eur. J. Pharmacol. 1970;11:139–154. doi: 10.1016/0014-2999(70)90043-9. [DOI] [PubMed] [Google Scholar]
- 29.Wilkinson I.V.L., Perkins K.J., Dugdale H., Moir L., Vuorinen A., Chatzopoulou M., Squire S.E., Monecke S., Lomow A., Geese M., et al. Chemical Proteomics and Phenotypic Profiling Identifies the Aryl Hydrocarbon Receptor as a Molecular Target of the Utrophin Modulator Ezutromid. Angew. Chem. Int. Ed. Engl. 2020;59:2420–2428. doi: 10.1002/anie.201912392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W., Hao Y., Zhang A., Yang W., Wei W., Wang G., Jia Z. miR-19a/b promotes EMT and proliferation in glioma cells via SEPT7-AKT-NF-kappaB pathway. Mol. Ther. Oncolytics. 2021;20:290–305. doi: 10.1016/j.omto.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Packer L.M., Geng X., Bonazzi V.F., Ju R.J., Mahon C.E., Cummings M.C., Stephenson S.A., Pollock P.M. PI3K Inhibitors Synergize with FGFR Inhibitors to Enhance Antitumor Responses in FGFR2(mutant) Endometrial Cancers. Mol. Cancer Therapeut. 2017;16:637–648. doi: 10.1158/1535-7163.MCT-16-0415. [DOI] [PubMed] [Google Scholar]
- 32.Sun W., Xie Z., Liu Y., Zhao D., Wu Z., Zhang D., Lv H., Tang S., Jin N., Jiang H., et al. JX06 Selectively Inhibits Pyruvate Dehydrogenase Kinase PDK1 by a Covalent Cysteine Modification. Cancer Res. 2015;75:4923–4936. doi: 10.1158/0008-5472.CAN-15-1023. [DOI] [PubMed] [Google Scholar]
- 33.Heudel P.E., Fabbro M., Roemer-Becuwe C., Kaminsky M.C., Arnaud A., Joly F., Roche-Forestier S., Meunier J., Foa C., You B., et al. Phase II study of the PI3K inhibitor BKM120 in patients with advanced or recurrent endometrial carcinoma: a stratified type I-type II study from the GINECO group. Br. J. Cancer. 2017;116:303–309. doi: 10.1038/bjc.2016.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai K., Killingsworth M.C., Lee C.S. Gene of the month: PIK3CA. J. Clin. Pathol. 2015;68:253–257. doi: 10.1136/jclinpath-2015-202885. [DOI] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas Research Network. Kandoth C., Schultz N., Cherniack A.D., Akbani R., Liu Y., Shen H., Robertson A.G., Pashtan I., Shen R. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y., Zhang T., Wu C., Xia Q., Xu D. ASIC1a mediates the drug resistance of human hepatocellular carcinoma via the Ca2+/PI3-kinase/AKT signaling pathway. Lab. Invest. 2017;97:53–69. doi: 10.1038/labinvest.2016.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foskett J.K., White C., Cheung K.-H., Mak D.-O.D. Inositol Trisphosphate Receptor Ca2+ Release Channels. Physiol. Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deb B.K., Pathak T., Hasan G. Store-independent modulation of Ca2+ entry through Orai by Septin 7. Nat. Commun. 2016;7 doi: 10.1038/ncomms11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kremer B.E., Haystead T., Macara I.G. Mammalian septins regulate microtubule stability through interaction with the microtubule-binding protein MAP4. Mol. Biol. Cell. 2005;16:4648–4659. doi: 10.1091/mbc.e05-03-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spiliotis E.T. Spatial effects - site-specific regulation of actin and microtubule organization by septin GTPases. J. Cell Sci. 2018;131 doi: 10.1242/jcs.207555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samuels Y., Ericson K. Oncogenic PI3K and its role in cancer. Curr. Opin. Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 42.Gustafson A.M., Soldi R., Anderlind C., Scholand M.B., Qian J., Zhang X., Cooper K., Walker D., McWilliams A., Liu G., et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci. Transl. Med. 2010;2 doi: 10.1126/scitranslmed.3000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill M.M., Hemmings B.A. Inhibition of protein kinase B/Akt. implications for cancer therapy. Pharmacol. Ther. 2002;93:243–251. doi: 10.1016/s0163-7258(02)00193-6. [DOI] [PubMed] [Google Scholar]
- 44.Fujishima K., Kiyonari H., Kurisu J., Hirano T., Kengaku M. Targeted disruption of Sept3, a heteromeric assembly partner of Sept5 and Sept7 in axons, has no effect on developing CNS neurons. J. Neurochem. 2007;102:77–92. doi: 10.1111/j.1471-4159.2007.04478.x. [DOI] [PubMed] [Google Scholar]
- 45.Spiliotis E.T., Hunt S.J., Hu Q., Kinoshita M., Nelson W.J. Epithelial polarity requires septin coupling of vesicle transport to polyglutamylated microtubules. J. Cell Biol. 2008;180:295–303. doi: 10.1083/jcb.200710039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagata T., Takahashi Y., Asai S., Ishii Y., Mugishima H., Suzuki T., Chin M., Harada K., Koshinaga S., Ishikawa K. The high level of hCDC10 gene expression in neuroblastoma may be associated with favorable characteristics of the tumor. J. Surg. Res. 2000;92:267–275. doi: 10.1006/jsre.2000.5918. [DOI] [PubMed] [Google Scholar]
- 47.Ottavia D., Andrea G., Aris R.B., Donatella C. Management of Endometrial Cancer: Molecular Identikit and Tailored Therapeutic Approach. Clin. Exp. Obstet. Gynecol. 2023;50:210. doi: 10.31083/j.ceog5010210. [DOI] [Google Scholar]
- 48.Di Donato V., Giannini A., Bogani G. Recent Advances in Endometrial Cancer Management. J. Clin. Med. 2023;12:2241. doi: 10.3390/jcm12062241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin P., Jiang J., Zhou L., Huang Z., Qin S., Chen H.N., Peng L., Zhang Z., Li B., Luo M., et al. Disrupting metformin adaptation of liver cancer cells by targeting the TOMM34/ATP5B axis. EMBO Mol. Med. 2022;14 doi: 10.15252/emmm.202216082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: RNA-Seq data have been deposited at GEO: GSE236228 and are publicly available as of the data of publication. Accession number is listed in the Key resources table.
-
•
Code: This paper does not report original code.
-
•
Other items: Additional information in this paper is available from the lead contact upon request.