

Abstract
In response to high levels of DNA damage, catalytic activation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP) triggers necrotic death because of rapid consumption of its substrate β-nicotinamide adenine dinucleotide and consequent depletion of ATP. We examined whether there are other consequences of PARP activation that could contribute to cell death. Here, we show that PARP activation reaction in vitro becomes acidic with release of protons during hydrolysis of β-nicotinamide adenine dinucleotide. In the cellular context, we show that Molt 3 cells respond to DNA damage by the alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) with a dose-dependent acidification within 30 min. Whereas acidification by 0.15 pH units induced by 10 μM MNNG is reversed within 1 h, 100 μM MNNG-induced acidification by 0.5–0.6 pH units is persistent up to 7 h. Acidification is a general DNA damage response because H2O2 exposure also acidifies Molt 3 cells, and MNNG causes acidification in Jurkat, U937, or HL-60 leukemia cells and in PARP+/+ fibroblasts. Acidification is significantly decreased in the presence of PARP inhibitors or in PARP−/− fibroblasts, suggesting a major role for PARP activation in acidification. Inhibition of proton export through ATP-dependent Na+/H+ exchanger is another major cause of acidification. Using the pH clamp method to either suppress or introduce changes in cellular pH, we show that brief acidification by 0.5–0.6 pH units may be a negative regulator of apoptosis while permitting necrotic death of cells with extensively damaged DNA.
In response to DNA damage, nuclear enzyme poly(ADP-ribose) polymerase (PARP or PARP-1, E.C. 2.4.2.30) is rapidly activated to form polymers of ADP-ribose from the substrate β-nicotinamide adenine dinucleotide (NAD) (1). PARP activation reaction participates in DNA repair and recovery after lower levels of DNA damage and cell death at higher levels of DNA damage (2–5). PARP overactivation in response to extensive DNA damage is suggested to cause cell death by depletion of NAD and consequent loss of ATP pools (6, 7). PARP activation as a cause of cell death is also evident from the remarkable resistance of PARP−/− mice to focal cerebral ischemia, myocardial infarctions, toxin-induced diabetes, parkinsonism, and inflammation, which are the diseases caused by pathologically high levels of DNA damage (2, 4). Of the two modes of cell death, apoptosis and necrosis, PARP activation is more specifically linked to necrotic death after DNA damage, because PARP−/− fibroblasts resist necrotic death induced by N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) (8), and PARP−/− mice resist necrotic death during hemorrhagic shock (9). PARP inhibitors also prevent necrotic death induced by oxidants in thymocytes (10) or epithelial cells of intestine (11), kidney (12), and pulmonary artery (13) while permitting oxidant-induced apoptotic death.
ATP depletion is the central mechanism by which PARP activation is suggested to participate in DNA damage-induced necrotic death (8). Here, we examined whether there are other consequences of PARP activation that could participate in DNA damage-induced cell death. PARP activation reaction produces 1 mol each of proton and nicotinamide for each mol of NAD consumed (1). Therefore, extensive PARP activation can consume a large amount of NAD and release a substantial amount of protons in a very short period; this may have the potential to cause cellular acidification after DNA damage. Indeed, oxidative damage that also activates PARP has earlier been shown to cause rapid acidification of certain cells that are pathophysiologically exposed to oxidants, such as cardiac myocytes or myoblasts (14), aortic endothelial cells (15, 16), renal epithelial cells (17), and hippocampal neurons (18). Neuronal acidification has also been reported in other models that indirectly implicate oxidant damage, such as cerebral ischemia (19) or treatment with N-methyl-D-glucamine (20). In the majority of these studies, acidification was attributed to inhibition of the proton export mainly through Na+/H+ exchangers (NHE) (15–17) or through the neuronal Ca2+/H+ exchanger (20). In other studies, acidification was suggested to be caused by passive influx of protons or release of protons from other subcellular compartments (17) or caused by oxidant-mediated inhibition of glycolysis and resultant hydrolysis of ATP (14). Whereas these studies focused on the role of oxidative damage in acidification, we examined whether acidification is a unique response to oxidants or whether it would occur with other DNA-damaging agents that activate PARP. Here, we report that various types of cells are acidified immediately after DNA damage by the alkylating agent MNNG, and PARP activation and inhibition of the ATP-dependent Na+/H+ exchanger are the major causes of acidification. We also show that rapid acidification after high levels of DNA damage can suppress apoptosis while permitting necrotic death.
Materials and Methods
Cells.
Molt 3, HL-60, Jurkat, and U937 cells (American Type Culture Collection) were cultured in RPMI medium 1640 (GIBCO) (21). PARP+/+ or PARP−/− mouse embryonal fibroblasts (obtained from Z. Q. Wang, International Agency for Research on Cancer, Lyon, France) were grown in DMEM low glucose medium (GIBCO) (22).
PARP Activation Reaction in Vitro.
Bovine hydroxylapatite-purified PARP (1,340 units/mg, Aparptosis, Quebec) was washed free of salts and buffer and activated in vitro, as described (23), except that Tris buffer was excluded and initial pH was adjusted to 7.4. The pH of the reaction was measured with a standard glass electrode. An aliquot of reaction was immunoblotted for PARP and polymer (see below).
Analysis of Intracellular pH.
Intracellular pH was analyzed by using fluorescent probe 2′,7′-bis-(2-carboxyethyl)-5-(6)-carboxyfluorescein, acetoxymethyl ester (BCECF, Molecular Probes) (24). In brief, cells were loaded with BCECF and treated with MNNG or H2O2 up to 1 h. For longer treatment, cells were treated first and then loaded with BCECF 15 min before harvesting. Cellular pH was analyzed by flow cytometry by using Coulter EPICS elite with excitation at 488 nm and ratio of emissions at 525 nm (pH sensitive) and 640 nm (pH insensitive). For each experiment, pH standard curves were established with a series of cells at pH 7.4, 7.1, 6.8, 6.5, and 6.3 units. Initial pH of the control cells was observed to be between 7.3 and 7.4 units.
Acid Loading of Cells by NH4Cl-Pulse Method.
BCECF-loaded Molt 3 were acidified by treatment with 20 mM
NH4Cl pulse (16, 17) for 10 min in low
Na+ buffer, washed, and suspended in
HCO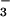 -free high Na+ buffer (24)
to allow acidification and pH recovery.
-free high Na+ buffer (24)
to allow acidification and pH recovery.
The pH Clamp Studies.
For clamping intracellular pH to 7.4 or 6.8, Molt 3 cells were suspended for 10 min in K+ calibration buffer at pH 7.4 or 6.8 (24) with (pH-clamped) or without (control without pH clamp) 2 μg/ml nigericin, and treated with MNNG for 1 h. Cells were washed and allowed to recover for 10 h in fresh medium before analyses of cell death.
Immunoblotting for PARP, Polymer-Modified Proteins, and Caspase 3.
Immunoblotting was carried out as described (22) with monoclonal anti-PARP (C-2–10, 1:10,000, Aparptosis), polyclonal antipolymer (LP96–10, 1:10,000, Aparptosis), monoclonal antipolymer (25) (10H, 1:1,000), or polyclonal anticaspase 3 (3-R#MF393 from D. Nicholson, Merck Frosst Canada, Montreal, 1:10,000).
Flow Cytometric Analysis of Mode of Cell Death.
Viable, apoptotic, and necrotic cells were identified by flow cytometric analyses of cells stained with 250 ng/ml Annexin V-FITC (Sigma) and 2.5 μg/ml propidium iodide, essentially as described (10).
Determination of NAD and ATP.
For NAD analysis, perchloric acid extracts of the cells were subjected to alcohol dehydrogenase-based microcycling assay (23). ATP was extracted from cells with 5% trichloroacetic acid and measured by using microcycling assay based on the Sigma ATP kit.
Results
Potential of PARP Activation Reaction to Cause Acidification.
DNA damage-activated PARP forms polymers of ADP-ribose from the substrate NAD and releases protons as a byproduct of the reaction. Hence, we examined whether pH would decrease with progress of PARP activation reaction in vitro (Fig. 1). Purified PARP was activated in vitro in the presence of nicked DNA in the assay mixture (23), from which Tris buffer was excluded, and initial pH was adjusted to 7.4. The pH of complete reaction mixture started decreasing after addition of NAD and reached ≈1 unit below the initial pH by 20 min (Fig. 1A). During this period, PARP (113 kDa) and two of its freeze-thaw fragments between 50 and 80 kDa (Fig. 1B, lane 3) were extensively modified with heterogeneous polymers of ADP-ribose (Fig. 1C, lane 4), resulting in their masking from PARP antibody (Fig. 1B, lane 4). Addition of 100 μM 1,5-dihydroxyisoquinoline (DHQ), an efficient inhibitor of PARP (26) completely suppressed acidification (Fig. 1A), as well as activation of PARP (lane 6 in Fig. 1 B and C). That acidification is a byproduct of reaction and not an essential requirement was demonstrated when addition of 100 mM Tris buffer, pH 7.4, completely suppressed acidification response (Fig. 1A) without affecting PARP activation (lane 8 in Fig. 1 B and C). Thus, protons released during PARP activation can potentially acidify the cells that are responding to DNA damage.
Figure 1.

Acidification during PARP activation reaction in vitro. (A) pH changes during PARP activation reaction. Purified PARP was activated for 20 min in the unbuffered complete assay (■), in which either 100 μM DHQ (▴) or 100 mM Tris, pH 7.4 (×) were added or PARP was excluded (○). Data (mean ± SD) was derived from three experiments, each carried out in duplicate. (B) PARP immunoblot. Samples from reactions in A at 0 and 20 min were immunoblotted for PARP. (C) Polymer immunoblot. Samples similar to B were immunoblotted with anti-polymer 10H. Blots in B and C represent one of three experiments with identical results.
MNNG-Induced Rapid Intracellular Acidification in Molt 3 and Other Cells.
MNNG is an alkylating agent that strongly activates PARP; therefore, it was examined whether MNNG-treated cells are acidified in the same time frame as PARP activation (Fig. 2). When exposed to high levels of DNA damage by 100 μM MNNG, intracellular pH of Molt 3 cells, measured by pH-sensitive dye, dropped rapidly from ≈7.3 to 6.6 within 1 h, with the major pH drop occurring in the first 30 min (Fig. 2A). Acidification was a reflection of the extent of DNA damage because it was dose-dependent from 10 to 100 μM MNNG (Fig. 2B), and it was also a general DNA damage response because exposure of cells to 300 μM H2O2 or 100 μM MNNG resulted in similar kinetics of acidification response (Fig. 2C). Acidification response to MNNG was also observed in other cell lines, namely, Jurkat, U937, and HL-60 (Fig. 2D) and also in PARP+/+ fibroblasts (see below). Thus, rapid intracellular acidification was a general DNA damage response that was not confined to specific damaging agent or a cell type.
Figure 2.

MNNG-induced acidification response. (A) Intracellular acidification in MNNG-treated Molt 3 cells. Changes in pH were monitored in BCECF-loaded control (○) or 100 μM MNNG-treated cells (□). Results (mean ± SD) were obtained from five experiments, each in triplicate. (B) Dose-dependent acidification response to MNNG. Molt 3 cells were treated as above with 10–100 μM MNNG for 30 min, and pH was measured. Results (mean ± SD) were obtained from two experiments, each in triplicate. (C) Acidification response to MNNG and H2O2. Molt 3 cells were treated for 1 h with 100 μM MNNG (○) or 300 μM H2O2 (□) as above, and pH changes were measured. Results (mean ± SD) were obtained from five experiments, each in triplicate. (D) MNNG-induced acidification in other cells. Jurkat, U937, and HL-60 cells were loaded with BCECF and treated with 100 μM MNNG for 30 min before analysis of pH. Results (mean ± SD) were obtained from two experiments, each in triplicate.
PARP Activation and Acidification in Response to DNA Damage with MNNG.
The extent of PARP activation depends on the level of DNA damage; therefore, we examined in greater detail PARP activation and acidification responses to 10 and 100 μM MNNG. PARP was weakly stimulated by 10 μM MNNG (Fig. 3A) with a correspondingly negligible drop in NAD and ATP levels (Fig. 3B). In contrast, PARP was strongly activated by 100 μM MNNG (Fig. 3A), resulting in a rapid and massive consumption of NAD and ATP (Fig. 3B). Thus, acidification and PARP activation were coincident after DNA damage.
Figure 3.

MNNG-induced PARP activation and acidification in Molt 3 cells. (A) Polymer immunoblot. Molt 3 cells were treated with 10 or 100 μM MNNG for a given time and immunoblotted with anti-polymer LP96–10. The blot represents one of the four experiments with identical results. (B) NAD and ATP depletion. Samples of Molt 3 cells, treated with 10 or 100 μM MNNG as above, were analyzed for NAD (○) or ATP (□). Results (mean ± SD) were obtained from four experiments, each in triplicate. (C) Time course of acidification. Molt 3 cells were treated with 10 (◊) or 100 (■) μM MNNG, and changes in pH were monitored by BCECF method up to 7 h. Results (mean ± SD) were obtained from four experiments, each in triplicate.
Cells are equipped to eliminate proton loads, and we examined whether MNNG-induced acidification was rapidly reversed (Fig. 3C). Cells treated with 10 μM MNNG rapidly reversed the initial acidification by 0.15 pH units within 1 h. In contrast, cells treated with 100 μM MNNG remained well below physiological pH by 0.3 units until 7 h, despite a small recovery after initial acidification by ≈0.5 pH units. The pH could not be accurately measured after 7 h in 100 μM MNNG-treated cells because of ongoing cell death (see below). Thus, rapid acidification that occurred in response to higher levels of DNA damage by MNNG was more persistent.
Decrease in Acidification Response by PARP Inhibition or Absence of PARP.
The role of PARP activation in acidification response to MNNG was examined by using PARP inhibitors in Molt 3 cells or by using PARP−/− cells (Fig. 4). Pretreatment of Molt 3 cells with PARP inhibitor DHQ prevented PARP activation (Fig. 4A) and also suppressed nearly 60–65% of the 30-min acidification response to 100 μM MNNG or 300 μM H2O2 (Fig. 4B). DHQ treatment by itself was responsible for a small amount of this residual acidification (Fig. 4B). DHQ was much more effective in inhibiting MNNG-induced acidification if it was added during the course of acidification (see below).
Figure 4.

Role of PARP in acidification response. (A) Suppression of PARP activation with DHQ. Molt 3 cells were exposed to 100 μM MNNG after 5-min pretreatment with 100 μM DHQ and immunoblotted for polymer with LP96–10. This blot represents one of the four experiments with identical results. (B) Suppression of acidification with PARP inhibitor. BCECF-loaded Molt 3 cells were exposed to 100 μM MNNG or 300 μM H2O2 with or without 5-min pretreatment with 100 μM DHQ, and changes in pH were measured at 30 min. Results (mean ± SD) were obtained from four experiments, each in triplicate. (C) MNNG-induced polymer synthesis in PARP+/+ and PARP−/− fibroblasts. Cells with two PARP genotypes were treated with 300 μM MNNG and immunoblotted with anti-polymer LP96–10. This blot represents one of the three experiments with identical results. (D) MNNG-induced acidification in PARP+/+ and PARP−/− fibroblasts. The pH changes in BCECF-loaded cells were measured at 30 min after exposure to 300 μM MNNG. Results (mean ± SD) were obtained from two experiments, each in triplicate.
PARP−/− cells respond to MNNG-induced DNA damage with a weak synthesis of polymer caused by action of PARP-2, a homolog of PARP (27, 28). Therefore, acidification response was compared in PARP+/+ and PARP−/− cells exposed to 300 μM MNNG, a level of DNA damage that stimulates both PARP and PARP-2. PARP+/+ cells rapidly activated PARP (Fig. 4C) and were acidified by 0.5 pH units within 30 min (Fig. 4D). In contrast, there was a weak accumulation of polymer-modified proteins (Fig. 4C) and very weak acidification response in PARP−/− cells (Fig. 4D). Thus, high levels of acidification required strong catalytic activation of PARP.
Role of PARP and NHE in MNNG-Induced Acidification Response.
Our results with PARP activation in vitro suggest that PARP activation may be directly contributing protons that cause acidification, whereas earlier studies suggested that inhibition of NHE is the principal cause of oxidant-induced acidification (15–17). Because NHE activity is reduced when cellular ATP levels are depleted (29), it has also been argued that PARP activation may be causing acidification only through ATP depletion-mediated inhibition of NHE (15). To distinguish between these two roles of PARP, we first determined the functional state of NHE in Molt 3 cells. This is measured by their capacity to restore pH after introduction of a proton load from NH4Cl (16, 17). Acid-loaded Molt 3 cells could stage a rapid recovery of pH within 15 min after removal of NH4Cl, and this recovery was suppressed by 5-N-ethyl-N-isopropyl amiloride (EIPA), a strong inhibitor of NHE (Fig. 5A). Thus, Molt 3 cells have an efficient NHE that can rapidly export a large flux of protons from NH4Cl. However, these cells do get acidified after MNNG exposure, suggesting that MNNG treatment could be inhibiting the proton-extruding capacity of NHE.
Figure 5.

Role of PARP and NHE in MNNG-induced acidification in Molt 3 cells. (A) pH recovery after acid loading with NH4Cl. BCECF-loaded cells were acidified with an NH4Cl pulse, and pH recovery was monitored in the absence (◊) or presence (□) of 10 μM of NHE inhibitor EIPA. Results (mean ± SD) were obtained from two experiments, each in quadruplicate. (B) Effect of inhibitors of PARP and NHE on MNNG-induced acidification. BCECF-loaded cells were treated with 100 μM MNNG for 30 min and incubated for 30 more min without any inhibitor (◊, M) or with 10 μM EIPA (□) or 100 μM DHQ (▵) before measurement of pH. Results (mean ± SD) were obtained from three experiments, each in triplicate. (C) Acidification effect of second MNNG treatment. BCECF-loaded Molt 3 cells were treated with 100 μM MNNG for 30 min before a second treatment for 30 min with 100 μM MNNG alone (◊, M) or with 10 μM EIPA (□) or 100 μM DHQ (▵). Results (mean ± SD) were obtained from three experiments, each in triplicate. (D) Lack of acidification by NHE inhibition per se. The pH changes in BCECF-loaded Molt 3 cells were monitored in the absence (◊) or presence (□) of 10 μM EIPA. Results (mean ± SD) were obtained from six experiments, each in triplicate.
Therefore, the functional state of NHE was examined at the peak of the acidification response to MNNG, i.e., at 30 min after exposure to 100 μM MNNG (Fig. 5B). At this stage, the initial acidification response was not complete, and cells acidified further by 0.1 pH units from 30 to 60 min. PARP inhibitor DHQ suppressed this acidification response, suggesting that PARP was actively contributing to acidification. If NHE was fully suppressed at this stage, then any further inhibition of NHE with EIPA should not make any difference to pH. However, there was a small increase in the acidification response by addition of EIPA, suggesting that even at the peak of the acidification response, NHE retained some function of extruding protons.
We then introduced additional DNA damage with a second exposure to 100 μM MNNG at 30 min after first exposure and analyzed acidification response with or without inhibitors of PARP and NHE (Fig. 5C). In absence of any inhibitor, there was additional acidification by 0.15 pH units (Fig. 5C), confirming that acidification was indeed a response to DNA damage. Addition of PARP inhibitor DHQ almost completely suppressed the renewed acidification response, strongly supporting the argument that PARP activation was the primary cause of this acidification. At this stage, NHE activity was fully suppressed because addition of NHE inhibitor EIPA could not increase the acidification response. This experiment clearly dissociated direct contribution of PARP activation as a proton source from its effect through NHE inhibition. If NHE inhibition was the only contribution of PARP toward acidification, then the second dose of MNNG could not have caused additional acidification because NHE was fully inhibited in these cells. However, renewed acidification after a second exposure to MNNG could be caused by PARP activation generating additional protons, which could not be exported from the cell.
Finally, we confirmed that NHE inhibition per se was incapable of causing acidification of Molt 3 cells up to 60 min without DNA damage by MNNG (Fig. 5D). Thus, even with total inhibition of NHE, normal cellular metabolism does not generate a sufficient amount of protons to cause acidification of Molt 3 cells. In contrast, when DNA damage was introduced with 100 μM MNNG after 5 min of treatment with EIPA, acidification response at 60 min was quite similar to that observed with MNNG alone, as shown in Fig. 2A. Therefore, once the cells were acidified by a metabolism, such as PARP activation, inhibition of NHE could play a crucial role in maintaining the acidified state by not exporting the protons.
Vacuolar H+-ATPase (30) and mitochondrial F0/F1 H+-ATPase (31) are the proton transporters that play a crucial role in acidification and cell death, and we examined their contribution to MNNG-induced acidification. Their inhibition with bafilomycin A1 or oligomycin, respectively, caused ≈10% inhibition in 100 μM MNNG-induced acidification response at 30 min (data not shown), suggesting a minor role for these two proton exchangers in MNNG-induced acidification. Collectively, our results demonstrate that PARP activation and inhibition of proton export by ATP-dependent NHE together account for most of the acidification response to MNNG.
Impact of MNNG-Induced Acidification on the Mode of Cell Death.
Our results with MNNG and earlier studies with oxidants clearly establish that DNA-damaged cells undergo rapid intracellular acidification; therefore, we examined whether acidification per se has any influence on the downstream events after DNA damage. Because acidosis or holding cells in acidic medium can induce cell death by apoptosis or necrosis (18, 32–34), impact of MNNG-induced acidification on the mode of cell death was examined by using the pH clamp method to either suppress or introduce acidification. Molt 3 cells were clamped at pH 7.4 and exposed to 100 μM MNNG to prevent the early acidification response. For comparison, another set of Molt 3 cells was clamped at pH 6.8 that mimics acidification following high levels of DNA damage, and these cells were exposed to a low level of DNA damage by 10 μM MNNG. In both models, pH clamps efficiently maintained the desired pH at 0, 15, 30, and 60 min after exposure to MNNG. The pH clamps did not significantly interfere with PARP activation, as PARP was weakly activated by 10 μM MNNG and strongly activated by 100 μM MNNG (Fig. 6A). We confirmed the extent of PARP activation with corresponding changes in the cellular NAD pools (data not shown). Hence, PARP activation was nearly identical in the cells with or without pH clamp, ensuring that PARP-mediated NAD and ATP depletion were dissociated from the acidification response.
Figure 6.

Impact of acidification on mode of cell death. (A) PARP activation in pH-clamped cells. Molt 3 cells were treated for 60 min with 10 μM MNNG without pH clamp, as in Fig. 3A, or with pH 6.8 clamp (lanes 1–5). Another set of cells was treated with 100 μM MNNG without pH clamp, as in Fig. 3A, or with pH 7.4 clamp (lanes 6–10). Samples were immunoblotted with antipolymer LP96–10. (B) Flow cytometry analysis of mode of cell death. Cells treated for 1 h with 10 or 100 μM MNNG with or without pH clamp were allowed to recover for 10 h, stained with annexin V-FITC and propidium iodide, and analyzed by flow cytometry. The viable cells were identified by low signals for both the dyes, whereas apoptotic cells were detected by exclusion of propidium iodide and staining with annexin V. In contrast, necrotic cells were detected by high uptake of both the dyes. (C) Caspase 3-immunoblot analysis of cell death. Cells treated as described in B were immunoblotted for caspase 3. All lanes marked C represent DMSO-treated controls, and etoposide-treated HL-60 cells were used as positive apoptosis control in both C and D (lane 9). (D) PARP immunoblot analysis of cell death. Cells treated as described in B were also immunoblotted for PARP. All data represent one of three experiments with identical results.
To examine the impact of initial acidification on eventual cell death, the pH clamp was lifted after 60 min, and cells were allowed to recover for 10 h in the culture medium. The specific mode of cell death by apoptosis or necrosis was examined by dual staining with propidium iodide and annexin V-FITC (Fig. 6B), activation of apoptotic caspase 3 (Fig. 6C), and formation of unique apoptotic or necrotic fragments of PARP (Fig. 6D) (21). The pH clamp procedure per se was not lethal. The pH-clamped control cells, like the untreated controls, were predominantly viable (Fig. 6B) with the absence of active caspase 3 (Fig. 6C, lanes 1, 3, 5, and 7) and presence of intact PARP (Fig. 6D, lanes 1, 3, 5, and 7). Treatment of cells with 10 μM MNNG without pH clamp induced apoptosis, identified by a shift of 16% cells to apoptotic fraction (Fig. 6B), activation of caspase 3 (Fig. 6C, lane 2), and formation of an 89-kDa apoptotic fragment of PARP (Fig. 6D, lane 2). Interestingly, forced acidification of these cells to pH 6.8 significantly suppressed apoptotic response with only 2% cells in apoptotic fraction (Fig. 6B), accompanied by a significant decrease in both activated caspase 3 (Fig. 6C, lane 4) and the 89-kDa fragment of PARP (Fig. 6D, lane 4).
Cells treated with 100 μM MNNG without pH clamp were undergoing necrotic death, with 53% necrotic and only 0.8% apoptotic cells (Fig. 6B). Absence of apoptosis and predominance of necrosis was confirmed by lack of activated caspase 3 (Fig. 6C, lane 6) and presence of a unique necrosis-associated ≈50-kDa fragment of PARP (Fig. 6D, lane 6) (21). When acidification was suppressed with a pH 7.4 clamp, 49% of the cells continued to undergo necrotic death. However, there was a 4-fold increase in apoptotic population (3.3% apoptosis) (Fig. 6B), which was evident from activation of caspase 3 (Fig. 6C, lane 8) and additional cleavage of PARP to the apoptotic 89-kDa fragment (Fig. 6D, lane 8). Thus, DNA damage-induced early acidification may be functioning as a negative regulator of apoptotic death in cells with damaged DNA.
Discussion
In the present study, we show that different types of leukemia cells or PARP+/+ fibroblasts exposed to alkylating DNA damage by MNNG undergo rapid intracellular acidification. Our results concur with and broaden the scope of earlier observations that oxidant exposure causes rapid acidification in specific cells from heart, kidney, or brain (14–20). Collectively, our results suggest that acidification is not confined to oxidant damage, but it may be a more general DNA damage response that is linked to PARP activation. Although cells readily recover from a smaller extent of acidification induced by lower levels of DNA damage, they do not recover from higher and persistent acidification induced by extensive DNA damage, which can influence subsequent cell death events.
We show that DNA damage-induced activation of PARP and inhibition of Na+/H+ exchangers together are the major causes of MNNG-induced acidification. This finding is in contrast to the earlier suggestion that inhibition of NHE is the main cause for oxidant-induced acidification (15–17). We clearly show that NHE inhibition alone cannot cause acidification in Molt 3 cells if it is not accompanied by DNA damage. However, NHE activity is significantly suppressed when cells are depleted of ATP, and hence at this stage, inhibition of NHE could play a critical role in maintaining acidic pH by not exporting protons. The results with PARP inhibitor and PARP−/− cells show that PARP plays a crucial dual role in MNNG-induced acidification. It initiates acidification after DNA damage by release of protons from NAD, as demonstrated in the in vitro PARP activation reaction, and it blocks proton export by NHE via depletion of ATP. Therefore, in response to lower levels of DNA damage, weaker PARP activation will cause smaller extent of acidification, and because NAD and ATP levels rapidly recover, acidification will be rapidly reversed by action of NHE. In contrast, extensive PARP activation in response to higher levels of DNA damage will cause a higher extent of acidification, and because ATP levels do not recover in these cells, prolonged inhibition of NHE will cause long-term acidification.
Other factors may play a minor role in MNNG-induced acidification, such as vacuolar proton-ATPase and mitochondrial F0/F1 proton-ATPase, which release protons from acidic vacuoles and mitochondria, respectively. Inhibition of glycolysis and consequent hydrolysis of ATP has also been suggested to cause oxidant-induced acidification (14). Because PARP activation also depletes ATP, inhibition of various ATP-driven reactions including other proton exporters could independently cause DNA damage-induced acidification. But we have shown that significant ATP depletion occurs at about 40 min, whereas acidification begins almost immediately after DNA damage.
It is interesting to compare the impact on the necrotic mode of cell death by PARP inhibitors, which suppress both acidification and ATP depletion, versus the pH clamp procedure, which suppresses only the acidification. PARP inhibitors prevent oxidant-induced necrotic death and force a switch from necrosis to apoptosis (10–13), and we confirmed this switch in mode of cell death in 100 μM MNNG-treated Molt 3 cells (data not shown). In contrast, suppression of only the acidification response while permitting PARP activation does not significantly suppress necrosis, but it does allow apoptotic death for some of the cells. Therefore, in response to necrotic DNA damage, ATP depletion is the major determinant of necrotic death (8), whereas acidification may have a supporting role of a negative regulator of apoptotic death, which was most clearly demonstrated with a pH 6.8 clamp during apoptosis induced by 10 μM MNNG.
It needs to be stressed that DNA damage-induced early acidification, which occurs during cell death by apoptosis or necrosis, is different from apoptosis-associated acidification, which occurs much later at 2–14 h along with caspase activation and apoptotic DNA fragmentation (35, 36). Other studies have addressed the role of acidification in cell death by holding the cells in acidic medium or by suppressing apoptotic acidification. In some of these studies, acidosis was shown to induce or facilitate apoptosis (18, 32, 33) or potentiate oxidant-induced death (34). In others, acidosis was shown to suppress perfusion-induced cell killing (37) or γ radiation-induced apoptosis (38). In these studies, acidification from pH 6.2 to 7.0 units was applied from a few minutes to several hours, and mode of death was not always clearly established. Our results suggest that DNA damage-induced early acidification suppresses apoptosis and is permissive for necrotic death.
Acknowledgments
We acknowledge the generous gift of PARP fibroblasts from Z. Wang, the 10H antibody from M. Miwa and A. Burkle, and the anticaspase antibody from D. Nicholson. E.B.A. is grateful to G. Poirier for his support during the preliminary discussions. This work was supported by Natural Sciences and Engineering Research Council Grant 155257-01 and Medical Research Council Grant MA 15023. G.M.S. is the recipient of the Chercheur-Boursier Award from Fonds de la Recherche en Santé du Québec.
Abbreviations
- PARP
poly(ADP-ribose) polymerase
- NAD
β-nicotinamide adenine dinucleotide
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
- NHE
Na+/H+ exchanger
- BCECF
2′,7′-bis-(2-carboxyethyl)-5-(6)-carboxyfluorescein
- DHQ
1,5-dihydroxyisoquinoline
- EIPA
5-N-ethyl-N-isopropyl amiloride
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Amé J C, Jacobson E L, Jacobson M K. In: From DNA Damage and Stress Signalling to Cell Death: Poly ADP-Ribosylation Reactions. de Murcia G, Shall S, editors. New York: Oxford Univ. Press; 2000. pp. 1–34. [Google Scholar]
- 2.Herceg Z, Wang Z Q. Mutat Res. 2001;477:97–110. doi: 10.1016/s0027-5107(01)00111-7. [DOI] [PubMed] [Google Scholar]
- 3.Shall S, de Murcia G. Mutat Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- 4.Pieper A A, Verma A, Zhang J, Snyder S H. Trends Pharmacol Sci. 1999;20:171–181. doi: 10.1016/s0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- 5.Le Rhun Y, Kirkland J B, Shah G M. Biochem Biophys Res Commun. 1998;245:1–10. doi: 10.1006/bbrc.1998.8257. [DOI] [PubMed] [Google Scholar]
- 6.Berger N A. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- 7.Schraufstatter I U, Hyslop P A, Hinshaw D B, Spragg R G, Sklar L A, Cochrane C G. Proc Natl Acad Sci USA. 1986;83:4908–4912. doi: 10.1073/pnas.83.13.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ha H C, Snyder S H. Proc Natl Acad Sci USA. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liaudet L, Soriano F G, Szabo E, Virag L, Mabley J G, Salzman A L, Szabo C. Proc Natl Acad Sci USA. 2000;97:10203–10208. doi: 10.1073/pnas.170226797. . (First Published August 22, 2000; 10.1073/pnas 170226797) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Virag L, Scott G S, Cuzzocrea S, Marmer D, Salzman A L, Szabo C. Immunology. 1998;94:345–355. doi: 10.1046/j.1365-2567.1998.00534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watson A J M, Askew J N, Benson R S P. Gastroenterology. 1995;109:472–482. doi: 10.1016/0016-5085(95)90335-6. [DOI] [PubMed] [Google Scholar]
- 12.Filipovic D M, Meng X, Reeves W B. Am J Physiol. 1999;277:F428–F436. doi: 10.1152/ajprenal.1999.277.3.F428. [DOI] [PubMed] [Google Scholar]
- 13.Walisser J A, Thies R L. Exp Cell Res. 1999;251:401–413. doi: 10.1006/excr.1999.4589. [DOI] [PubMed] [Google Scholar]
- 14.Wu M L, Tsai K L, Wang S M, Wu J C, Wang B S, Lee Y T. Circ Res. 1996;78:564–572. doi: 10.1161/01.res.78.4.564. [DOI] [PubMed] [Google Scholar]
- 15.Hu Q, Xia Y, Corda S, Zweier J L, Ziegelstein R C. Circ Res. 1998;83:644–651. doi: 10.1161/01.res.83.6.644. [DOI] [PubMed] [Google Scholar]
- 16.Cutaia M, Parks N. Am J Physiol. 1994;267:L649–L659. doi: 10.1152/ajplung.1994.267.6.L649. [DOI] [PubMed] [Google Scholar]
- 17.Kaufman D S, Goligorsky M S, Nord E P, Graber M L. Arch Biochem Biophys. 1993;302:245–254. doi: 10.1006/abbi.1993.1206. [DOI] [PubMed] [Google Scholar]
- 18.Vincent A M, TenBroeke M, Maiese K. J Neurobiol. 1999;40:171–184. doi: 10.1002/(sici)1097-4695(199908)40:2<171::aid-neu4>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 19.Siesjo B K, Katsura K, Kristian T. Adv Neurol. 1996;71:209–233. [PubMed] [Google Scholar]
- 20.Wu M L, Chen J H, Chen W H, Chen Y J, Chu K C. Am J Physiol. 1999;277:C717–C727. doi: 10.1152/ajpcell.1999.277.4.C717. [DOI] [PubMed] [Google Scholar]
- 21.Shah G M, Shah R G, Poirier G G. Biochem Biophys Res Commun. 1996;229:838–844. doi: 10.1006/bbrc.1996.1889. [DOI] [PubMed] [Google Scholar]
- 22.Halappanavar S S, Le Rhun Y, Mounir S, Martins M, Huot J, Earnshaw W C, Shah G M. J Biol Chem. 1999;274:37097–37104. doi: 10.1074/jbc.274.52.37097. [DOI] [PubMed] [Google Scholar]
- 23.Shah G M, Poirier D, Duchaine C, Brochu G, Desnoyers S, Lagueux J, Verreault A, Hoflack J C, Kirkland J B, Poirier G G. Anal Biochem. 1995;227:1–13. doi: 10.1006/abio.1995.1245. [DOI] [PubMed] [Google Scholar]
- 24.Chow S, Hedley D. In: Current Protocols in Cytometry. Robinson J P, Darzynkiewcz Z, Dean P N, Orfao A, Rabinovitch P S, Stewert C C, Tanke H J, Wheeless L L, editors. New York: Wiley; 1997. pp. 9.3.1–9.3.10. [Google Scholar]
- 25.Kawamitsu H, Hoshino H, Okada H, Miwa M, Momoi H, Sugimura T. Biochemistry. 1984;23:3771–3777. doi: 10.1021/bi00311a032. [DOI] [PubMed] [Google Scholar]
- 26.Banasik M, Komura H, Shimoyama M, Ueda K. J Biol Chem. 1992;267:1569–1575. [PubMed] [Google Scholar]
- 27.Shieh W M, Amé J C, Wilson M V, Wang Z Q, Koh D W, Jacobson M K, Jacobson E L. J Biol Chem. 1998;273:30069–30072. doi: 10.1074/jbc.273.46.30069. [DOI] [PubMed] [Google Scholar]
- 28.Ame J C, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Murcia J M, de Murcia G. J Biol Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 29.Brown S E, Heming T A, Benedict C R, Bidani A. Am J Physiol. 1991;261:C954–C963. doi: 10.1152/ajpcell.1991.261.6.C954. [DOI] [PubMed] [Google Scholar]
- 30.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. Cancer Res. 2001;61:439–444. [PubMed] [Google Scholar]
- 31.Matsuyama S, Llopis J, Deveraux Q L, Tsien R Y, Reed J C. Nat Cell Biol. 2000;2:318–325. doi: 10.1038/35014006. [DOI] [PubMed] [Google Scholar]
- 32.Ding D, Moskowitz S I, Li R, Lee S B, Esteban M, Tomaselli K, Chan J, Bergold P J. Exp Neurol. 2000;162:1–12. doi: 10.1006/exnr.2000.7226. [DOI] [PubMed] [Google Scholar]
- 33.Park H J, Lyons J C, Ohtsubo T, Song C W. Br J Cancer. 1999;80:1892–1897. doi: 10.1038/sj.bjc.6690617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ying W, Han S K, Miller J W, Swanson R A. J Neurochem. 1999;73:1549–1556. doi: 10.1046/j.1471-4159.1999.0731549.x. [DOI] [PubMed] [Google Scholar]
- 35.Gottlieb R A, Nordberg J, Skowronski E, Babior B M. Proc Natl Acad Sci USA. 1996;93:654–658. doi: 10.1073/pnas.93.2.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf C M, Reynolds J E, Morana S J, Eastman A. Exp Cell Res. 1997;230:22–27. doi: 10.1006/excr.1996.3401. [DOI] [PubMed] [Google Scholar]
- 37.Currin R T, Gores G J, Thurman R G, Lemasters J J. FASEB J. 1991;5:207–210. doi: 10.1096/fasebj.5.2.2004664. [DOI] [PubMed] [Google Scholar]
- 38.Park H, Lyons J C, Griffin R J, Lim B U, Song C W. Radiat Res. 2000;153:295–304. doi: 10.1667/0033-7587(2000)153[0295:aaccpi]2.0.co;2. [DOI] [PubMed] [Google Scholar]