

Abstract
Sequence evolution of the reverse transcriptase (RT) gene in retrotransposons belonging to the Ty1-copia class was studied in 11 plant species. Phylogenetic reconstruction of the evolutionary history of RT sequences indicated a strong pattern of purifying selection, manifested as high ratios of third to first plus second codon position substitutions, and low ratios of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site, especially in internal portions of the element phylogenies. Evidence of purifying selection was most pronounced in plant species with low estimated copy numbers of Ty1-copia elements. This finding is consistent with the hypothesis that high element turnover rates (e.g., caused by high rates of element loss and selection against high element copy number) favors elements capable of transposition. Simulations of RT sequence evolution were conducted to help verify the logical validity of this conclusion. The results argue that it is incorrect to assume that low copy numbers of transposable elements are the product of reduced levels of element activity.
Retrotransposons are mobile genetic elements that replicate by reverse transcription (1). They are widespread in plant and animal genomes and are thought to be important contributors to genome evolution by causing a broad spectrum of mutations, reorganizing genomes, and contributing to the physical size of the host genome (2). The advent of PCR methods for amplifying conserved regions of retrotransposons (3, 4) has led to the proliferation of large numbers of molecular level studies of these elements, especially of Ty1-copia class of retrotransposons in plants (5–9). Many investigations have focused on determining the chromosomal position of retroelements by using in situ hybridization and on copy number estimates through Southern blot and slot blot analyses. It is now appreciated that a large fraction of the repeat portions of many plant genomes are comprised of retroelements (10–12). The copy number of copia-like retrotransposons in plants has been estimated to vary over 4 orders of magnitude, from several hundred elements per genome in Arabidopis thaliana to around one million in Vicia faba (1).
One region of these elements, that corresponding to a portion of the reverse transcriptase (RT) gene, has been the subject of many analyses of sequence evolution. RT sequences in plants exhibit a high degree of diversity (1), and the level of sequence heterogeneity is correlated with the total number of elements per genome in some plant groups (13). To account for these observations, Flavell et al. (13) proposed that retrotransposon copy number has historically experienced varying degrees of selective constraint in different host species. For example, in host species where smaller genomes have been favored, perhaps as a consequence of selection for rapid cell division (14, 15), genomic numbers of retrotransposons may have become reduced. In contrast, in host species where selection against retroelements has been relaxed, the expansion of element copy number may have occurred. According to this hypothesis, when there has been selection for reduced numbers of retroelements, defective elements, which are incapable of transposition, should be rapidly eliminated from the genome compared with transposition-proficient elements. The latter are more likely to escape selective removal as a result of their ability to increase in numbers through transposition. Thus, when examining sequence evolution of genes involved in transposition, the model predicts that purifying selection (e.g., as evidenced by low ratios of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site, and low incidence of stop codons in sampled RT sequences) should be most pronounced in elements that occur within host genomes harboring relatively few elements (16).
Purifying selection of transposable element (TE) sequences has, in fact, been documented in a number of cases, but it has rarely been related to the population biology of the elements. For example, by conducting phylogenetic analyses of retroelements belonging to the non-long terminal repeat group Helena in Drosophila, Petrov and Hartl (17) were able detect a pattern of selection against nonsynonymous substitutions in active element lineages. In yeast, a similar examination of the pattern of nucleotide substitution in the phylogeny of Ty1 and Ty2 retroelements suggested that selection against nonsynonymous substitutions has occurred in active element lineages (16). Although these investigations demonstrate that inferences about TE population history can be made by examining patterns of sequence evolution, there have been no comparative evolutionary investigations of element sequences in host genomes harboring many versus few TEs. In this article we examine patterns of nucleotide substitution in the evolutionary histories of Ty1-copia retrotransposons that occur in the genomes of different plant species, using data published in GenBank, supplemented with additional copia-RT sequence data obtained in the lab. Relative levels of past purifying selection in RT sequences are inferred from these analyses. The comparison focuses especially on contrasts between plant genomes exhibiting low versus high Ty1-copia element copy number. Our findings are interpreted with the aid of simulation models that examine how copia-RT sequences evolve when forces that reduce TE number vary.
Materials and Methods
Copia-RT Sequence Data for Plant Species.
Copia elements in plant species were chosen for analysis because of the wide range of copy numbers reported. For all species except A. thaliana and Zea mays, copia-RT sequences were extracted from GenBank between November 1, 2000 and February 15, 2001 (Table 1). The RT sequence data were obtained originally by different investigators through direct sequencing of PCR-amplified portion of the RT gene (3, 4). Because of the potential for selective amplification by PCR, it is possible that the RT sequences available on GenBank represent a nonrandom subset of the entire population of RT sequences in the host genomes analyzed.
Table 1.
Copia-RT sequences sampled and Jukes–Cantor corrected estimates of nucleotide diversity (πJC)
| Host genome | N* | GenBank accession number | πJC | Reference |
|---|---|---|---|---|
| A. thaliana | 43 | TE Database† | 0.76 | TE Database† |
| H. vulgare | 22 | AJ241322–343 | 0.68 | 18 |
| L. esculentum | 25 | AJ228804–805, D12844–845, AF072637–656 | 0.56 | 19 |
| O. sativa | 51 | Z75496–518, AB017978–18000, M94492 | 0.54 | 20 |
| P. abies | 51 | AJ288018–053, AJ288171–181, AJ290660–676, AJ224363–368 | 0.53 | N. Friesen, personal communication, 8 |
| P. sativum | 31 | AJ405217–247 | 0.80 | 21 |
| V. faba | 27 | AJ239201–207, AJ239227 | 0.68 | 6 |
| V. melanops | 25 | AJ239468–494 | 0.68 | 6 |
| V. sativa | 27 | AJ239495–521 | 0.74 | 6 |
| T. aestivum | 74 | AJ241094–115, D90618–671 | 0.49 | 22 |
| Z. mays | 28 | D12830, AF398186–213 | 0.55 | 37, this study |
Number of sequences analyzed.
For A. thaliana, RT sequences were obtained for copia-like elements that were previously identified by sequence similarity searches of the Arabidopsis Genome Project (23). For studies in maize, the number of RT sequences available on GenBank was insufficient for our study, and so PCR cloning was used to obtain additional information for this species. DNA was extracted from Z. mays cv. Seneca horizon with a Qiagen (Chatsworth, CA) DNAEasy plant mini-kit. Amplification of the RT-conserved domain was done following the protocol described by Voytas et al. (4). PCR products were ligated into pCR 2.1 and subcloned into INV-F′-TOP10 competent cells (Invitrogen). Positive colonies were selected, and the plasmids were extracted by alkaline lysis. Plasmids were digested with the appropriate restriction enzyme for cutting out the inserted PCR product of approximately 280–300 bp. Plasmid DNA was purified with QIAprep Spin miniprep kits (Qiagen). Sequencing reactions were done with a Sequitherm Excell II-Kit LC (Epicentre Technologies, Madison, WI), using labeled M13 primers from Li-Cor (Lincoln, NE). Sequencing was performed with a Li-Cor-4200 Long ReadIR automated sequencer.
Copy Number Estimates for Ty1-copia Sequences.
Copy number estimates for Ty1-copia elements were obtained from published reports and are based on results obtained by using slot blots, Southern blots, and hybridization to bacterial artificial chromosome genomic libraries (Table 2). Slot blots are the most accurate of these techniques, because the estimates are calibrated by using known concentrations of DNA (6). Southern blots are most appropriate when estimating lower copy numbers, in the range of hundreds or fewer elements per genome (24). Hybridization to bacterial artificial chromosome genomic libraries gives estimates of copy number by enumerating the number of colonies to which the RT probe hybridizes, but the method cannot distinguish the presence of more than one element in each colony. For A. thaliana, the existence of genome sequence and sequence similarity searches that have identified most known TEs in this species (23) allowed us to directly estimate the number of Ty1-copia elements in noncentromeric regions.
Table 2.
Copy number estimates for Ty1-copia elements
| Species | Copy number | Method of estimation | Reference |
|---|---|---|---|
| O. sativa | 100 | Hybridization to BAC library | 20 |
| A. thaliana | 310 | Sequence similarity | TE Database† |
| V. melanops | 1,000 | Slot blot | 6 |
| L. esculentum | 2,500 | Slot blot | 19 |
| P. sativum | 4,000 | Hybridization to BAC library | 21 |
| V. sativa | 5,000 | Slot blot | 6 |
| Z. mays | 50,000 | Southern blot | 1 |
| T. aestivum | 80,000 | Slot blot | 37 |
| H. vulgare | 196,000 | Slot blot | 37 |
| P. abies | 0.5–1.0 × 106* | Southern blot | 38 |
| V. faba | 106 | Slot blot | 6 |
Species listed in order of increasing copy number. BAC, bacterial artificial chromosome.
Based on the estimate that 10–20% of the genome is composed of copia-like elements.
Because of the problems of comparing copy number across species (e.g., hybridization conditions that vary among investigations), the copy number estimates presented here are viewed as qualitative—capable of distinguishing high versus low copy numbers for copia-like elements. The comparisons below, therefore, are based on classifying the copy numbers as “low” (≤ 10,000 elements per genome) or as “high” (> 10,000 elements genome), which splits the plant species included in the study into two roughly equal-sized classes. Additional analyses were performed based on the relative ranking of the copy number estimates.
Sequence Alignment, Polymorphism Analysis, and Phylogenetic Reconstruction.
For elements from each plant species, multiple sequence alignments were done with clustalx (25), using the default gap insertion/extension penalties. Further alignment was done with the program pileup (GCG, version 10.0), using the translated amino acid product described by Xiong and Eickbush (26) and Voytas et al. (4) as a guide. Consensus sequences were obtained with the program consensus (GCG, version 10.0). The program dnasp version 3.0 (27) was used to calculate Jukes–Cantor corrected estimates of nucleotide diversity of the RT sequences.
Phylogenetic reconstruction of the aligned sequences was done by using maximum parsimony and maximum likelihood, with PAUP* 4.0b8 (28), using the HKY85 model (29) of nucleotide substitution. For maximum parsimony tree reconstruction, nucleotide positions were accorded equal weight, and heuristic searches were performed with tree bisection-reconnection branch swapping and 100 random stepwise addition replicates. The score of the best tree was retained, and an additional heuristic search with tree bisection-reconnection branch swapping was done, filtering all candidate trees for minimum score.
Analysis of Synonymous and Nonsynonymous Sequence Evolution.
As discussed by Petrov and Hartl (17), when sampled RT sequences are derived from different elements, nucleotide substitutions that fall in the internal branches of the element phylogeny (i.e., shared by two or more element sequences) most likely have occurred in a transpositionally active lineage of elements. On the other hand, nucleotide substitutions in the terminal branches of the phylogeny could have occurred either in an actively transposing lineage, or in a lineage in which there has been no transposition. Thus, nucleotide substitutions in genes that are involved in transposition and that map to internal branches of the element phylogeny are expected to be more highly constrained by the action of natural selection on the RT sequence than those occurring along terminal branches. For this reason, the analysis of purifying selection described below focuses on sequence evolution along the internal branches.
Maximum parsimony was used to reconstruct sequences at internal nodes of the phylogeny and to map the nucleotide changes occurring in internal and terminal branches (17). Base substitutions were classified as first, second, and third codon position changes. Because most third-position substitutions are synonymous, whereas most first- and second-position substitutions are nonsynonymous, an excess of third-position substitutions suggests that the sequence in question is evolving under selective constraint. Deviations from equal numbers of changes at the three codon positions were evaluated by χ2 tests.
Codon-based analysis of sequence evolution, as implemented in the program codeml in the package paml (phylogenetic analysis by maximum likelihood), version 3.0a (30), was used to estimate the ratio of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site, dN/dS. This procedure estimates the ratio dN/dS for the entire phylogeny of the elements or for specific branches. The maximum-likelihood trees found by using paup was used in this portion of the analysis. Different tree topologies were compared by using the Kishino–Hasegawa test (31).
A potential problem in estimating dN/dS ratios for divergent sequences such as the RT loci analyzed in this study is the possibility that multiple substitutions per site may lead to saturation of the estimate of dS (i.e., the estimate of dS will be less than its actual value) (32). Estimation of dN/dS, ratios using a phylogenetic approach, however, helps to ameliorate this problem, as estimates are obtained along shorter segments of the phylogenetic tree connecting the divergent sequences from one another, and when there are many such sequences per tree (as in the data analyzed here), there are many relatively short branch segments per tree.
The ratio dN/dS was estimated: (i) separately for internal and terminal branches; and (ii) globally (single dN/dS for the entire tree) as described by Yang (30). The log likelihood ratio test was applied to determine whether separate estimates of dN/dS for internal and terminal branches led to a significant improvement in the likelihood. The test was also used to determine whether dN/dS ratios differ from 1.0; i.e., log likelihoods were compared when dN/dS ratios were estimated and when dN/dS was constrained to a value of 1.0. The test statistic is 2ΔL = 2 (Li − Lj), where i and j (i > j) are the number of dN/dS ratios estimated, is distributed as χ2 with i-j degree of freedom. When the likelihood ratio test statistic for the comparison of global dN/dS ratios versus separate internal and terminal dN/dS ratios was insignificant, the global dN/dS ratio was interpreted to be the best estimate for both the internal and terminal dN/dS ratio. Simulation studies have shown that the power of the likelihood ratio test to detect non-neutral evolution peaks at between low to moderate and high levels of sequence divergence (33), as in the case of the RT sequences analyzed here.
In addition to examining patterns of nonsynonymous and synonymous nucleotide changes in the RT sequences, the number of stop codons per sequence was enumerated for the different species studied.
Simulations.
Simulation of TE dynamics followed the procedure outlined by
Charlesworth and Charlesworth (34). Briefly, a population of size
N diploid individuals, each with a genome consisting of
L unlinked TE insertion sites per haploid genome, was
assumed. The initial number of TEs per genome was determined randomly
by using a Poisson distribution with a mean of E elements
per diploid genome. These elements were distributed at random positions
among the 2L sites per genome. Selection, gamete
production and mating were then simulated. Specifically, the fitness of
the ith individual was assumed to equal
wi = 1 −
sn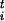 , where
s represents the selective effect of each insertion,
ni is the number of insertions in the
ith individual, and t is an exponent with a value
greater than one—a stable equilibrium copy number of TEs occurs only
when elements interact synergistically to determine host fitness (34).
After mating, a random number of Poisson-distributed replicative
transposition and loss events (e.g., as may occur through long terminal
repeat–long terminal repeat recombination) were simulated,
assuming per element probabilities of transposition and loss of
U and V, respectively. The cycle began anew with
selection, as described above. The simulation of TE dynamics was
checked for accuracy by comparing the results with analytical findings
and other published simulation results (34).
, where
s represents the selective effect of each insertion,
ni is the number of insertions in the
ith individual, and t is an exponent with a value
greater than one—a stable equilibrium copy number of TEs occurs only
when elements interact synergistically to determine host fitness (34).
After mating, a random number of Poisson-distributed replicative
transposition and loss events (e.g., as may occur through long terminal
repeat–long terminal repeat recombination) were simulated,
assuming per element probabilities of transposition and loss of
U and V, respectively. The cycle began anew with
selection, as described above. The simulation of TE dynamics was
checked for accuracy by comparing the results with analytical findings
and other published simulation results (34).
To examine how RT sequences evolve when simulation conditions lead to different equilibrium copy numbers, sequence evolution was incorporated into the dynamical simulation described above. The initial complement of TEs harbored by each individual was assumed to consist of a single, 99-bp coding (33 codon) segment of RT. The sequence was assumed to code for a functional RT product. Each generation, before transposition, elements were allowed to mutate at random with a per-element probability of mutation of υ. Mutation could occur with equal probability at any of the three base positions in the codon. Nonsynonymous mutations in the RT sequence were assumed to impair transposition by reducing the probability that the mutated element transposes by mp, where m is the number of nonsynonymous mutations in the RT sequence, and p is the reduction in transposition probability per nonsynonymous mutation. The simulation procedure was verified for accuracy by comparing the results obtained for selectively neutral RT sequence evolution (i.e., no penalty for nonsynonymous mutations) against the analytical-derived probability of identity of two randomly chosen elements at equilibrium Φ = 1/(1 + 2Ncυ) (35), where c is the mean copy number of elements per genome.
RT sequence evolution was compared under simulation conditions that produced contrasting equilibrium copy numbers of retrotransponsons (e.g., high versus low rates of retrotransposon loss, and strong versus weak selection against retrotransposon copy number). In the present model, it is assumed that these conditions are element-specific. RT sequences were sampled to determine frequency distributions of numbers of nonsynonymous differences between element sequences present in the equilibrium population of retrotransposons versus the initial RT sequence at generation 0.
Results
Variation in RT Sequences and Copy Number of Ty1-copia Elements.
Data for 22–74 copia-RT sequences per plant species were available on GenBank (Table 1). The sequences were heterogeneous both within and between plant species. Nevertheless, several highly conserved motifs were present in all sequences studied (Fig. 1). The conserved motifs are similar to those observed in the copia-RT sequence analyzed from Drosophila (36) and in other plant species (4) (Fig. 1). Nucleotide diversity of the RT sequences was high in all species studied (Table 1).
Figure 1.

Amino acid similarity among the conserved RT domain of Ty1-copia elements investigated in this study. x indicates position with no consensus amino acid. − indicates single base gaps introduced for alignment. Dots separating the two conserved motifs represent a gap of 14 nucleotide base residues that are not conserved among the species.
The copy number estimates for Ty1-copia elements ranges from several hundred per genome in A. thaliana and Oryza sativa, to several hundred thousand in Picea abies and V. faba (Table 2).
Patterns of copia-RT Sequence evolution.
Maximum parsimony reconstruction of copia-RT sequence evolution revealed excesses of third-position substitutions in both the internal and terminal portions of the element phylogenies of all host species except that of V. faba (Fig. 2), where no third-position excess was found along the terminal branches. Concordant with the analyses of third- to first- + second-position substitutions, are the estimated dN/dS, ratios, which are all significantly less than 1.0 by the likelihood ratio test (χ2 > 100, P < 0.001 for all comparisons). The dN/dS ratios range from 0.023 to 0.414 for the internal portion of the phylogenies and from 0.102 to 0.414 for the terminal portions. In eight of 11 species, the likelihood ratio test revealed that dN/dS ratios for internal portions of the element phylogeny are significantly less than those for the terminal portions (Table 3). The values of dN and dS, along the different branch segments or each tree, are given as supporting information, which is published on the PNAS web site, www.pnas.org.
Figure 2.

Distribution of internal branch (black bars) and terminal branch (open bars) substitutions among the first, second, and third codon positions of the conserved copia-RT sequence in three representative plant species differing in copy numbers of Ty1-copia elements.
Table 3.
Maximum-likelihood estimates of dN/dS ratios
| Species | dN/dS (Internal) | dN/dS (Terminal) | 2ΔL |
|---|---|---|---|
| O. sativa | 0.038 | 0.252 | 51.10** |
| A. thaliana | 0.023 | 0.102 | 15.51** |
| V. melanops | 0.026 | 0.383 | 15.61** |
| L. esculentum | 0.094 | 0.264 | 13.10** |
| P. sativum | 0.027 | 0.227 | 32.19** |
| V. sativa | 0.144 | 0.144 | 0.94 |
| Z. mays | 0.073 | 0.189 | 8.40** |
| T. aestivum | 0.150 | 0.341 | 20.58** |
| H. vulgare | 0.217 | 0.217 | 0.61 |
| P. abies | 0.127 | 0.391 | 28.01** |
| V. faba | 0.414 | 0.414 | 0.26 |
Species listed in order of increasing copy number of Ty-copia elements.
, P < 0.001;
, P < 0.01.
In all of the species investigated, some of the copia-RT sequences sampled contain stop codons, but the proportion varies widely between species (Table 4). A. thaliana has the smallest proportion of stop codons in the set of sequences analyzed (0.07), whereas V. faba has the largest (0.80) (Table 4).
Table 4.
Stop codons in the copia-RT sequences analyzed
| Species | Number of sequences | Proportion of sequences containing stop codons |
|---|---|---|
| O. sativa | 51 | 0.25 |
| A. thaliana | 42 | 0.07 |
| V. melanops | 27 | 0.22 |
| L. esculentum | 25 | 0.12 |
| P. sativum | 31 | 0.48 |
| V. sativa | 27 | 0.19 |
| Z. mays | 28 | 0.36 |
| T. aestivum | 74 | 0.42 |
| H. vulgare | 23 | 0.30 |
| P. abies | 51 | 0.41 |
| V. faba | 25 | 0.80 |
Species listed in order of increasing copy number of Ty1-copia elements.
Sequence Evolution and Copy Number of Ty1-copia Elements.
The internal dN/dS ratios for the low copy number species group, consisting of O. sativa, A. thaliana, Vicia melanops, Lycopersicon esculentum, Pisum sativa, and Vicia sativa (mean = 0.06) are significantly smaller than the internal dN/dS ratios for the high copy number species group (mean = 0.20), consisting of Z. mays, Triticum aestivum, Hordeum vulgare, P. abies, and V. faba (Mann–Whitney u test, P < 0.05). Likewise, the proportion of copia-RT sequences containing stop codons is significantly smaller in the low (mean = 0.22) versus high copy number (mean = 0.46) group (Mann–Whitney u test, P < 0.05).
Both the internal dN/dS ratios and the proportion of sequences containing stop codons increase with the estimated copy number of Ty1-copia elements per genome (Fig. 3). The rank correlation between the internal dN/dS ratio and estimated copy number is significant (Spearman rs = 0.83, P < 0.01), as is the rank correlation between the proportion of copia-RT sequences containing stop codons and estimated copy number (Spearman rs = 0.71, P < 0.025).
Figure 3.

Scatter plots of estimated dN/dS ratios for internal portions of copia-RT phylogenies versus estimated copy numbers of Ty1-copia elements in 11 plant species (●), and observed proportions of copia-RT sequences containing stop codons versus estimated copy number (○).
Simulation Results.
RT sequence evolution was compared under simulation conditions that produced a range of equilibrium copy numbers of retrotransposons. In all cases, equilibrium copy numbers were obtained after several hundred generations (Fig. 4). When nonsynonymous mutations were allowed to cause reductions in the probability of transposition, the frequency distribution of the proportion of nucleotide changes that are nonsynonymous contrasted strongly under lower versus higher copy number conditions (Fig. 4). In particular, under lower copy number equilibria, the standing population of retrotransposons contained RT sequences with relatively few nonsynonymous changes compared with populations under higher copy number equilibria.
Figure 4.

Simulated retrotransposon dynamics and sequence evolution. Copy number versus generation under: (A) high excision rate (V = 0.005) and stronger selection (t = 1.5) or (B) low excision rate (V = 0.00001) and weaker selection (t = 1.25). Proportion of nucleotide changes that are nonsynonymous under: (C) high excision and stronger selection (parameter values as in A) and (D) low excision and weaker selection (parameter values as in B). Illustrated are copy number versus generation, and patterns of substitution for 10 different simulation trials per parameter combination. Other simulation parameters used were identical in all runs and were: n = 50 individuals, L = 500 insertion sites, E = 10 TEs per genome at start of simulation, transposition rate U = 0.01, selection coefficient s = 0.0005, per element probability of mutation υ = 0.001, reduction in probability per nonsynonymous mutation P = 0.2. Histograms of proportions of nonsynonymous changes are based on sampling the haploid complement of elements from 10 individuals per trial at generation 10,000.
Discussion
Sequences of Ty1-copia Elements.
There is a large degree of sequence conservation among the RT genes examined in this study. The conserved motifs illustrated in Fig. 1 have been detected in most copia-RT sequences in plants (4). This sequence conservation is consistent with the evidence of purifying selection observed for RT sequences in all of the species investigated here, manifested as significant excesses of third-position substitutions in the parsimony-based analysis of sequence evolution, and by the maximum-likelihood estimates of dN/dS ratios < 1.0, obtained from the codon-based analysis (Table 3).
As expected, purifying selection is most pronounced along the internal branches of the phylogenies (Table 3). Strong purifying selection in RT sequences has also been inferred in the case of Ty1 elements in yeast genome by Jordan and McDonald (16), who interpreted it as arising from rapid turnover of elements. They noted that the yeast genome possesses mechanisms (e.g., intraelement long terminal repeat recombination) that can repress and eliminate Ty1 elements.
Stop codons, as detected here in a proportion of the RT sequences analyzed, have been noted to be relatively frequent in plant retrotransposon coding sequences (7) (Table 4). According to the complete Ty1-copia data from A. thaliana, examination of the RT sequences alone may underestimate the fraction of elements that are defective. In A. thaliana, 7% of the partial RT sequences contain stop codons (Table 4). But, if all Ty1-copia elements that contain stop codons or lack the full RT sequence are analyzed, 61% of copia-like retrotransposons in Arabidopsis would be classified as defective. Unfortunately, because sequence data from the other 10 host plant species studied here do not typically include portions of the Ty1-copia elements that lie outside the RT region, we cannot be certain of the total fraction of the elements in these species that are defective.
Ty1-copia Sequence Evolution and Copy Numbers.
Among the different plant species compared in this study, the estimated copy number of Ty1-copia elements ranges over 4 orders of magnitude. The average dN/dS in the internal portions of the element phylogenies is significantly smaller in low compared with high copy number species. As well, there is a significant rank correlation between the estimated dN/dS in the internal portions of the element phylogenies and the respective copy number estimates for the host species. The low copy number species have fewer stop codons in the RT sequences compared with high copy number species, and there is a significant rank correlation between the proportion of sequences containing stop codons and the estimated copy numbers.
The pattern of more pronounced purifying selection in RT sequences in low copy number genomes is unlikely to be an artifact of high levels of nucleotide diversity among the RT sequences. In particular, some of the most divergent RT sequences, where saturation of the estimate of dS might be expected to lead to underestimation of the dN/dS ratio, are found in the low copy number species (see supporting information on the PNAS web site). Thus the dN/dS ratios for these species may be even smaller than reported here, and the pattern of small dN/dS ratios in the low copy number species may be even stronger than actually observed.
The findings are consistent with the hypothesis that RT sequences in plant genomes that harbor fewer Ty1-copia elements have evolved under a higher degree of selective constraint compared with those in species with higher copy numbers, an interpretation that is bolstered by the simulation results. The latter show that when there is strong selection against individuals harboring many elements, and/or high per-element loss rates leading to low copy numbers of elements, the vast majority of elements that comprise the equilibrium population of retrotransposons contain few nonsynonymous mutations and are transpositionally competent. This is in contrast to simulation conditions where there is weaker selection against individuals with many elements and/or low rates of element loss, which allows both a larger element population per genome, as well as one with many nonsynonymous mutations.
Another possible reason RT sequences appear to evolve under lower selective constraint in host genomes with many elements may pertain to coevolutionary coexistence between nonautonoumous and autonomous elements. That is, in species with higher copy numbers of Ty1-copia elements, a large fraction of the elements may contain mutations in the RT sequence or in other functional portions of the sequence, but nevertheless, transpose in the presence of reverse transcriptase produced by other (autonomous) elements in the genome (1). If Ty1-copia elements are, in fact, capable of trans-activation, this could reduce the degree of selective constraint on the RT gene. The coexistence of defective elements with wild-type elements is supported by theoretical analysis (39).
Conclusions.
It has sometimes been argued that copy numbers of TEs can be interpreted as a direct reflection of activity levels—i.e., elements present in high copy numbers are equated with elements that have high transposition rates (20, 21). The findings in this study suggest that this view is incorrect. Both the pattern of stronger purifying selection of RT sequences in genomes with fewer elements, and the findings from simulated RT sequence evolution suggest that there can be the strong selection for conservation of function in elements that are present in relatively few copies per genome. That is to say, low copy numbers may reflect an underlying dynamic equilibirum, such that only the most transpositionally active elements are capable of long-term persistence. This does not, however, argue against the possibility that genomes may also contain remnant element sequences that are the product of their coancestry with other taxa, particularly in cases where these sequences have been co-opted to serve other functions (40, 41).
Supplementary Material
Acknowledgments
We thank Anne Bruneau, Tom Bureau, Amar Kumar, Boris Legault, Aaron Windsor, and Steve Wright for advice and assistance. This work was supported by an Operating Grant from the Natural Sciences and Engineering Research Council of Canada to D.J.S. and a scholarship from the Mexican government to A.N.Q.
Abbreviations
- RT
reverse transcriptase
- TE
transposable element
Footnotes
References
- 1.Kumar A, Bennetzen J L. Annu Rev Genet. 1999;33:479–572. doi: 10.1146/annurev.genet.33.1.479. [DOI] [PubMed] [Google Scholar]
- 2.Kidwell M G, Lisch D. Proc Natl Acad Sci USA. 1997;94:7704–7711. doi: 10.1073/pnas.94.15.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flavell A J, Dunbar E, Anderson R, Pearce S R, Hartley R, Kumar A. Nucleic Acids Res. 1992;20:3639–3644. doi: 10.1093/nar/20.14.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voytas D F, Cummings M P, Komeczny A, Ausbel F M, Rodermel S R. Proc Natl Acad Sci USA. 1992;89:7124–7128. doi: 10.1073/pnas.89.15.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.VanderWeil P L, Voytas D F, Wendel J F. J Mol Evol. 1993;36:429–447. doi: 10.1007/BF02406720. [DOI] [PubMed] [Google Scholar]
- 6.Pearce S R, Harrison G, Li D, Heslop-Harrison J S, Kumar A, Flavell A J. Mol Gen Genet. 1996;250:305–315. doi: 10.1007/BF02174388. [DOI] [PubMed] [Google Scholar]
- 7.Brandes A, Heslop-Harrison J S, Kamm A, Kubis S, Doudrick R L, Schmidt T. Plant Mol Biol. 1997;33:11–21. doi: 10.1023/a:1005797222148. [DOI] [PubMed] [Google Scholar]
- 8.Stuart-Rogers C, Flavell A J. Mol Biol Evol. 2001;18:155–163. doi: 10.1093/oxfordjournals.molbev.a003789. [DOI] [PubMed] [Google Scholar]
- 9.Smyth D R, Kalitsis P, Joseph J L, Sentry J W. Proc Natl Acad Sci USA. 1989;86:5015–5019. doi: 10.1073/pnas.86.13.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.SanMiguel P, Tikhonov A, Jin Y K, Motchoulskaia N, Zakharov D, MelakeBerhan A, Springer P S, Edwards K J, Lee M, Avramora Z, et al. Science. 1996;274:765–768. doi: 10.1126/science.274.5288.765. [DOI] [PubMed] [Google Scholar]
- 11.Suoniemi A, Tanskanen J, Schulman A. Plant J. 1998;13:699–705. doi: 10.1046/j.1365-313x.1998.00071.x. [DOI] [PubMed] [Google Scholar]
- 12.Heslop-Harrison J S, Brandes A, Taketa S, Schmidt T, Vershinin A V, Alkhimova E G, Kamm A, Doudrick R L, Schwarzacher T, Katsiotis A, et al. Genetica. 1997;100:197–204. [PubMed] [Google Scholar]
- 13.Flavell A J, Pearce S R, Heslop-Harrison J S, Kumar A. Genetica. 1997;100:185–195. [PubMed] [Google Scholar]
- 14.Vant'Hoff J, Sparrow A H. Proc Natl Acad Sci USA. 1963;49:897–902. doi: 10.1073/pnas.49.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavalier-Smith T. J Cell Sci. 1978;34:247–278. doi: 10.1242/jcs.34.1.247. [DOI] [PubMed] [Google Scholar]
- 16.Jordan I K, McDonald J F. J Mol Evol. 1999;49:352–357. doi: 10.1007/pl00006558. [DOI] [PubMed] [Google Scholar]
- 17.Petrov D A, Hartl D L. Gene. 1997;205:279–289. doi: 10.1016/s0378-1119(97)00516-7. [DOI] [PubMed] [Google Scholar]
- 18.Vicient C M, Suoniemi A, Anamthawat-Jonsson J, Tanskanen A, Beharav E, Nevo E, Schulman A. Plant Cell. 1999;11:1769–1784. doi: 10.1105/tpc.11.9.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogers S A, Paul K P. Genome. 2000;43:887–894. doi: 10.1139/g00-056. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Liu N, Peng K, Zhang Q. Proc Natl Acad Sci USA. 1999;96:6824–6828. doi: 10.1073/pnas.96.12.6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pearce S R, Knox M, Ellis T H N, Flavell A J, Kumar A. Mol Gen Genet. 2000;263:898–907. doi: 10.1007/s004380000257. [DOI] [PubMed] [Google Scholar]
- 22.Matsuoka Y, Tsunewaki K. Mol Biol Evol. 1996;13:1384–1392. doi: 10.1093/oxfordjournals.molbev.a025585. [DOI] [PubMed] [Google Scholar]
- 23.Le Q H, Wright S, Yu Z, Bureau T. Proc Natl Acad Sci USA. 2000;97:7376–7381. doi: 10.1073/pnas.97.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuoka Y, Tsunewaki K. Mol Biol Evol. 1999;16:208–217. doi: 10.1093/oxfordjournals.molbev.a026103. [DOI] [PubMed] [Google Scholar]
- 25.Higgins N P, Bleasby A J, Fuchs R. Comput Appl Biosci. 1992;8:189–191. doi: 10.1093/bioinformatics/8.2.189. [DOI] [PubMed] [Google Scholar]
- 26.Xiong Y, Eickbush T E. EMBO J. 1990;9:3354–3362. doi: 10.1002/j.1460-2075.1990.tb07536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rozas J, Rozas R. Bioinformatics. 1999;15:174–175. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- 28.Swofford D L. paup*, Phylogenetic Analysis Using Parsimony (*and Other Methods) Sunderland, MA: Sinauer; 1998. , Version 4.0b8. [Google Scholar]
- 29.Hasegawa M, Kishino H, Yano T. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 30.Yang Z. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- 31.Kishino H, Hasegawa M. J Mol Evol. 1989;29:170–179. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- 32.Li W-H. Molecular Evolution. Sunderland, MA: Sinauer; 1997. [Google Scholar]
- 33.Anisomova M, Bielawski J P, Yang Z. Mol Biol Evol. 2001;18:1585–1592. doi: 10.1093/oxfordjournals.molbev.a003945. [DOI] [PubMed] [Google Scholar]
- 34.Charlesworth B, Charlesworth D. Genet Res. 1983;42:1–27. [Google Scholar]
- 35.Slatkin M. Genetics. 1985;110:145–158. doi: 10.1093/genetics/110.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mount S M, Rubin G M. Mol Cell Biol. 1985;5:1630–1638. doi: 10.1128/mcb.5.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu K B, Somerville S. Genome. 1996;39:1159–1168. doi: 10.1139/g96-146. [DOI] [PubMed] [Google Scholar]
- 38.L'Homme Y, Séguin A, Tremblay F M. Genome. 2000;43:1084–1089. [PubMed] [Google Scholar]
- 39.Nee S, Maynard Smith J. Parasitology. 1990;100:5–18. [Google Scholar]
- 40.Wessler S, Bureau T E, White S E. Curr Opin Genet Dev. 1995;5:814–821. doi: 10.1016/0959-437x(95)80016-x. [DOI] [PubMed] [Google Scholar]
- 41.White S E, Habera L F, Wessler S R. Proc Natl Acad Sci USA. 1994;91:11792–11796. doi: 10.1073/pnas.91.25.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.