

Abstract
Molecular photoswitches produce light‐controlled changes at the nanometer scale and can therefore be used to alter the states and behavior of materials in a truly bottom‐up fashion. Here an escalating photonic complexity of material property control with light is shown using a recently developed aza‐diarylethene in combination with hemiindigo (HI) photoswitches. First, aza‐diarylethene can be used as a photoswitch in polystyrene (PS) to reversibly inscribe relief‐type 3D structures into PS. Second, aza‐diarylethene can further be used as a photoinitiator for light‐induced polymerization of methyl acrylate (MA), demonstrating for the first time light‐controlled chemical reactivity control with its zwitterionic switching state. Third, aza‐diarylethene and HIs are implemented into aza‐diarylethene polymerized MA, generating photochromic polymers. At the fourth level, a binary mixture allows to synergize aza‐diarylethene‐induced photopolymerization with localized photochromism changes of the simultaneously entrapped functional HI. With such multilevel light response, the utility of this particular photoswitch combination for applications in advanced photonic materials is demonstrated.
Keywords: aza‐diarylethene, hemiindigo, light‐responsive materials, photochromism, photopolymerization
Four levels of advanced photoresponsive materials are reported using aza‐diarylethene and hemiindigo photoswitches. Starting from blending photochromes with polymers, a novel photopolymerization by aza‐diarylethene is demonstrated next, which can be combined with subsequent photochrome addition. Finally, synergistic and simultaneous photopolymerization and photochrome localization in a multi‐photoresponsive materials application is realized.

1. Introduction
Applications of molecular photoswitches[ 1 , 2 ] in materials are of high interest because multiple properties can be controlled with nanoscale precision by simple outside light irradiation stimuli.[ 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 ] Consequentially, the resulting light‐responsive materials give rise to smart and programmable behavior, the potential of which is just starting to be tapped. A very potent yet simple approach in this regard is an implementation of photoswitches into transparent polymers, endowing the latter with photochromism.[ 12 , 13 ] This allows for a reversible inscription of information with light and resulting applicability in optical memories, cryptographics, displays, or programmable filters.[ 14 , 15 , 16 , 17 ] When geometrical or electronic changes of the molecular photoswitches lead to corresponding changes within the embedding material, light control over its shape, density, aggregation state, or wettability (to name a few options) can be achieved.[ 7 , 18 , 19 , 20 , 21 , 22 , 23 ] There are virtually limitless applications that result in such cases including soft robotics,[ 24 , 25 , 26 ] haptic topological material changes,[ 27 , 28 ] or self‐healing.[ 29 , 30 , 31 ] Photoswitches can further be used as molecular photoinitiators for polymerization and thus open up applications in 3D printing,[ 32 , 33 , 34 , 35 , 36 , 37 ] for 4D materials with dynamically controllable changes,[ 6 , 38 ] or for sequence‐precise information polymers[ 39 ] (again to just name a few). In the overwhelming cases, the most common types of photoswitches are used for materials applications i.e., azobenzenes,[ 40 , 41 , 42 ] diarylethenes,[ 43 ] spiropyranes,[ 44 , 45 ] or stilbenes.[ 46 , 47 ] The latter are interesting for materials application in their own right in the more elaborate form of molecular machines.[ 48 , 49 , 50 ]
In this work we explore two very recent additions to the realm of photoswitches, hemiindigo (HI)[ 51 , 52 , 53 , 54 ] (derivatives 1–8, Figure 1a) and aza‐diarylethene 9 [ 55 ] (Figure 1b), for their potential use in advanced light‐responsive materials. HIs belong to the emerging class of indigoid photoswitches,[ 56 , 57 , 58 , 59 ] which in general show excellent switching properties, high thermal stability, and broadly tunable absorptions in the visible,[ 60 , 61 , 62 , 63 , 64 , 65 ] red,[ 51 , 54 , 66 , 67 , 68 , 69 , 70 ] and near infrared[ 71 , 72 ] part of the electromagnetic spectrum. Aza‐diarylethene 9 is a heterocyclic variant of diarylethenes and allows reversibly forming a CN double bond photochemically under zwitterion formation and aromatization.[ 55 ] Here we scrutinize the potential of HI and aza‐diarylethene for light‐responsive materials applications and deliver four levels of advances in this regard. At level one we start with photochromic polymers and 3D‐reversible relief photoimprinting by physically mixing our novel aza‐diarylethene 9 photoswitch with ready‐made polystyrene (PS) polymers (Figure 1c, level one). At level two, we use aza‐diarylethene 9 for light‐induced radical polymerization initiation for the first time (Figure 1c, level two). At level three we take the resulting polymers and turn them into photochromic materials by subsequent addition of compounds 1–7, and 9 (Figure 1c, level three). Finally, at level four, we synergize photocontrolled polymerization and localized photochromism simultaneously in an unprecedented multi‐photoresponsive materials application (Figure 1c, level four).
Figure 1.

Advanced light‐controlled materials using HIs 1–8 and aza‐diarylethene 9 as photoresponsive components. a) Schematic overview of HI 1–8 switching modes. b) Schematic overview of aza‐diarylethene 9 switching modes. c) Four levels of advanced photoresponsive materials using HI and aza‐diarylethene. Level one: Mixing of aza‐diarylethene 9 with polystyrene (PS) enables reversible photochromic inscription and 3D relief imprinting. Level two: Photopolymerization of methyl acrylate (MA) using aza‐diarylethene 9 as photoinitiator. Level three: Sequential multi‐photoresponses by first photopolymerization of MA with aza‐diarylethene 9 followed by addition of HIs to the resulting poly‐(methyl)‐acrylate (PMA) delivering photochromic materials. Level four: Simultaneous multi‐photohotochromic behavior in a dual mixture of aza‐diarylethene 9, HI 7, and MA. Selective photopolymerization initiated with aza‐diarylethene 9 yields a colored transparent PMA polymer in which HI 7 photochromism is retained and localized.
2. Results and Discussion
2.1. Level One: Photoswitching of Aza‐Diarylethene 9 in PS
Recently, we have reported compound 9 undergoing a reversible electrocyclic ring closure mechanism with concomitant C‐S bond cleavage leading to a charged zwitterionic species by UV light irradiation (Figure 1b).[ 55 ] In this work, we turned our attention to aza‐diarylethene 9 and its photoresponse in a polymer context. The physical addition of photochromes into a ready‐made polymeric matrix is a well‐established application for photoswitches allowing for optical inscription of information in a spatially resolved manner.[ 12 , 13 ] However, the rigid matrix of polymers can preclude proper photoswitching depending on the particular polymer and photochromic molecule. For aza‐diarylethene 9 such an application has not been tested before and thus serves as an entry‐point into more advanced photonic functions of our novel photoswitch. First, 9 was embedded into polystyrene (PS) and also in this case reversible photoresponse with light of 365 nm is observed allowing localization of photoswitching and inscribing information into the material (Figure 2a). Moderate color contrasts are observed as a result of the photoswitching from open 9 to closed Z‐9, which allows one to distinguish the inscription with the naked eye. Upon diluting the inscribed polymer with benzene at ambient temperature, the inscription can be erased and a transparent colorless material is received again upon drying. Another round of photoinscription directly showed reversible photoswitching of 9 in polystyrene (Figure 2b). From the corresponding UV/vis spectroscopic analysis of 9 photoswitching in PS (Figure 2c, yellow coloration) we observed that enrichment of closed Z‐9 as well as the charge separation of the zwitterionic state are less pronounced compared to MeOH solution, which explains the reduced color contrast in the polymer. Upon very long irradiation durations some photodegradation, correlated to noticeable color loss (gray dotted), occurs. Additionally, in MeOH solution, closed Z‐9 can further undergo light‐induced Z/E photoisomerization upon longer irradiation durations with light of 365 nm to an extent of up to 3%, which leads to a diminishing of the yellow color and corresponding lower intensity of the visible absorption band in the UV/vis spectrum. Further accumulation of the closed E‐9 isomer is possible through several irradiation cycles at ambient temperature. Given the only pale‐yellow color observed upon photoswitching of 9 in PS and the incomplete recovery of the colorless open 9 form after dilution, we assume the resulting photoproducts in PS to contain a noticeable contribution of the thermally more stable ring closed E‐9 isomer.
Figure 2.
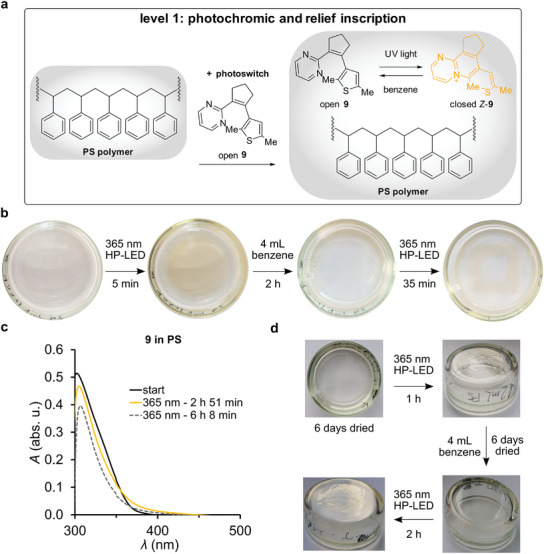
Level one photoresponsive materials. a) The addition of aza‐diarylethene 9 to PS enables localized reversible photochromism and 3D relief inscription in a solid material. b) Photochromic polymer with 9 in PS showing reversible writing and erasing. c) Photoswitching of open 9 (black) to closed 9 isomers (yellow) in PS. d) Reversible photoinduced 3D relief formation upon photoswitching of 9. The FAU University Seal was inscribed into the material in a haptic way using a photomask.
A very interesting behavior is observed upon irradiation of 9 using a photomask with very narrow inscription lines on PS, which leads to a persistent 3D relief formation in the irradiated areas. The specific molecular cause for this behavior is currently under investigation in our laboratory, but a strong local accumulation of charged ring closed 9 species and concomitant localized polymer adjustments and packing changes seem likely to be important. Evidence for the presence of charged 9 isomers is directly obtained from the localized color changes, which coincide with the 3D structured polymer areas. Such behavior is rare but highly sought‐after for photochromic materials[ 27 , 28 ] and can be used to inscribe information in a haptic way instead of relying on optical changes, which is of potential interest for applications in braille displays or reprogrammable stamps, for example. After diluting the material with benzene and allowing the chromophore to thermally isomerize to the open 9 form at ambient temperature, the imprint is erased and a smooth surface is obtained into which a different 3D relief can be inscribed again (Figure 2d). With this series of experiments reversibility of the 3D inscription process is demonstrated opening unique application areas for aza‐diarylethene photoswitches.
2.2. Level Two: Photoinduced Polymerization of MA by Aza‐Diarylethene 9
As described above, photoswitching of aza‐diarylethene 9 induces the formation of an aromatized zwitterionic state. This zwitterion inherits a positively charged pyrido[1,2‐a]pyrimidin‐5‐ium ring and also a highly nucleophilic thiolate functional group (Figure 1b). Light irradiation thus leads to the formation of a pronounced chemical reactivity and we were wondering if starting from the zwitterionic form we could generate radicals initiating radical polymerization upon continued light irradiation similar to zwitterionic merocyanines.[ 32 , 73 , 74 , 75 ] In this way, light could be used as an external trigger ultimately controlling the formation of a solid material in a timed fashion. We projected that photoswitching of 9 would first lead to the formation of the thiolate, which would then form radicals and be able to initialize radical methyl acrylate (MA) polymerization by an initial attack on the MA Michael system. The resulting radicals would then polymerize the material under continuous carbon‐carbon bond formation yielding poly‐(methyl)‐acrylate (PMA) (Figure 3a). We tested different mixtures of aza‐diarylethene 9 and MA and found suitable conditions for the range between 0.4 and 13 mg of 9 mL−1 MA. To our delight, irradiation with light of 365 nm (260 mW) or 367 nm (1 W) led to effective photopolymerization in under four hours. For example, at a concentration of 0.7 mg 9 mL−1 MA a solid yellow material was obtained within 2 h 40 min of 365 nm light irradiation (Figure 3b). Upon subsequent polymer storage in the dark, a post‐curing effect was observed. The same irradiation conditions in the absence of 9 did not lead to any visible polymerization and only liquid MA monomers remained. Only a much longer light irradiation duration of more than 17 h with 365 nm (260 mW) ultimately resulted in radical polymerization of MA (see Figure S16, Supporting Information). Additionally, we wanted to understand if the polymerization‐initiating event was triggered by the excited state of open 9 or by the excited state of closed Z‐9. We therefore irradiated a sample containing 0.7 mg 9 mL−1 MA simultaneously with light of 365 (260 mW) nm and 450 nm (400 mW). Full polymerization occurred within 1 h 5 min (Figure S19, Supporting Information). The polymerization speed thus was more than doubled compared to irradiation with solely 365 nm light. As open 9 does not absorb at 450 nm and closed Z‐9 absorbs both at 365 and 450 nm, we therefore assume the polymerization event is triggered by the photoproduct of 9, the closed Z‐9 isomer. If going to concentrations above 13 mg 9 mL−1 MA or below 0.4 mg 9 mL−1 MA no polymerization occurred in under 4 h of light irradiation with 365 or 367 nm most likely because of radical self‐/quenching effects or too low relative concentration of radicals versus MA monomers.
Figure 3.

Level two photoresponsive materials. a) Aza‐diarylethene 9 as photoinitiator for radical polymerization of MA. b) Irradiation of two samples with 365 nm light for 2 h 40 min (or with 365 nm + 450 nm light for 1 h 5 min). The left sample vial contains 0.72 mg of open 9 in 1 mL MA, and the right sample vial contains solely 1 mL MA. After the specified irradiation duration, the sample containing compound 9 shows the characteristic color of the closed Z‐9 isomer and is polymerized. The sample containing solely MA did not polymerize. After storing both samples for 14 h in the dark at 23 °C, a post‐curing effect happened to the compound 9 carrying sample. The sample containing solely MA did not show any changes.
To prove the radical polymerization mechanism, an equimolar amount of 2,2,6,6‐tetramethyl(piperidin‐1‐yl)oxyl (TEMPO, 0.4 mg mL−1) was added to a solution of 9 (0.7 mg mL−1) in MA and the mixture was irradiated for 2 h 40 min with light of 365 nm (260 mW, Figure S18, Supporting Information). TEMPO is a known radical quencher stopping radical polymerization of acrylic acids. Indeed, in the presence of TEMPO, no polymerization occurred, evidencing the radical polymerization mechanism photoinitiated by 9. Further, NMR experiments revealed the consumption of closed Z ‐9 during the polymerization process resulting in a reduced amount of remaining open 9 after PMA polymerization is complete (Figures S20–S22, Supporting Information). In this specific photopolymerization application, aza‐diarylethene 9 was thus established as a viable molecular photoinitiator demonstrating for the first time the direct and chemical use of the photoproducts of 9.
2.3. Level Three Part 1: Sequential Photopolymerization and Photochrome 9 Addition
We further elevated the polymeric application of aza‐diarylethene 9 by using it in two different capacities for the same material in a sequential fashion (Figure 4a). First, MA was polymerized with 9 as photoinitiator in lower concentrations (0.03 mg 9 mL−1 MA) to yield an almost colorless and transparent PMA material. Monitoring of the polymerization by UV/vis spectroscopy revealed further details of this process (Figure 4b). Starting from open 9 in MA (Figure 4b, black graph), irradiation with light of 365 nm (260 mW) leads to the formation of closed Z‐9 (Figure 4b, yellow graph) first. Then, the polymerization process starts as indicated by a loss of absorption in the spectral region between 400 and 500 nm. After complete polymerization, only the remaining open 9 is detectable within the PMA matrix (Figure 4b, dark gray graph; Figure S23, Supporting Information). These UV/vis spectroscopy observations therefore correspond very well to those made using NMR spectroscopy described above. Next, after finishing the polymerization, 9 was added anew to the PMA matrix as a photochromic component. When analyzing the photoswitching behavior in the transparent material by UV/vis spectroscopy, a somewhat similar behavior to polar protic MeOH solution is observed.[ 55 ] In MeOH, open 9 undergoes a concomitant ring closure, aromatization, and charge‐separating zwitterion formation upon irradiation with light of 365 nm. The resulting closed Z‐9 isomer is colored yellow with a significant absorption increase in the visible region ≈450 nm. In the PMA application, the UV/vis spectra of open 9 (Figure 4c) reveal similar behavior. After irradiation with light of 275 nm also a broad absorption band appears at ≈450 nm indicating the zwitterionic closed Z‐9 species (Figure 4c, yellow graph). The zwitterionic state is more pronounced in PMA (Figure 4c) than it is in PS (Figure 2c), which can be explained by the polarity and rigidity difference of the two polymers. Photodegradation appeared only after irradiation durations between 1 h 48 min and 3 h 23 min with energy‐rich 275 nm light. The viable photoswitching of closed Z‐9 in PMA is also visible by the naked eye via localized light yellow color changes (Figure 4d). The reversibility of aza‐diarylethene 9 T‐type photoswitching in PMA was proven by diluting the polymer with CH2Cl2, followed by evaporating the solvent, which resulted in decolorization to almost colorless again (Figure 4d). As for photoisomerization of 9 in PS, also in PMA a contribution of closed E‐1 enrichment is possible, hampering complete discoloration upon dilution.
Figure 4.

Level three photoresponsive materials part 1: a) Sequential photopolymerization and photochromism introduction using aza‐diarylethene 9. Aza‐diarylethene 9 was used first as a photoinitiator for MA polymerization and subsequently fresh 9 was added again as photochrome to the obtained PMA. b) UV/vis spectra showing irradiation of open 9 (black) with light of 365 nm (260 mW) leads to closed Z‐9 generation (gray to yellow) followed by consumption of closed Z‐9 (yellow dotted) in the course of the polymerization progress until full polymerization occurred and only remaining open 9 (dark gray) is visible. The related UV/vis cuvette is also depicted, as well as a reference cuvette containing only MA after irradiation with light of 365 nm for 6 h 36 min. c) UV/vis absorption spectra recorded during photoswitching of open 9 (black) to closed Z‐9 (gray to yellow) in PMA with light of 275 nm during 1 h 48 min. After 3 h 23 min photodegradation occurred (gray dotted). d) Reversible T‐type photoswitching in PMA with light of 365 nm from open 9 to closed Z‐9 (yellow square) and reversion to open 9 after the addition of CH2Cl2 at 23 °C.
2.4. Level Three Part 2: Sequential Photopolymerization and HI Photochrome Addition
In part 2 of level three, we combined the photopolymerization capacity of aza‐diarylethene 9 with the photochromism of HIs in a sequential manner (Figure 5 ). We have recently reported on photochromic PS polymers using newly developed diaryl‐HI photoswitches (Figure S9, Supporting Information) as the light‐active component.[ 54 ] We could demonstrate that their photochromism generating reversible E/Z photoisomerization can be retained in the solid material through different external stimuli such as light and acid, giving rise to marked contrast changes between red and purple. To advance both, HI applicability and complexity of the here presented smart material applications, we now combined their pronounced photochromism with the photoinitiator functions of aza‐diarylethene 9, again in a sequential fashion (Figure 5a). To this end, we introduced the series of HIs 1–7 (Figure 5b) into PMA, which was separately photopolymerized by aza‐diarylethene 9 before. To this end, the photoinitiated polymerized PMA was diluted in CH2Cl2, mixed with HIs 1–7, and subsequently, the solvent was evaporated, leading to highly colored and transparent photochromic materials. Photoswitching within the transparent polymers could be followed directly by the naked eye as well as with UV/vis spectroscopy and compared to solution behavior (see Figures S29–S50, Supporting Information). When investigating the different derivatives 1–7, marked differences could be observed depending on the molecular setups as visible by the UV/vis spectra and corresponding photochromic polymers (Figure 5c–i).
Figure 5.

Level three photoresponsive materials part 2. a) Sequential MA photopolymerization using aza‐diarylethene 9 followed by photochromism introduction by adding HIs 1–7 to the finished PMA. UV/vis spectroscopic monitoring of the localized light responses of HI photoswitches 1–7 within photochromic PMA polymers is shown together with the corresponding photochromic PMA/HI polymers in petri dishes. The inner squares within the petri dishes contain enriched bathochromic isomers, while the surroundings contain the hypsochromic isomers. b) Schematic representations of the molecular structures of HIs 1–7. c) Photochromic polymer with HI 1. d) Photochromic polymer with HI 2. e) Photochromic polymer with HI 3. f) Photochromic polymer with HI 4. g) Photochromic polymer with HI 5. h) Photochromic polymer with HI 6. i) Photochromic polymer with HI 7.
Mono‐arylated HIs such as 1 are strongly photochromic in solution with maxima separation between the two switching isomers reaching more than 30 nm in monomeric MA. However, within the polymer matrix, the photochromism is markedly reduced, and photodegradation occurs soon after light irradiation (Figure 5c). HI, derivative 2 inheriting increasing electron donation at the stilbene‐fragment does not deliver a significantly better absorption band separation between the E/Z isomers (Figure 5d) but no photodegradation is visible from the spectra. However, visible color paling in the PMA material still indicates photodegradation after prolonged irradiation durations with light of 470 nm. For HI derivative 3 (Figure 5e), which differs from compound 2 by a chlorine substituent in ortho‐position to the dimethylaniline and para‐position to the methoxy substituents, again photodegradation occurs soon after irradiating the samples. Irradiating the very electron‐rich julolidine substituted HI 4 did not lead to photodegradation, but again color differences are not very pronounced (Figure 5f).
From analyzing the different UV/vis measurements of compounds 1–4, some conclusions can be drawn. It is evident that by moving from solution to polymer, a spectral broadening for the individual E and Z isomers results in an increased spectral overlap. This leads to a reduced addressability of the hypsochromic isomers. In the case of the bathochromic isomers, the absorption flanks in the redshifted spectral area are still separated enough, preserving the addressability of the bathochromic isomers similar to that in solution. This effect is the reason for the reduced photochromism achievable in polymers. On the other side, photodegradation effects are drastically increased by moving monoaryl‐HIs from solvent to PMA polymer.
Diaryl‐HIs are more bathochromically shifted in their absorption than monoaryl‐HIs and deliver strong photochromism, pronounced color contrasts between the isomers, and pronounced band separation. Additionally, the second aryl substituent at the stilbene fragment and substitution of the indoxyl‐nitrogen lead to photoswitches that are highly robust against photodegradation in solution and in polymer applications.
When employing diary‐HIs 4–7 as photochromic components in PMA, robust photoswitching is seen in all cases. Their inherent pronounced photochromism and larger absorption band separation translate favorably into the PMA matrix. In these cases, the very good color contrasts between red and purple remain, allowing to reversibly inscribe information with robust performance into the transparent material (Figure 5g–i). The best visible performance was achieved by diaryl‐HI 7, which delivered an easily distinguishable contrast between deep red and purple as exemplified in Figure 5i. Reversibility was demonstrated by erasing inscribed information with red light and rewriting new information with green light irradiation (Figure S51, Supporting Information). Note that HI 8 was not used at this level but rather at level one as photochrome in PS (see Supporting Information).
2.5. Level Four: Synergistic Polymerization of MA by Aza‐Diarylethene 9 in the Presence of HI 7
Finally, we strived for a synergistic combination of our photoresponsive molecular tools within the same advanced material by using them simultaneously during different light‐controlled processes (Figure 6a). For this purpose, we combined HI 7 and aza‐diarylethene 9 in a mixture with MA monomers (m/m 1:1.6:1.6 = 7:9:MA). This mixture remains liquid in the absence of light but can be photopolymerized upon 365 nm (2.8 W) irradiation to form a transparent yet strongly red‐colored solid material (Figure 6b). The remaining and unchanged red color directly shows that 365 nm irradiation, zwitterion formation of 9, as well as the polymerization reactions do not interfere with the HI 7 photoswitch, effectively uncoupling the aza‐diarylethene photoresponsive function from the HI function. Interestingly, HI 7 is not destroyed in the presence of a radical polymerization, which is clearly in line with the robustness of indoxyl nitrogen‐substituted diaryl‐hemiindigos.[ 54 ] After successful polymerization, the sample was two times irradiated with light of 490 and 625 nm demonstrating the retained HI photoswitchability, before the red solid material was transferred into a flat petri dish and irradiated again with red and green light through a photomask. With the longer wavelength light, the photoswitching function of HI 7 was addressed, leading to well‐visible photochromism and corresponding contrast changes with spatial resolution. Taken together, this multiphoton response establishes a highly advantageous synergistic use of two photoswitches for vastly different purposes within the same smart material application. The first light‐responsive component (aza‐diarylethene 9) solidifies the material upon UV light irradiation and in the course of this polymerization traps the second light‐responsive component (HI photoswitch 7) locally by inhibiting its diffusion. As a result, the HI photoswitch 7 can then be addressed with spatial resolution, leading to the possibility of reversible information inscription and erasure with visible and red light. Specific and non‐interfering molecular photoresponses are thus creating a multi‐photoresponsive material application in which light‐induced chemical reactions and reversible photoswitching are synergized.
Figure 6.

Level four photoresponsive materials. a) MA photopolymerization using aza‐diarylethene 9 in the presence of HI 7, which retains its photochromic function and allows for spatially resolved light‐addressing. b) Upon 365 nm irradiation, 9 undergoes zwitterion formation. Prolonged irradiation unleashes radical formation that initiates MA polymerization via the initial attack of the Michael system. HI 7 is not affected during this polymerization process and stays fully functional proving its exceptional robustness. Reversible visible light photoswitching with 490 and 625 nm of 7 in the resulting polymer allows to inscribe and erase information afterwards several times.
3. Conclusion
In summary, we establish here advanced photoresponsive materials with escalating levels of complexity. First, we established transparent light‐responsive photochromic polymers where a single photoswitch component is added to a ready‐made polymer and reversibly alternated to inscribe and erase information. The thus localized photoswitching of aza‐diarylethene 9 was used for photochromic changes as well as for reversible inscription of 3D relief type structures into the polymers, which broadens the applicability of molecular light‐responsiveness to include haptic material changes. Second, the capacity for photoinduced radical polymerization initiated by light irradiation of aza‐diarylethene 9 was harnessed for the first time. In this way, the solidification of a monomeric liquid could be brought under light control in a timed fashion with the aid of a novel molecular photoinitiator. Third, a new batch of aza‐diarylethene 9 or different HIs were mixed with the newly photoinitiated PMA to generate smart polymers that are rich in color contrast, retain sufficient photochromism, and are robust against photodamage. Fourth, the photoinitiator function of 9 was then finally synergized in situ with the reversible photochromism changes of HI 7 to obtain a multilevel simultaneous photoresponsive materials application. Here both light‐responsive components work together to enable full photocontrol over solidification, localization of the photoresponse, and reversible information inscription into the resulting material. With these results, we establish a new way for integrating different elements of photocontrol into smart material applications allowing to time and localize specific responses by light irradiation. Further studies to refine and enhance photocontrol are currently ongoing in our laboratories.
4. Experimental Section
Information about materials and general methods is given in Chapter S1 (Supporting Information). Details about the synthesis and characterization of the photoswitches are given in Chapter S2 (Supporting Information). Details about photochromic and relief inscription of photochromes in PS (level 1) as well as related sample preparation and analytics are given in Chapter S3 (Supporting Information). Details about photopolymerization of MA by aza‐diarylethene 9 (level 2) as well as related sample preparation and analytics are given in Chapter S4 (Supporting Information). Details about sequential photopolymerization and photochrome addition (level 3) as well as related sample preparation and analytics are given in Chapter S5 (Supporting Information). Details about simultaneous photopolymerization and photochrome localization (level 4) as well as related sample preparation and analytics are given in Chapter S6 (Supporting Information).
Processing Details
The absorbances of photochromes within polymer matrixes were analyzed via UV/vis spectroscopy on quartz plates and were set to zero at a wavelength of 600 or 800 nm where the photochromes do not show absorbances. UV/vis experiments on quartz plates were conducted on a Varian Cary 5000 UV/Vis‐NIR spectrophotometer comprised of two lamps for sample analysis. At 350 nm, the instrument changed from one lamp to another, which was visible through an offset in the measured absorption spectra that was pre‐processed if necessary. Instrument‐related outliers were deleted from spectra. Otherwise, no pre‐processing of data was carried out.
Polymerization and Photoisomerization Details
For polymerization experiments LEDs with a power output of 260 mW (365 nm) or 400 mW (450 nm) were used. For the reversible photochromic inscription into PS or PMA with compound 9 as a photochrome in petri dishes and for photopolymerization experiments with compound 9 at very low concentrations, as for the level 4 photopolymerization experiment, a 365 nm UHP LED from Prismatix (2.8 W*) was used. For the photoisomerization and photopolymerization experiments of compound 9 in UV/vis cuvettes or quartz plates with PS, PMA, and MA as matrices, the 260 mW (365 nm) LED was used. A high‐power 1 W 367 nm LED was used for photopolymerization experiments in NMR tubes. The 275 nm (19 mW*), 450 nm (400 mW) to 625 nm (920 mW*) LEDs used for photoisomerization experiments were low‐power LEDs. More details about the polymerization and photoisomerization conditions are given in the Supporting Information. *manufacturer information.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
H.D. thanks the Deutsche Forschungsgemeinschaft (DFG) for an Emmy Noether fellowship (DU 1414/1‐2). This project has also received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (PHOTOMECH, grant agreement No 101001794).
Open access funding enabled and organized by Projekt DEAL.
Sacherer M., Dube H., Combined Photopolymerization and Localized Photochromism by Aza‐Diarylethene and Hemiindigo Synergy. Adv. Mater. 2024, 37, 2411223. 10.1002/adma.202411223
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Feringa B. L., Browne W. R., Molecular Switches, 2nd ed., Wiley‐VCH, Weinheim, Germany: 2011. [Google Scholar]
- 2. Pianowski Z. L., Molecular Photoswitches. Chemistry,Properties, and Applications, Wiley‐VCH, Weinheim, Germany: 2022. [Google Scholar]
- 3. Goulet‐Hanssens A., Eisenreich F., Hecht S., Adv. Mater. 2020, 32, 1905966. [DOI] [PubMed] [Google Scholar]
- 4. Boelke J., Hecht S., Adv. Opt. Mater. 2019, 7, 1900404. [Google Scholar]
- 5. Lu P., Ahn D., Yunis R., Delafresnaye L., Corrigan N., Boyer C., Barner‐Kowollik C., Page Z. A., Matter 2021, 4, 2172. [Google Scholar]
- 6. Spiegel C. A., Hippler M., Münchinger A., Bastmeyer M., Barner‐Kowollik C., Wegener M., Blasco E., Adv. Funct. Mater. 2020, 30, 1907615. [Google Scholar]
- 7. Xu F., Feringa B. L., Adv. Mater. 2023, 35, 2204413. [DOI] [PubMed] [Google Scholar]
- 8. Danowski W., Leeuwen T. V., Browne W. R., Feringa B. L., Nanoscale Adv. 2021, 3, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pianowski Z. L., Chem.‐Eur. J. 2019, 25, 5128. [DOI] [PubMed] [Google Scholar]
- 10. Thaggard G. C., Haimerl J., Park K. C., Lim J., Fischer R. A., Maldeni Kankanamalage B. K. P., Yarbrough B. J., Wilson G. R., Shustova N. B., J. Am. Chem. Soc. 2022, 144, 23249. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J., Zou Q., Tian H., Adv. Mater. 2013, 25, 378. [DOI] [PubMed] [Google Scholar]
- 12. McArdle C. B., Applied Photochromic Polymer Systems, 1st ed., Springer, New York: 1992. [Google Scholar]
- 13. Zou J., Liao J., He Y., Zhang T., Xiao Y., Wang H., Shen M., Yu T., Huang W., Research 2024, 7, 0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meng X., Lin S., Chen S., Shen X., Guo D., Guo J., ChemPlusChem. 2024, 89, 202300700. [DOI] [PubMed] [Google Scholar]
- 15. Xi G., Sheng L., Zhang S. X.‐A., Mater. Sci. Eng. R: Rep. 2024, 158, 100774. [Google Scholar]
- 16. Andreasson J., Pischel U., Chem. Soc. Rev. 2018, 47, 2266. [DOI] [PubMed] [Google Scholar]
- 17. Wang X., Xu B., Tian W., Acc. Mater. Res. 2023, 4, 311. [Google Scholar]
- 18. Imato K., Kaneda N., Ooyama Y., Polym. J. 2024, 56, 269. [Google Scholar]
- 19. Lei M., Wang Q., Gu R., Qu D. H., Respons. Mater. 2024, 2, 20230027. [Google Scholar]
- 20. Truong V. X., Barner‐Kowollik C., Trends Chem. 2022, 4, 291. [Google Scholar]
- 21. Pang X., Lv J. A., Zhu C., Qin L., Yu Y., Adv. Mater. 2019, 31, 1904224. [DOI] [PubMed] [Google Scholar]
- 22. Nie H., Self J. L., Kuenstler A. S., Hayward R. C., Read de Alaniz J., Adv. Opt. Mater. 2019, 7, 1900224. [Google Scholar]
- 23. Zhang Q., Qu D. H., Tian H., Adv. Opt. Mater. 2019, 7, 1900033. [Google Scholar]
- 24. Jiang J., Xu S., Ma H., Li C., Huang Z., Mater Today Bio 2023, 20, 100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu C., Lu Y., Jiang L., Yu Y., Adv. Funct. Mater. 2021, 31, 2009835. [Google Scholar]
- 26. Shen Z., Chen F., Zhu X., Yong K. T., Gu G., J. Mater. Chem. B 2020, 8, 8972. [DOI] [PubMed] [Google Scholar]
- 27. Liu D., Broer D. J., Liq. Cryst. Rev. 2013, 1, 20. [Google Scholar]
- 28. Feng W., Liu D., Broer D. J., Small Struct. 2020, 2, 2000107. [Google Scholar]
- 29. Amaral A. J. R., Pasparakis G., Polym. Chem. 2017, 8, 6464. [Google Scholar]
- 30. Lugger S. J. D., Houben S. J. A., Foelen Y., Debije M. G., Schenning A., Mulder D. J., Chem. Rev. 2022, 122, 4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou S. W., Yu C., Chen M., Shi C. Y., Gu R., Qu D. H., Smart Mol. 2023, 1, 20220009. [Google Scholar]
- 32. Regehly M., Garmshausen Y., Reuter M., Konig N. F., Israel E., Kelly D. P., Chou C. Y., Koch K., Asfari B., Hecht S., Nature 2020, 588, 620. [DOI] [PubMed] [Google Scholar]
- 33. Stuwe L., Geiger M., Rollgen F., Heinze T., Reuter M., Wessling M., Hecht S., Linkhorst J., Adv. Mater. 2024, 36, 2306716. [DOI] [PubMed] [Google Scholar]
- 34. Hahn V., Rietz P., Hermann F., Müller P., Barner‐Kowollik C., Schlöder T., Wenzel W., Blasco E., Wegener M., Nat. Photonics 2022, 16, 784. [Google Scholar]
- 35. Gauci S. C., Vranic A., Blasco E., Brase S., Wegener M., Barner‐Kowollik C., Adv. Mater. 2024, 36, 2306468. [DOI] [PubMed] [Google Scholar]
- 36. Ehrmann K., Barner‐Kowollik C., J. Am. Chem. Soc. 2023, 145, 24438. [DOI] [PubMed] [Google Scholar]
- 37. Bao Y., Macromol. Rapid Commun. 2022, 43, 2200202. [DOI] [PubMed] [Google Scholar]
- 38. Wan X., Xiao Z., Tian Y., Chen M., Liu F., Wang D., Liu Y., Bartolo P., Yan C., Shi Y., Zhao R. R., Qi H. J., Zhou K., Adv. Mater. 2024, 36, 2312263. [DOI] [PubMed] [Google Scholar]
- 39. Eisenreich F., Kathan M., Dallmann A., Ihrig S. P., Schwaar T., Schmidt B. M., Hecht S., Nat. Catal. 2018, 1, 516. [Google Scholar]
- 40. Jerca F. A., Jerca V. V., Hoogenboom R., Nat. Rev. Chem. 2022, 6, 51. [DOI] [PubMed] [Google Scholar]
- 41. Crespi S., Simeth N. A., König B., Nat. Rev. Chem. 2019, 3, 133. [Google Scholar]
- 42. Bandara H. M., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809. [DOI] [PubMed] [Google Scholar]
- 43. Irie M., Fukaminato T., Matsuda K., Kobatake S., Chem. Rev. 2014, 114, 12174. [DOI] [PubMed] [Google Scholar]
- 44. Kortekaas L., Browne W. R., Chem. Soc. Rev. 2019, 48, 3406. [DOI] [PubMed] [Google Scholar]
- 45. Lukyanov B. S., Lukyanova M. B., Chem. Heterocycl. Compd. 2005, 41, 281. [Google Scholar]
- 46. Waldeck D. H., Chem. Rev. 1991, 91, 415. [Google Scholar]
- 47. Villaron D., Wezenberg S. J., Angew. Chem., Int. Ed. 2020, 59, 13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chau A. K., Leung F. K., Adv. Colloid Interface Sci. 2023, 315, 102892. [DOI] [PubMed] [Google Scholar]
- 49. Krause S., Feringa B. L., Nat. Rev. Chem. 2020, 4, 550. [Google Scholar]
- 50. Dattler D., Fuks G., Heiser J., Moulin E., Perrot A., Yao X., Giuseppone N., Chem. Rev. 2020, 120, 310. [DOI] [PubMed] [Google Scholar]
- 51. Petermayer C., Thumser S., Kink F., Mayer P., Dube H., J. Am. Chem. Soc. 2017, 139, 15060. [DOI] [PubMed] [Google Scholar]
- 52. Petermayer C., Dube H., J. Am. Chem. Soc. 2018, 140, 13558. [DOI] [PubMed] [Google Scholar]
- 53. Carrascosa E., Petermayer C., Scholz M. S., Bull J. N., Dube H., Bieske E. J., ChemPhysChem. 2020, 21, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sacherer M., Hampel F., Dube H., Nat. Commun. 2023, 14, 4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sacherer M., Gracheva S., Maid H., Placht C., Hampel F., Dube H., J. Am. Chem. Soc. 2024, 146, 9575. [DOI] [PubMed] [Google Scholar]
- 56. Petermayer C., Dube H., Acc. Chem. Res. 2018, 51, 1153. [DOI] [PubMed] [Google Scholar]
- 57. Wiedbrauk S., Dube H., Tetrahedron Lett. 2015, 56, 4266. [Google Scholar]
- 58. Bartelmann T., Dube H., Molecular Photoswitches: Chemistry, Properties, and Applications, Wiley, Hoboken, NJ, USA: 2022, ch.13. [Google Scholar]
- 59. Huang C. D., Hecht S., Chem. ‐ Eur. J. 2023, 29, 202300981. [Google Scholar]
- 60. Zitzmann M., Fröhling M., Dube H., Angew. Chem., Int. Ed. 2024, 63, 202318767. [DOI] [PubMed] [Google Scholar]
- 61. Köttner L., Wolff F., Mayer P., Zanin E., Dube H., J. Am. Chem. Soc. 2024, 146, 1894. [DOI] [PubMed] [Google Scholar]
- 62. Josef V., Hampel F., Dube H., Angew. Chem., Int. Ed. 2022, 61, 202210855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Berdnikova D. V., Steup S., Bolte M., Suta M., Chem.‐Eur. J. 2023, 29, 202300356. [DOI] [PubMed] [Google Scholar]
- 64. Cordes T., Schadendorf T., Rück‐Braun K., Zinth W., Chem. Phys. Lett. 2008, 455, 197. [DOI] [PubMed] [Google Scholar]
- 65. Ikegami M., Arai T., Bull. Chem. Soc. Jpn. 2003, 76, 1783. [Google Scholar]
- 66. Huber L. A., Peter M., Dube H., ChemPhotoChem. 2018, 2, 458. [Google Scholar]
- 67. Zweig J. E., Newhouse T. R., J. Am. Chem. Soc. 2017, 139, 10956. [DOI] [PubMed] [Google Scholar]
- 68. Kink F., Collado M. P., Wiedbrauk S., Mayer P., Dube H., Chem.‐Eur. J. 2017, 23, 6237. [DOI] [PubMed] [Google Scholar]
- 69. Huang C. Y., Bonasera A., Hristov L., Garmshausen Y., Schmidt B. M., Jacquemin D., Hecht S., J. Am. Chem. Soc. 2017, 139, 15205. [DOI] [PubMed] [Google Scholar]
- 70. Hoorens M. W. H., Medved M., Laurent A. D., Di Donato M., Fanetti S., Slappendel L., Hilbers M., Feringa B. L., Jan Buma W., Szymanski W., Nat. Commun. 2019, 10, 2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Köttner L., Ciekalski E., Dube H., Angew. Chem., Int. Ed. 2023, 62, 202312955. [DOI] [PubMed] [Google Scholar]
- 72. Thumser S., Köttner L., Hoffmann N., Mayer P., Dube H., J. Am. Chem. Soc. 2021, 143, 18251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dumur F., Eur. Polym. J. 2024, 203, 112697. [Google Scholar]
- 74. Yagi S., Maeda K., Nakazumi H., J. Mater. Chem. 1999, 9, 2991. [Google Scholar]
- 75. Ichimura K., Sakuragi M., J. Polym. Sci. Polym. Lett. Ed. 1988, 26, 185. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.