

Conspectus
Classical education in organic chemistry and catalysis, not least my own, has centered on two electron transformations, from nucleophilic attack to oxidative addition. The focus on two electron chemistry is well-founded, as this brand of chemistry has enabled incredible feats of synthesis, from development of life-saving pharmaceuticals to production of ubiquitous commodity chemicals. With that said, this approach is in many ways complementary to the approach of nature, where enzymes frequently make use of single electron “radical” steps to achieve challenging reactions with exceptional selectivity, including light detection and C─H hydroxylation. While the power of radical elementary steps is undeniable, fundamental understanding of -and ability to apply- these in catalysis remains underdeveloped, constraining the palette with which chemists can make new reactions.
Motivation to remedy this traditional underemphasis on radical catalysis has been intensified by the runaway success of outer-sphere photoredox catalysis, not only confirming the versatility of radicals in anthropogenic catalysis but also teaching the value of robust and well-understood catalytic cycles for reaction design. Indeed, I would argue the success of outer-sphere photoredox catalysis has been fueled by strong fundamental understanding of its underlying radical elementary steps, with consideration of single electron transfer (SET) energetics allowing new reactions to be designed de novo with enviable confidence. However, outer-sphere photoredox catalysis is an outlier in this regard, with other mechanistic approaches remaining underexplored.
Our research group is part of a growing movement to expand the vocabulary of synthetic radical catalysis beyond the traditional outer-sphere photoredox SET manifold, assembling new cycles comprised of hydrogen atom transfer (HAT), light-induced homolysis (LIH), and radical ligand transfer (RLT) steps in new combinations to achieve challenging transformations. These efforts have been made possible by ever-growing understanding of these radical elementary steps and discovery of catalyst systems with significant mechanistic flexibility, most recently iron/thiol (Fe/S) cocatalysis.
In this account, I will focus on our efforts applying HAT and LIH steps in Fe/S cocatalysis, sharing broad guidelines we have found helpful for using these steps and demonstrating how they can be combined to make new reactions using three case studies: radical hydrogenation (HAT + HAT), decarboxylative protonation (LIH + HAT), and alkene hydrofluoroalkylation (LIH + HAT, with an intervening radical alkene addition). These efforts have highlighted the importance of several key parameters, including bond dissociation energy (BDE) and radical polarity, and I hope our findings similarly provide a valuable framework to others designing new radical catalytic reactions.
Graphical Abstract

Introduction
Electrons often move in pairs. The double-barbed curly arrow is one of the first formalisms taught to aspiring organic chemists,5-8 in its simplest iteration denoting the movement of two electrons from a filled “HOMO” orbital to an empty “LUMO” orbital to form (and sometimes break) a bond. Mastering this fundamental principle of reactivity opens the possibility to describe a multitude of powerful synthetic reactions, from humble nucleophilic substitutions to doubly diastereoselective aldol reactions, many of which have been key steps in the synthesis of the most complex natural products and transformational pharmaceutical compounds. Similarly, organometallic chemists spend considerable time mastering “canonical” organometallic elementary steps9,10 consisting of two electron movements, including oxidative addition, reductive elimination, transmetallation, migratory insertion, de-insertion, coordination, and decoordination, which collectively can be used to describe (and design) a wide array of mechanistically-intriguing, synthetically-versatile, and industrially-critical transformations including alkene hydroformylation and myriad palladium-catalyzed cross couplings (Fig. 1a).11,12
Fig. 1.

Representative catalytic reaction using 2e− steps I learned in undergraduate chemistry (A), and ubiquitous catalytic reactions using radical, 1e− steps in nature (B)
In contrast, comparatively little time is spent on the single-electron single-barbed curly arrow in many introductory organic chemistry courses, with foundational radical transformations (hydrogen atom transfer, alkene addition etc) having little (and sometimes no) coverage. Similarly, introductory organometallic chemistry has often focused on late transition metals that often adhere to canonical organometallic steps,10 with early metals prone to a range of single electron transfer (SET), radical capture, and otherwise non-two electron steps13,14 often relegated to a brief discussion at the end of the course and minimally connected to catalysis. This was my experience,15 at least, and after finishing my introductory chemistry education, I came away with the distinct impression that “useful” chemistry goes through two electron steps and single electron steps are to be avoided or, at best, tolerated if two electron steps aren’t feasible, and this was doubly so for catalysis.16
Of course, this is not to say powerful radical reactions had not been invented and used by chemists long before I was in the classroom. Beautiful stoichiometric radical transformations, from the Hoffman-Löffler-Freytag reaction17-19 (with early work beginning in 1881!) of N-halo alkylamines to form cyclic amines via a N─X homolysis-initiated 1,5-Hydrogen Atom Transfer (HAT)/halogen atom transfer (XAT) radical chain C─H halogenation to the innovative Barton-McCombie Deoxygenation20 allowing alcohol removal via a radical chain reduction of the corresponding xanthate, have played a key and growing role in synthetic strategy. While many of these chain processes have perfused the graduate organic chemistry curriculum,21 truly catalytic reactions were again in short supply during my classroom training.
A notable chemist that received a very different education is Nature, whose billions of years of evolution has taught it to combine single electron steps relentlessly in the beautiful and complex machinery of life.13,22,23 Often, these reactions make use of the redox promiscuity of earth abundant metals to perform a controlled series of canonical single electron steps, from the HAT and radical ligand transfer (RLT)24 “radical rebound” reactivity of iron in cytochrome P45025,26 and non-heme oxygenases/halogenases27,28 to the light induced homolysis (LIH)29,30 and hydrogen atom transfer (HAT) reactivity of cobalt in the photodetection enzyme CarH (Fig. 1b).31,32 Radical reactivity also dominates in environmental chemistry, where such ubiquitous reactions as autooxidation and photochemical degradation of organics proceed through radical intermediates and enjoy significant acceleration via first row metal catalysis.33,34 If we look beyond the confines of the laboratory, radical catalysis is everywhere with mechanistic variety galore.
As I proceeded through my graduate and postdoctoral training, it was exciting to see an exponential growth in appreciation (and synthetic application) of radical reactions,16,35,36 with radical catalysis particularly experiencing a surge in interest over the last ~20 years.14,37-46 Several factors have undoubtedly driven this development, with better understanding of reactivity rules being key to enabling exquisitely selective reactions. Indeed, the intervening years have increasingly showed the historic view of “uncontrollable” radicals was more so a lack of understanding of how to control reactions, rather than inherent uncontrollability.35 Another factor has been the emergence of radical photocatalysis, with photoredox catalysis becoming a mainstay of organic methodology development.38-44,46 While the inherent reactivity of photoredox catalysis, allowing for radicals to be oxidatively or reductively generated from a wide variety of radical precursors, has been essential for its adoption, I would argue an equally important facet has been the robustness and fundamental understanding of the catalytic cycle (Fig. 2).38 Indeed, many photoredox catalytic reactions proceed through 1) photoexcitation of a well-defined catalyst, 2) outer-sphere electron transfer (OSET) between the excited state catalyst and a substrate to generate a radical and an oxidized or reduced catalyst, and 3) OSET between the oxidized or reduced catalyst and another species to regenerate the starting catalyst in its initial oxidation state.
Fig. 2.

Annotated representative photoredox catalytic cycle with conceptual strengths and the challenge its success invites for our group.
This simple, 3-step cycle allows for granular design of catalytic reactions, with measurement of substrate redox potentials permitting effective catalysis to be predicted based on corresponding excited and ground state potentials of the catalysts.38-44,46 Further, the well-behaved nature of this three-stage cycle positions photoredox catalysis to be combined with many different cocatalysts,46 including nickel organometallic catalysis.37 Indeed, our lab has also taken advantage of the well-behaved nature of outer-sphere photoredox catalysis to enable dual-catalytic reaction with a manganese radical ligand transfer (RLT) cocatalyst for alkene difunctionalization.47
While outer-sphere photoredox catalysis will doubtless continue to be a pillar of synthetic design, we nonetheless believe that additional mechanistic approaches are needed to unlock the full potential of radical catalysis. This belief led to the animating question for our research group: can we, as chemists, design catalytic methods beyond the mechanistic confines of precedented radical reactions? We hypothesized this will be possible via a twofold, interconnected approach: 1) increasing understanding of radical elementary steps beyond OSET in practical catalysis settings, allowing heuristics to be developed, and 2) demonstrating how this understanding can be applied to develop useful synthetic reactions. Toward realizing this goal, we have found cocatalytic systems of Fe and thiols (Fe/S) to be particularly flexible in accommodating different elementary steps1-4,48 and able to yield intriguing insights into the underlying mechanisms across both thermal and photochemical reactions.
Here, we will summarize a handful of useful design rules and heuristics for using radical elementary steps gleaned from our studies of Fe/S cocatalysis and demonstrate their application in several case studies.
1. Increased Understanding of Single Electron Steps in Catalytic Settings
Hydrogen Atom Transfer (HAT)
Hydrogen atom transfer (HAT) can be succinctly defined as a special case of proton-coupled electron transfer (PCET) with the simultaneous movement of one proton and one electron from one orbital to another, breaking one element-hydrogen bond and forming a new one.49 From the perspective of catalysis, one can imagine two different directions of HAT: transfer of the hydrogen atom from catalyst to substrate (donor HAT catalysis) and the inverse (acceptor HAT catalysis). We (and many others) have found both cases to be of significant utility in designing catalytic reactions. Some design heuristics that we have found useful are shown below.
HAT from Catalyst to Substrate (Donor HAT Catalysis)
Amongst the most important considerations of successful HAT from catalyst to substrate is the bond dissociation energy (BDE) of the catalyst─H bond, as exothermicity of HAT not only affects favorability of transfer but also has a striking impact on the reaction rate in many cases, with more exothermic HAT being more rapid and the overall behavior being well-described by Marcus Theory.50-52 Of course, the strength of the incipient substrate─H bond must also be considered in these cases, with weaker desired substrate─H bonds requiring commensurately weaker catalyst─H bonds. Regarding the practical use of donor HAT catalysts in synthetic organic chemistry, I would argue two important regimes are BDEcatalyst─H > 52 kcal•mol−1 (Fig. 3) and < 40 kcal•mol−1 (Fig. 4).
Fig. 3.

An example of strong (>52 kcal•mol−1) BDEcatalyst─H reactivity (A) and key characteristics: stability to hydrogen evolution (B) and effect of radical polarity (C)
Fig. 4.

An example of weak (<40 kcal•mol−1) BDEcatalyst─H reactivity (A) and key characteristics: instability to hydrogen evolution (B) and effect of radical polarity (C)
One reason for the importance of the upper range is purely practical: above ~52 kcal•mol−1, the HAT catalyst is thermodynamically stable to spontaneous hydrogen evolution (BDEH─H = 104 kcal•mol−1 with additional contributions from entropy),53 allowing for the active species to be present in high concentrations (Fig. 3b). Further, the relatively high strength of the catalyst─H bond limits the number of substrate functionalities that can be reduced, with formation of an exceptionally strong substrate─H bond required for catalysis. In many catalytic systems, including those developed in our lab,1-4,48,54 these high BDEcatalyst─H HAT donors are typically used to quench alkyl radicals to form strong (~100 kcal•mol−1)53 C─H bonds, resulting in the formation of a closed shell product.
Prototypical catalysts in this regime are thiols (BDES─H ~80-85 kcal•mol−1),53,55,56 though metal hydrides can also be found in this range and have been demonstrated in catalysis.57-61 In the case of thiols, the thiyl byproduct of HAT is commonly reduced via either electron transfer and proton transfer occurring stepwise or a concerted proton-coupled electron transfer (PCET), allowing the catalyst─H species to be regenerated reductively.55,56
In addition to bond strength, radical polarity matching is also important when considering reaction rate and selectivity,62-64 enabling for otherwise-challenging multicomponent reactions or selective activations to occur. Simply put, radical polarity matching posits that (similar to ionic reactions) radical reactions will occur more rapidly between species of opposite electron polarity, with electron rich “nucleophilic” HAT donors reacting more rapidly with electron deficient “electrophilic” radicals than electron rich “nucleophilic” radicals and vice versa. It has been proposed that the this HAT polarity matching effect arises from an asynchronous electron and proton transfer in the HAT transition state, with polar effects that increase the favorability of the proton and electron transfer components similarly stabilizing the transition state.64
In the realm of strong BDEcatalyst─H HAT donors, it is known that thiols function as electron deficient HAT donors, meaning that the electron rich cyclohexyl radical will be reduced via HAT much more rapidly than the electron deficient trifluoromethyl radical (Fig. 3c), despite the fact that the fluoroform C─H bond (106 kcal•mol−1)65 is significantly stronger than the cyclohexane C─H bond (~98 kcal•mol−1 for a linear methylene C─H).53 This property can be used to enable polarity reversal catalysis (PRC).62 and multicomponent reactions, including alkene hydrofluoroalkylation (case study 3, below).4
Below ~52 kcal•mol−1, the catalyst─H is unstable to hydrogen evolution,53 presenting a challenge due to this competitive process (Fig. 4b).66 While catalysis is known for species with effective catalyst─H BDEs > 40 kcal•mol−1 and < 52 kcal•mol−1 (e.g. cycloisomerization using cobaloxime catalysis),57,58 a synthetically-useful threshold is BDEcatalyst─H < 40 kcal•mol−1, as this places the strength of the catalyst─H bond comparable or below the typical C─H bond adjacent to a radical.48,66 A striking implication of this strength is that HAT from catalyst─H to unactivated alkenes is favored, allowing direct generation of carbon-centered radicals under mild conditions (Fig. 4). This reactivity is the basis of metal-catalyzed HAT (MHAT) catalysis and has been used for numerous radical hydrofunctionalization reactions, especially those using cobalt and iron catalysts.66-68
A notable quirk of many MHAT hydrofunctionalization reactions is the use of hydride (H─) reagents as the hydrogen atom (H•) donor, requiring a single electron oxidant (e.g. TBHP or electrophilic fluorine sources) to return the post-HAT metal center to the oxidation state capable of accepting the hydride ligand, effectively generating H• from H─ and oxidant.67,68 Simple molecular oxygen can also function as the terminal oxidant for MHAT in alkene hydration reactions; however, this oxidant is challenging to use in other hydrofunctionalizations due to its rapid capture of carbon-centered radicals via C─O bond formation.67,69 While this overall approach is effective for many reactions, it would be more redox efficient to avoid stoichiometric co-oxidants for MHAT reactions. This could be accomplished by reoxidizing the metal center using a catalytically-generated oxidant.
With respect to polarity in the weak HAT donor regime, FeIII─H species often behave as “electrophilic” HAT donors, leading to rapid reduction of electron rich, unactivated alkenes (e.g. 1,1-disubstituted aliphatic alkenes) and sluggish reaction of electron-deficient alkenes (e.g. acrylates).66,70-72 This difference in reactivity has been used for alkene-alkene cross coupling, where rapid MHAT to an unactivated alkene generates an electron-rich “nucleophilic” alkyl radical which can then undergo polarity-matched addition to an electron deficient acrylate, allowing selective cross-reaction (Fig. 4c).
HAT from Substrate to Catalyst (Acceptor HAT)
Like all reactions, HAT can also occur in the microscopic reverse, with a catalyst accepting H• from an organic substrate. As we have not leveraged this reactivity using Fe/S cocatalysis, we will not focus on this case in this review. However, we have found this to be incredibly useful and have leveraged both excited state photo-HAT73 and ground state acceptor HAT of both closed shell74 and open shell75 substrates to achieve C─H azidation and alkene formation. Several strong reviews are available in these areas for the interested reader.48,76-80
Light Induced Homolysis (LIH)
One of the first radical reactions I learned in introductory organic chemistry was the homolytic photolysis of Cl2 to liberate two Cl• which could then activate alkanes via HAT and, eventually, form alkyl chlorides (Fig. 5a).8 While this particular reaction is challenging to execute on the laboratory scale (not least due to use of Cl2!),81 it indelibly connected radical photochemistry with bond homolysis and generation of reactive radical intermediates in my mind. I recall wondering what other bonds could be broken using light energy, and whether less arduous precursors could liberate reactive intermediates like Cl•.
Fig. 5.

My first exposure to light induced homolysis (LIH, A) and the analogous (and much more synthetically tractable) reactivity of metal anion salts (B)
Fortuitously, pioneers including Kochi82 and Shul’Pin83,84 had considered this same fundamental question years before, discovering that simple inorganic salts such as CuCl2 and FeCl3 could undergo light-induced homolysis (LIH)29 to liberate Cl• which could then participate in its characteristic reactions, including alkane functionalization and alkene dichlorination (Fig. 5b). These processes are often proposed to occur via ligand-to-metal charge transfer (LMCT),30 though elucidating the precise mechanism(s) of LIH in these systems is ongoing. Already, it is known that the lifetimes of these photoexcited states are exceptionally short,29,30,85 a liability for outer-sphere photoredox catalysis due to the need for sufficient lifetime to enable bimolecular reaction;38 however, this is a non-issue for LIH reactions as these are intramolecular, opening the door to the use of first row transition metals (e.g. Fe)83,85,86 typically avoided due to their short excited state lifetimes outside of specific ligand sets. Interestingly, this LIH reaction is balanced by single electron reduction of the metal, opening the door to catalysis if a suitable oxidative process could be paired with this LIH step.29,30
As per usual, chemists were preceded by nature in this reactivity as demonstrated by beautiful work from Drennan,31,32 who found the photo-step of light detection enzyme CarH is LIH of the Co(III)─C bond of adenosyl VB12, generating an alkyl radical that then undergoes acceptor HAT to Co(II) to generate an alkene and Co(III)─H (see Fig. 1b above!). This intriguing reactivity has underpinned many recent efforts in VB12 photocatalysis, including from our group,22,54,75,87 and several recent reviews are available in this vein.88-90
The true potential of LIH as a synthetic step is hinted at above: many [M]─X bonds can be photolyzed beyond [M]─Cl,29,30 with beautiful work demonstrating steps including decarboxylation of metal carboxylates,3,4,91-98 oxyl radical generation,99 and azide radical generation (Fig. 5b).100-102 These reactions are remarkable from a synthetic standpoint, as they allow a wide variety of radicals to be generated using simple metal salts (e.g. Cu(II) and Fe(III)) and anionic radical precursors (e.g. Cl, carboxylate, alkoxide, azide), avoiding the use of reactive activators such as hypervalent iodine reagents103 common in historic generation of these species. Interestingly, the facility of LIH substrate activation appears to vary based on metal; for example, decarboxylation of sp2 acids (e.g. benzoic acids) proceeds readily with Cu94-96 and decarboxylation of fluorocarboxylic acids (e.g. trifluoroacetic acid) occurs with great facility using Fe.4,104 The idiosyncratic reactivity of LIH precursors with diverse substrates and impacts of ligands, solvents, and light source is an active area of investigation and we anticipate additional heuristics emerging over the coming years. While many of these reactions have used stoichiometric metal,94,95,97,98 some have also shown how privileged oxidants such as electrophilic fluorine sources96,101 and nitrate91,100 can permit catalysis, opening the door for inner-sphere photocatalysis. Additionally, our work in the realm of radical hydrogenation1,2,48 has shown a suitable thiyl oxidant equivalent can be generated in situ with thiol cocatalysis, enabling intriguing dual catalytic reactions including decarboxylative protonation3 and alkene hydrofluoroalkylation4 with no external oxidant (See below).
2. Demonstrations of New Combinations of Single Electron Steps in Catalytic Reactions
Case 1: Donor HAT + Donor HAT for Alkene Hydrogenation
An early question in our research group posed by Dr. Venkatesh Kattamuri was whether we could combine HAT Donor catalysts in both weak (<40 kcal•mol−1) and strong (~80 kcal•mol−1) catalyst─H reactivity regimes to achieve efficient alkene hydrogenation using a radical mechanism. We were inspired by beautiful early work by Shenvi105,106 and Herzon107,108 who had shown that weak BDEcatalyst─H donors could catalytically hydrogenate alkenes via a stepwise HAT radical mechanism (Fig. 6a). These early studies demonstrated some significant advantages for HAT hydrogenation, including complementary (trans) stereoselectivity and functional group tolerance (e.g. vinyl halides) compared to organometallic methods.105-108
Fig. 6.

Mechanistic design and proposed catalytic cycles for radical hydrogenation via both metal-catalyzed hydrogen atom transfer (MHAT, A and B) and cooperative hydrogen atom transfer (cHAT, C and D)
Mechanistically, both reactions are thought to go through a Mukaiyama-type MHAT cycle,66-68 where HAT to the alkene from a weak M(III)─H generates an alkyl radical and reduced M(II). Reoxidation by a stoichiometric single electron oxidant in the presence of a H─ donor (typically a silane, e.g. PhSiH3) then regenerates the M(III)─H state, which can reduce the alkyl radical to the desired alkane (Fig. 6b). Thus, two turns of the MHAT cycle allow alkene hydrogenation.
As discussed above, the strengths of bonds being formed are quite different (33 kcal•mol−1 and 100 kcal•mol−1 for cyclohexene to cyclohexane),48 making it energetically-feasible for each donor HAT to be done by a different catalyst, for example an Fe(III)─H (BDEFe─H ~17 kcal•mol−1)72 to form the weaker bond and PhS─H (BDES─H 84 kcal•mol−1)53 to form the stronger one (Fig. 6c). In considering the stoichiometric byproducts of successive donor HAT from Fe(III)─H and PhS─H, we realized that we would (presumably) form reducing Fe(II)72 and oxidizing PhS•55,56 in situ, which might productively interact in the presence of H─ (e.g. PhSiH3) and H+ (e.g. EtOH) to regenerate our starting catalyst─H species in the absence of a stoichiometric oxidant (Fig. 6d). Further, since each H• is derived from an orthogonal reagent, this design allows for selective deuteration using reagent control that is not possible using prior MHAT methods.109
Happily, we found this reaction design to be viable,1 and were able to hydrogenate a range of alkenes with largely recapitulated functional group tolerance compared to prior MHAT hydrogenations.105-108 Further, we were able to achieve selective and controllable hydrodeuteration using orthogonal reagents, opening the door to isotope labeling applications (Fig. 7a).
Fig. 7.
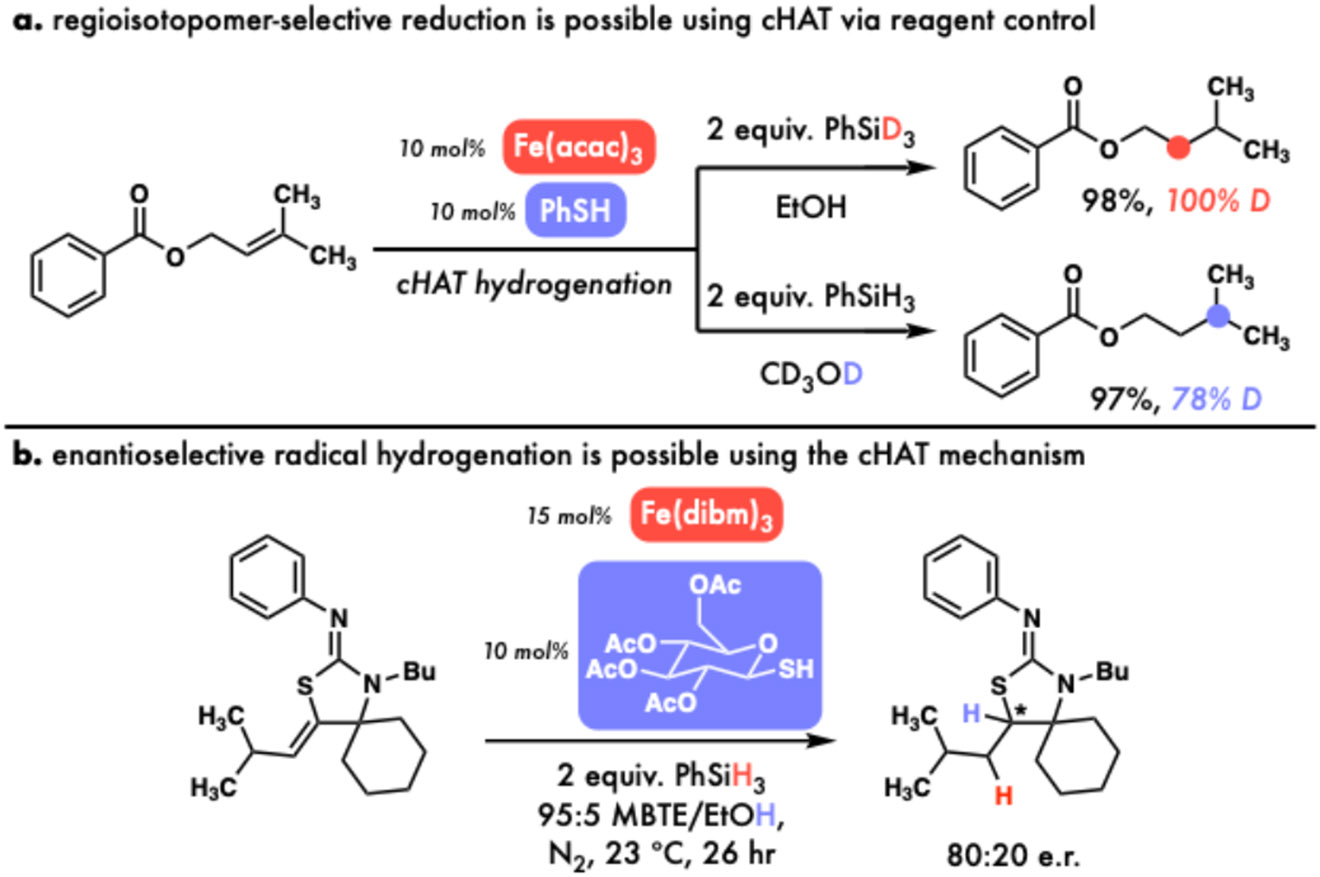
The cHAT mechanism provides new synthetic opportunities, including regioselective isotopomer synthesis (A) and asymmetric radical hydrogenation (B)
An interesting further implication of this dual catalytic design is the opportunity to tune each catalyst independently, with pioneering work from Roberts62,110,111 and recent innovations from Miller,112 Ye,113 and Maimone114 in asymmetric HAT using chiral thiols suggesting that a similar approach might enable the first enantioselective radical alkene hydrogenation. Sarah Buzsaki from our group and Savannah Mason from Scott Miller’s group sought to explore this in what grew to be a large and enriching collaboration, finding that chiral thiols could indeed induce asymmetric radical hydrogenation, with thioglycosides such as those used by Roberts providing the highest levels of enantioselectivity and reactivity observed thus far (Fig. 7b), though ample room for improvement remains.2
In the course of this collaboration, we were lucky to engage the expertise of Pat Holland and his group to simultaneously investigate the mechanism in finer detail, focusing on the redox interaction of the two catalysts which we had been unable to probe in our initial study.1 Intriguingly, computational, spectroscopic, and intermediate studies support this turnover occurring via a PCET process72 involving a thiol/Fe(II) complex to the alkyl radical intermediate, directly forming the thiolate/Fe(III) successor complex in the same step as HAT from the strong catalyst─H HAT donor, at least in aprotic, polar media (Fig. 8a).2 This finding not only increases fundamental understanding of thiol/Fe redox interaction in putative catalytic cycles, but also suggests that Fe might be involved in the second HAT step, providing another means of tuning enantioselectivity. Indeed, we were able to preliminarily explore this possibility and found a small, though nonzero, improvement in enantioinduction with a bulkier (though still achiral) ligand on Fe, supporting participation in the enantiodetermining step, though additional work is needed to fully investigate the origin this effect (Fig. 8b).
Fig. 8.

Further study implicates the Strong BDES─H HAT to occur via proton-coupled electron transfer from an FeII/thiol complex, resulting in effective bond weakening (A). This hypothesis is consistent with a small change in enantioselectivity based on achiral Fe ligand identity, suggesting Fe might participate in the enantiodetermining Strong BDES─H HAT step (B).
Case 2: LIH + Donor HAT for Decarboxylative Protonation
Having established the mutual compatibility of Fe and thiol HAT radical cocatalysis in our group and building on a parallel investigation of LIH/HAT cascades using VB12,22,54,75,87 Yen-Chu Lu wondered whether the multifaceted reactivity of Fe could be leveraged to enable new photocatalytic reactions in conjunction with donor HAT. In particular, he was inspired by beautiful early photocatalysis studies by Jin92,93 and others85,86,100 showing that Fe(III) carboxylates readily undergo LIH (presumably via LMCT)30 to perpetrate radical decarboxylation, liberating an alkyl radical able to perform Giese-type addition to electron deficient olefins92 or Minisci-type substitution of aromatic rings.93 The byproduct of this photoreaction is Fe(II), similar to the post-HAT Fe(II) species we encountered in our radical cHAT hydrogenation,1,48 leading Yen-Chu to consider whether a thiyl radical, generated in situ via thiol donor HAT (or the PCET process described above)2 might similarly enable reoxidation of Fe(II) to Fe(III) and reduction of thiyl to thiol (in the presence of H+) and permit photocatalysis coupled to HAT catalysis (Fig. 9).
Fig. 9.

Mechanistic design (A) and proposed catalytic cycle (B) for decarboxylative protonation via tandem LIH/HAT using Fe/S cocatalysis with example conditions and results (C).
Toward testing our hypothesis that Fe/S cocatalysis could also be effective photochemically, Yen-Chu realized that the simplest possible reaction we could build using LIH and HAT would be decarboxylative protonation of carboxylic acids, or the “deletion” of carboxylic acid group through replacement with a C─H bond (Fig. 9a).3 In a happy synergy, this reaction also happens to be quite useful (and challenging) synthetically, often requiring pre-activation of the acid (e.g. as a thiohydroxamate ester, as per the Barton Decarboxylation)115 and treatment with stoichiometric HAT reagents (e.g. Bu3Sn─H). Yen-Chu went to the lab and rapidly found that minor modifications of the Fe/S hydrogenation system, most notably absence of silane, use of slightly different Fe and thiol sources, and biphasic conditions, allowed the reaction to proceed in good efficiency (Fig. 9c). Interestingly, a ligand was also found to accelerate the reaction; however, this acceleration effect is relatively modest (< 2x). Consistent with prior work on LIH decarboxylation,29,30 our Fe-based system is ineffective for decarboxylating Csp2 acids (e.g. benzoic acids), a contrast to recent Cu-based LIH decarboxylative functionalizations.94-96 Further, the method is surprisingly scalable in batch configuration, allowing for gram-scale synthesis of decarboxylated products.
Mechanistic investigation is consistent with the broad mechanistic features of our initial design,3 with rearrangement and TEMPO studies supporting intermediacy of radicals and kinetics studies showing the reaction to require light and be light-limited in many cases, a result consistent with radical decarboxylation via LIH. Interestingly, a primary KIE was observed when performing deuterium atom transfer (DAT) instead of HAT for a substrate forming a relatively weak (~90 kcal•mol−1) bond and not observed when forming a relatively strong (~100 kcal•mol−1) bond, showing this step can exhibit turnover limiting character in cases where there is relatively little enthalpic driving force for HAT (Fig. 10).3 This result is consistent with the work of Mayer showing HAT rates can be strongly impacted by thermodynamic driving force and be represented using a Marcus-type relation.50-52 Together, these results taught us that the Fe/S cocatalyst system is extensible to photocatalysis; however, both the LIH and HAT step can exhibit rate limiting character depending on the substrate architecture, an important consideration for designing reactions in this space.
Fig. 10.
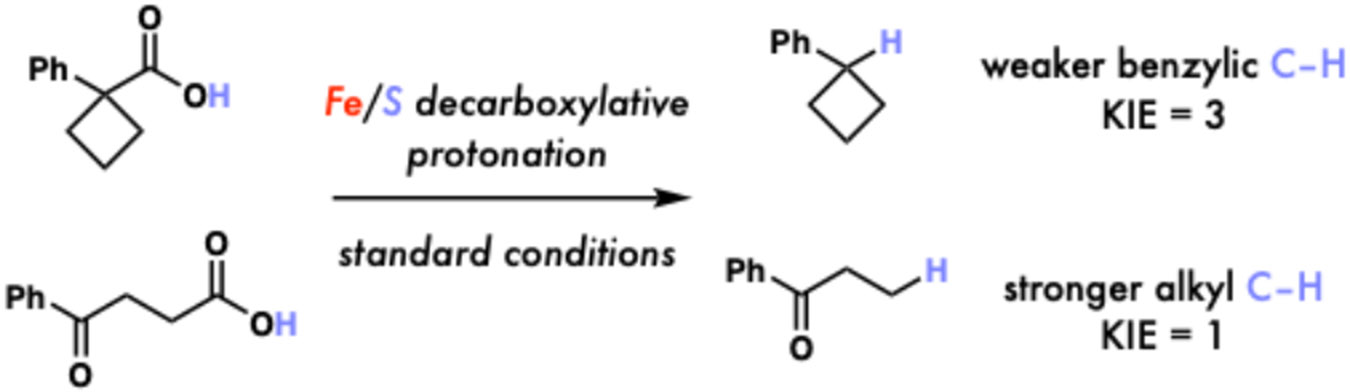
Kinetic isotope effect (KIE) results vary based on substrate identity, suggesting strength of substrate C─H bond must be considered in reaction design.
Case 3: LIH + Donor HAT for Alkene Hydrofluoroalkylation
Yen-Chu began discussing Fe/S photocatalysis with another graduate student in our lab, Kang-Jie (Harry) Bian, and they hypothesized that the broad Csp3 decarboxylation of Fe LIH photocatalysis should enable even recalcitrant perfluoroalkyl carboxylic acids (PFCAs)116 to be activated, allowing for versatile perfluoroalkyl radicals (e.g. •CF3)117 to be generated directly from commodity PFCAs (e.g. trifluoroacetic acid, TFA). This LIH approach could be particularly appealing in a logistical sense as PFCAs are exceptionally stable (so stable, in fact, that they are a major environmental contaminant)116 and easily handled liquids. Further, PFCAs are commercially available in a wide variety of structures, presenting many operational advantages over the bespoke synthetic reagents (e.g. Togni’s hypervalent iodine reagent)118 or gaseous/low boiling perfluoroalkyl bromides/iodides commonly used in previous methods.117
Aside from synthetic ease, a key opportunity from this PFCA decarboxylation would be inclusion of an additional elementary step in a Fe/S cocatalytic cycle: radical addition to an alkene (Fig. 11a and 11b).119,120 We hypothesized that this additional step would be possible due to radical polarity matching as discussed in Fig 2c above, with the putative electron deficient perlfluoroalkyl radicals63 being slowly reduced by electron deficient thiol HAT donors from polarity mismatching.62 This slow HAT direct rate would allow sufficient lifetime of the perfluoroalkyl radical to add (in a polarity matched fashion, Fig. 11a) to electron-rich alkenes, generating electron-rich alkyl successor radicals which could then be rapidly reduced by a thiol HAT donor in a polarity-matched fashion. Thus, we could “stitch” an alkene in between the LIH and HAT steps to enable both C─C and C─H bond formation. Outside of this addition step, the proposed catalytic cycle of this Fe/S system remains unchanged from the decarboxylative protonation reaction.3 Happily, we found this hypothesis to be borne out in the reaction flask, allowing for hydrofluoroalkylation of alkenes using Fe/S cocatalysis (example conditions and results in Fig. 11c).4 However, in a somewhat perplexing departure from the decarboxylative protonation,3 this reaction functions most efficiently in monophasic solution and is significantly impeded by inclusion of ligands that were beneficial in the previous case. The basis of these differences is non-obvious and we conclude at interim that ligand effects remain largely empirical in Fe LIH catalysis.
Fig. 11.

Mechanistic design (A) and proposed catalytic cycle (B) for alkene hydrofluoroalkylation via tandem LIH/HAT using Fe/S cocatalysis with example conditions and results (C). Radical polarity is particularly critical for success of this reaction.
Aside from illustrating the importance of polarity matching in radical catalytic design, this reaction further confirmed the generality of Fe LIH catalysis for Csp3 decarboxylation, allowing a wide range of commercial fluorocarboxylic acids, from TFA to perfluorooctanoic acid (PFOA), to serve as fluoroalkyl donors,4 suggesting that we have not yet found the limit of this activation mode. We continue to explore this in our ongoing LIH catalysis program.
Conclusion and Outlook
Together, we have identified useful design rules for using radical elementary steps including HAT and LIH in catalysis and applied these to rationally construct a number of unprecedented radical catalytic cycles using Fe/S cocatalysis. Not only have these cycles allowed us to further refine our understanding of these steps in catalytic settings, but also achieve synthetically-relevant transformations, including asymmetric hydrogenation, decarboxylative protonation, and alkene hydrofluoroalkylation.
We envision this work as a step toward a future where single electron catalysis is co-equal to two-electron catalysis in both understanding and use cases, though much work remains. Particularly, identification of alternative robust catalyst systems similar to Fe/S able to accommodate many different radical elementary steps will be essential to exploring new mechanistic space. With that said, immediate questions certainly remain in Fe/S cocatalysis, including understanding the impacts of ligand on LIH and how to leverage PCET turnover of putative Fe(II)–thiol species for higher efficiency and enantioselectivity in reductive reactions. Additionally, our experience has shown radical polarity to be a key, though often underappreciated, ingredient for selective catalysis. Increased understanding of how to modulate polarity matching will doubtless accelerate development of selective single electron catalysis.
We are resolved to continue working toward this goal and hope these efforts similarly empower the catalysis community to construct increasingly diverse radical catalytic reactions for challenging and valuable product architectures.
Acknowledgements
I acknowledge financial support from CPRIT (RR190025), NIH (R35GM142738), the Welch Foundation (C-2085), RCSA (CS-CSA-2023-007), and ACS-PRF (62397-DNI1), and Eli Lilly and Company (A-36829). I am a CPRIT Scholar in Cancer Research. Finally, I acknowledge a fantastic collection of students and collaborators who have driven all discussed chemistry and brought their creativity, enthusiasm, and persistence to our lab.
Biography
Julian G. West received his B.Sc.H. from University of British Columbia, Vancouver (with Glenn Sammis) in 2013 and Ph.D. from Princeton University (with Erik Sorensen) in 2017. He pursued a postdoc at Caltech (with Harry Gray and Brian Stoltz) from 2017-2019 and began his independent career at Rice in 2019 where he has been lucky to work with some fantastic mentees.
Footnotes
The author declares no competing financial interest.
References
- (1). Kattamuri PV; West JG Hydrogenation of Alkenes via Cooperative Hydrogen Atom Transfer. J. Am. Chem. Soc 2020, 142 (45), 19316–19326. 10.1021/jacs.0c09544. This paper introduces Fe/S cocatalysis in the context of alkene hydrogenation via cooperative hydrogen atom transfer (cHAT). Lessons from this first study have been critical in further development of Fe/S cocatalysis and our understanding of HAT reactions.
- (2). Buzsaki SR; Mason SM; Kattamuri PV; Serviano JMI; Rodriguez DN; Wilson CV; Hood DM; Ellefsen JD; Lu Y-C; Kan J; West JG; Miller SJ; Holland PL Fe/Thiol Cooperative Hydrogen Atom Transfer Olefin Hydrogenation: Mechanistic Insights That Inform Enantioselective Catalysis. J. Am. Chem. Soc 2024, 146 (25), 17296–17310. 10.1021/jacs.4c04047. This paper further explored the mechanism of Fe/S hydrogenation and leveraged these insights to enable the first asymmetric radical alkene hydrogenation.
- (3). Lu Y-C; West JG Chemoselective Decarboxylative Protonation Enabled by Cooperative Earth-Abundant Element Catalysis. Angew. Chem. Int. Ed 2023, 62 (3), e202213055. 10.1002/anie.202213055. This paper was our first foray into photocatalysis using Fe/S and provided a preliminary mechanistic framework for developing future reactions using a combination of light-induced homolysis (LIH) and HAT.
- (4). Bian K-J; Lu Y-C; Nemoto D; Kao S-C; Chen X; West JG Photocatalytic Hydrofluoroalkylation of Alkenes with Carboxylic Acids. Nat. Chem 2023, 15 (12), 1683–1692. 10.1038/s41557-023-01365-0. This paper leverages our insights from decarboxylative protonation using Fe/S photocatalysis in conjunction with radical polarity considerations to allow decarboxylative hydrofluoroalkylation of alkenes, teaching how radical polarity considerations can enable more complex reaction schemes.
- (5).Loudon GM; Parise J Organic Chemistry, Seventh edition.; Macmillan Learning: Austin, 2021. [Google Scholar]
- (6).Clayden J; Greeves N; Warren S Organic Chemistry, 2nd ed.; Oxford university press: Oxford, 2012. [Google Scholar]
- (7).Klein DR Organic Chemistry as a Second Language: First Semester Topics, 5e ed.; Wiley: Hoboken, NJ, 2020. [Google Scholar]
- (8).Ciufolini MA Chemistry 203: Organic Chemistry I, 2010. [Google Scholar]
- (9).Koszinowski K. Observation and Characterization of Single Elementary Reactions of Organometallics. Organometallics 2024, 43 (3), 205–218. 10.1021/acs.organomet.3c00415. [DOI] [Google Scholar]
- (10).Cotton FA; Wilkinson G; Murillo C. a.; Bochmann M Advanced Inorganic Chemistry, 6th Edition; Wiley-Interscience: New York, 1999. [Google Scholar]
- (11).Organometallic Chemistry in Industry: A Practical Approach; Colacot TJ, Johansson Seechurn CCC, Grubbs RH, Eds.; Wiley-VCH: Weinheim, 2020. [Google Scholar]
- (12).Parshall GW; Putscher RE Organometallic Chemistry and Catalysis in Industry. J. Chem. Educ 1986, 63 (3), 189. 10.1021/ed063p189. [DOI] [Google Scholar]
- (13).van Leest NP; Epping RFJ; van Vliet KM; Lankelma M; van den Heuvel EJ; Heijtbrink N; Broersen R; de Bruin B Chapter Two - Single-Electron Elementary Steps in Homogeneous Organometallic Catalysis. In Advances in Organometallic Chemistry; Pérez PJ, Stone FGA, West R, Eds.; Academic Press, 2018; Vol. 70, pp 71–180. 10.1016/bs.adomc.2018.07.002. [DOI] [Google Scholar]
- (14).van Leest NP; de Zwart FJ; Zhou M; de Bruin B Controlling Radical-Type Single-Electron Elementary Steps in Catalysis with Redox-Active Ligands and Substrates. JACS Au 2021, 1 (8), 1101–1115. 10.1021/jacsau.1c00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Legzdins P. Chemistry 310: D and F Block - Chemistry and Applications, 2012. [Google Scholar]
- (16).Zard SZ Radicals in Action: A Festival of Radical Transformations. Org. Lett 2017, 19 (6), 1257–1269. 10.1021/acs.orglett.7b00531. [DOI] [PubMed] [Google Scholar]
- (17).Hofmann AW Ueber Die Einwirkung Des Broms in Alkalischer Lösung Auf Amide. Berichte Dtsch. Chem. Ges 1881, 14 (2), 2725–2736. 10.1002/cber.188101402242. [DOI] [Google Scholar]
- (18).Hofmann AW Zur Kenntniss Der Coniin-Gruppe. Berichte Dtsch. Chem. Ges 1885, 18 (1), 5–23. 10.1002/cber.18850180103. [DOI] [Google Scholar]
- (19).Löffler K; Freytag C Über Eine Neue Bildungsweise von N-Alkylierten Pyrrolidinen. Berichte Dtsch. Chem. Ges 1909, 42 (3), 3427–3431. 10.1002/cber.19090420377. [DOI] [Google Scholar]
- (20).Barton DHR; McCombie SW A New Method for the Deoxygenation of Secondary Alcohols. J. Chem. Soc. Perkin 1 1975, No. 16, 1574–1585. 10.1039/P19750001574. [DOI] [Google Scholar]
- (21).Sammis GM Chemistry 411: Advanced Organic Chemistry, 2012. [Google Scholar]
- (22).West JG A Blueprint for Green Chemists: Lessons from Nature for Sustainable Synthesis. Pure Appl. Chem 2021, 93 (5), 537–549. 10.1515/pac-2021-0107. [DOI] [Google Scholar]
- (23).Bullock RM; Chen JG; Gagliardi L; Chirik PJ; Farha OK; Hendon CH; Jones CW; Keith JA; Klosin J; Minteer SD; Morris RH; Radosevich AT; Rauchfuss TB; Strotman NA; Vojvodic A; Ward TR; Yang JY; Surendranath Y Using Nature’s Blueprint to Expand Catalysis with Earth-Abundant Metals. Science 2020, 369 (6505). 10.1126/science.abc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jr DTN; Bian K-J; Kao S-C; West JG Radical Ligand Transfer: A General Strategy for Radical Functionalization. Beilstein J. Org. Chem 2023, 19 (1), 1225–1233. 10.3762/bjoc.19.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Huang X; Groves JT Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C─H Activation. J. Biol. Inorg. Chem 2017, 22 (2), 185–207. 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Groves JT Enzymatic C─H Bond Activation: Using Push to Get Pull. Nat. Chem 2014, 6 (2), 89–91. 10.1038/nchem.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Que L. The Road to Non-Heme Oxoferryls and Beyond. Acc. Chem. Res 2007, 40 (7), 493–500. 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- (28).Que L Jr; Tolman WB Biologically Inspired Oxidation Catalysis. Nature 2008, 455 (7211), 333–340. 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- (29).Abderrazak Y; Bhattacharyya A; Reiser O Visible-Light-Induced Homolysis of Earth-Abundant Metal-Substrate Complexes: A Complementary Activation Strategy in Photoredox Catalysis. Angew. Chem. Int. Ed 2021, 60 (39), 21100–21115. 10.1002/anie.202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Juliá F. Ligand-to-Metal Charge Transfer (LMCT) Photochemistry at 3d-Metal Complexes: An Emerging Tool for Sustainable Organic Synthesis. ChemCatChem 2022, 14 (19), e202200916. 10.1002/cctc.202200916. [DOI] [Google Scholar]
- (31).Jost M; Fernández-Zapata J; Polanco MC; Ortiz-Guerrero JM; Chen PYT; Kang G; Padmanabhan S; Elías-Arnanz M; Drennan CL Structural Basis for Gene Regulation by a B12-Dependent Photoreceptor. Nature 2015, 526 (7574), 536–541. 10.1038/nature14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jost M; Simpson JH; Drennan CL The Transcription Factor CarH Safeguards Use of Adenosylcobalamin as a Light Sensor by Altering the Photolysis Products. Biochemistry 2015, 54 (21), 3231–3234. 10.1021/acs.biochem.5b00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gen M; Zhang R; Li Y; Chan CK Multiphase Photochemistry of Iron-Chloride Containing Particles as a Source of Aqueous Chlorine Radicals and Its Effect on Sulfate Production. Environ. Sci. Technol 2020, 54 (16), 9862–9871. 10.1021/acs.est.0c01540. [DOI] [PubMed] [Google Scholar]
- (34).Stahl J; König B A Survey of the Iron Ligand-to-Metal Charge Transfer Chemistry in Water. Green Chem. 2024, 26 (6), 3058–3071. 10.1039/D3GC04595A. [DOI] [Google Scholar]
- (35).Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc 2016, 138 (39), 12692–12714. 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Stephenson CRJ; Studer A; Curran DP The Renaissance of Organic Radical Chemistry – Deja vu All over Again. Beilstein J. Org. Chem 2013, 9, 2778–2780. 10.3762/bjoc.9.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lee W-CC; Zhang XP Metalloradical Catalysis: General Approach for Controlling Reactivity and Selectivity of Homolytic Radical Reactions. Angew. Chem. Int. Ed 2024, 63 (20), e202320243. 10.1002/anie.202320243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Arias-Rotondo DM; McCusker JK The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev 2016, 45 (21), 5803–5820. 10.1039/C6CS00526H. [DOI] [PubMed] [Google Scholar]
- (39).Beatty JW; Stephenson CRJ Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res 2015, 48 (5), 1474–1484. 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2022, 122 (2), 1485–1542. 10.1021/acs.chemrev.1c00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Margrey KA; Nicewicz DA A General Approach to Catalytic Alkene Anti-Markovnikov Hydrofunctionalization Reactions via Acridinium Photoredox Catalysis. Acc. Chem. Res 2016, 49 (9), 1997–2006. 10.1021/acs.accounts.6b00304. [DOI] [PubMed] [Google Scholar]
- (42).Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem Soc Rev 2011, 40 (1), 102–113. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- (43).Shaw MH; Twilton J; Macmillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113 (7), 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nagib DA Asymmetric Catalysis in Radical Chemistry. Chem. Rev 2022, 122 (21), 15989–15992. 10.1021/acs.chemrev.2c00622. [DOI] [PubMed] [Google Scholar]
- (46).Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bian K-J; Nemoto DT Jr; Kao S-C; He Y; Li Y; Wang X-S; West JG Modular Difunctionalization of Unactivated Alkenes through Bio-Inspired Radical Ligand Transfer Catalysis. J. Am. Chem. Soc 144 (26), 11810–11821. 10.1021/jacs.2c04188. [DOI] [PubMed] [Google Scholar]
- (48).Kattamuri PV; West JG Cooperative Hydrogen Atom Transfer: From Theory to Applications. Synlett 2021, 32 (12), 1179–1186. 10.1055/a-1463-9527. [DOI] [Google Scholar]
- (49).Buzsaki SR; Bian K-J; West JG HAT Lessons Help Hydrogen Hop, Skip, and Jump. Trends Chem. 2022, 4 (12), 1062–1064. 10.1016/j.trechm.2022.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res 2011, 44, 36–46. 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Mayer JM A Simple Marcus-Theory Type Model for Hydrogen Atom Transfer/Proton-Coupled Electron Transfer. J. Phys. Chem. Lett 2011, 2 (12), 1481–1489. 10.1021/jz200021y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Roth JP; Yoder JC; Won T; Mayer JM Application of the Marcus Cross Relation to Hydrogen Atom Transfer Reactions. Science 2001, 294, 2524–2526. 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- (53).Luo YR Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, 2007. 10.1201/9781420007282. [DOI] [Google Scholar]
- (54).Funk BE; Pauze M; Lu Y-C; Moser AJ; Wolf G; West JG Vitamin B12 and Hydrogen Atom Transfer Cooperative Catalysis as a Hydride Nucleophile Mimic in Epoxide Ring Opening. Cell Rep. Phys. Sci 2023, 4 (4), 101372. 10.1016/j.xcrp.2023.101372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Thiols as Powerful Atom Transfer Catalyst: Opportunities in Photoredox-Mediated Reactions. Adv. Synth. Catal 2023, 365 (14), 2299–2309. 10.1002/adsc.202300535. [DOI] [Google Scholar]
- (56).Dénès F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114 (5), 2587–2693. 10.1021/cr400441m. [DOI] [PubMed] [Google Scholar]
- (57).Choi J; Tang L; Norton JR Kinetics of Hydrogen Atom Transfer from (H5-C5H5)Cr(CO)3H to Various Olefins: Influence of Olefin Structure. J. Am. Chem. Soc 2007, 129 (1), 234–240. 10.1021/ja066325i. [DOI] [PubMed] [Google Scholar]
- (58).Li G; Pulling ME; Estes DP; Norton JR Cobaloxime-Mediated Radical Cyclization under H2: Evidence for Formation of a Co─H Bond and Kinetics of H• Transfer. J Am Chem Soc 2014, 134, 14662–14662. [DOI] [PubMed] [Google Scholar]
- (59).Li G; Han A; Pulling ME; Estes DP; Norton JR Evidence for Formation of a Co-H Bond from (H2O) 2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc 2012, 134 (36), 14662–14665. 10.1021/ja306037w. [DOI] [PubMed] [Google Scholar]
- (60).Bullock RM; Samsel EG Hydrogen Atom Transfer Reactions of Transition-Metal Hydrides. Kinetics and Mechanism of the Hydrogenation of a-Cyclopropylstyrene by Metal Carbonyl Hydrides. J. Am. Chem. Soc 1990, 112, 6886–6898. 10.1007/s41061-016-0018-2. [DOI] [Google Scholar]
- (61).Wiedner ES; Chambers MB; Pitman CL; Bullock RM; Miller AJM; Appel AM Thermodynamic Hydricity of Transition Metal Hydrides. Chem. Rev 2016, 116 (15), 8655–8692. 10.1021/acs.chemrev.6b00168. [DOI] [PubMed] [Google Scholar]
- (62).Roberts P, Polarity-Reversal B Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem. Soc. Rev, 1999, 28, 25–35. 10.1039/A804291H. [DOI] [Google Scholar]
- (63).Parsaee F; Senarathna MC; Kannangara PB; Alexander SN; Arche PDE; Welin ER Radical Philicity and Its Role in Selective Organic Transformations. Nat. Rev. Chem 2021, 5 (7), 486–499. 10.1038/s41570-021-00284-3. [DOI] [PubMed] [Google Scholar]
- (64).Groff BD; Koronkiewicz B; Mayer JM Polar Effects in Hydrogen Atom Transfer Reactions from a Proton-Coupled Electron Transfer (PCET) Perspective: Abstractions from Toluenes. J. Org. Chem 2023, 88 (23), 16259–16269. 10.1021/acs.joc.3c01748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Amphlett JC; Coomber JW; Whittle E The C-H Bond Dissociation Energy in Fluoroform. J. Phys. Chem 1966, 70 (2), 593–594. 10.1021/j100874a507. [DOI] [Google Scholar]
- (66).Shevick SL; Wilson CV; Kotesova S; Kim D; Holland PL; Shenvi RA Catalytic Hydrogen Atom Transfer to Alkenes: A Roadmap for Metal Hydrides and Radicals. Chem. Sci 2020, 11 (46), 12401–12422. 10.1039/D0SC04112B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Crossley SWM; Obradors C; Martinez RM; Shenvi RA Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev 2016, 116 (15), 8912–9000. 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Green SA; Crossley SWM; Matos JLM; Vásquez-Céspedes S; Shevick SL; Shenvi RA The High Chemofidelity of Metal-Catalyzed Hydrogen Atom Transfer. Acc. Chem. Res 2018, 51 (11), 2628–2640. 10.1021/acs.accounts.8b00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Mukaiyama T; Yamada T Recent Advances in Aerobic Oxygenation. Bull. Chem. Soc. Jpn 1995. 10.1246/bcsj.68.17. [DOI] [Google Scholar]
- (70).Lo JC; Kim D; Pan CM; Edwards JT; Yabe Y; Gui J; Qin T; Gutiérrez S; Giacoboni J; Smith MW; Holland PL; Baran PS Fe-Catalyzed C-C Bond Construction from Olefins via Radicals. J. Am. Chem. Soc 2017, 139 (6), 2484–2503. 10.1021/jacs.6b13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Lo JC; Gui J; Yabe Y; Pan C-M; Baran PS Functionalized Olefin Cross-Coupling to Construct Carbon–Carbon Bonds. Nature 2014, 516 (7531), 343–348. 10.1038/nature14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Kim D; Rahaman SMW; Mercado BQ; Poli R; Holland PL Roles of Iron Complexes in Catalytic Radical Alkene Cross-Coupling: A Computational and Mechanistic Study. J. Am. Chem. Soc 2019, 141, 7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Lu Y-C; Kao S-C; West JG Decatungstate-Photocatalysed C(Sp3)─H Azidation. Chem. Commun 2022, 58 (31), 4869–4872. 10.1039/D2CC00425A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Hooson J; Tran HN; Bian K-J; West JG Simple, Catalytic C(Sp3)─H Azidation Using the C─H Donor as the Limiting Reagent. Chem. Commun 2024, 60, 3705–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Bam R; Pollatos AS; Moser AJ; West JG Mild Olefin Formation via Bio-Inspired Vitamin B12 Photocatalysis. Chem. Sci 2021, 12 (5), 1736–1744. 10.1039/D0SC05925K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Capaldo L; Ravelli D Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem 2017, 2017 (15), 2056–2071. 10.1002/ejoc.201601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Capaldo L; Ravelli D; Fagnoni M Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C─H Bonds Elaboration. Chem. Rev 2021. 10.1021/acs.chemrev.1c00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).West JG; Sorensen EJ Development of a Bio-Inspired Dual Catalytic System for Alkane Dehydrogenation. Isr. J. Chem 2017, 57 (3–4), 259–269. 10.1002/ijch.201600115. [DOI] [Google Scholar]
- (79).Cartwright KC; Davies AM; Tunge JA Cobaloxime-Catalyzed Hydrogen Evolution in Photoredox-Facilitated Small-Molecule Functionalization. Eur. J. Org. Chem 2020, 2020 (10), 1245–1258. 10.1002/ejoc.201901170. [DOI] [Google Scholar]
- (80).Gridnev AA; Ittel SD Catalytic Chain Transfer in Free-Radical Polymerizations. Chem Rev 2001, 101, 3611–3659. 10.1021/cr9901236. [DOI] [PubMed] [Google Scholar]
- (81).Achanta S; Jordt S-E Toxic Effects of Chlorine Gas and Potential Treatments: A Literature Review. Toxicol. Mech. Methods 2021, 31 (4), 244–256. 10.1080/15376516.2019.1669244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Kochi JK Photolyses of Metal Compounds: Cupric Chloride in Organic Media. J. Am. Chem. Soc 1962, 84 (11), 2121–2127. 10.1021/ja00870a025. [DOI] [Google Scholar]
- (83).Shul’pin GB; Nizova GV Photo-Oxidation of Cyclohexane by Atmospheric Oxygen in Acetonitrile, Catalysed by Chloride Complexes of Iron, Copper and Gold. Pet. Chem 1993, 33, 107–112. [Google Scholar]
- (84).Shilov AE; Shul’pin GB Activation of C-H Bonds by Metal Complexes. Chem. Rev 1997, 97 (8), 2879–2932. 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
- (85).de Groot LHM; Ilic A; Schwarz J; Wärnmark K Iron Photoredox Catalysis–Past, Present, and Future. J. Am. Chem. Soc 2023, 145 (17), 9369–9388. 10.1021/jacs.3c01000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Chen J; Browne WR Photochemistry of Iron Complexes. Coord. Chem. Rev 2018, 374, 15–35. 10.1016/j.ccr.2018.06.008. [DOI] [Google Scholar]
- (87).Moser AJ; Funk BE; West JG Vitamin B12 in Photocatalysis – An Underexplored Frontier in Cooperative Catalysis. ChemCatChem 2024, 16 (7), e202301231. 10.1002/cctc.202301231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Giedyk M; Goliszewska K; Gryko D Vitamin B12 Catalysed Reactions. Chem. Soc. Rev 2015, 44 (11), 3391–3404. 10.1039/c5cs00165j. [DOI] [PubMed] [Google Scholar]
- (89).Giedyk M; Gryko D Vitamin B12: An Efficient Cobalt Catalyst for Sustainable Generation of Radical Species. Chem Catal. 2022, 2 (7), 1534–1548. 10.1016/j.checat.2022.05.004. [DOI] [Google Scholar]
- (90).Wdowik T; Gryko D C─C Bond Forming Reactions Enabled by Vitamin B12─Opportunities and Challenges. ACS Catal. 2022, 12 (11), 6517–6531. 10.1021/acscatal.2c01596. [DOI] [Google Scholar]
- (91).Kao S-C; Bian K-J; Chen X-W; Chen Y; Martí AA; West JG Photochemical Iron-Catalyzed Decarboxylative Azidation via the Merger of Ligand-to-Metal Charge Transfer and Radical Ligand Transfer Catalysis. Chem Catal. 2023, 3 (6), 100603. 10.1016/j.checat.2023.100603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Feng G; Wang X; Jin J Decarboxylative C─C and C─N Bond Formation by Ligand-Accelerated Iron Photocatalysis. Eur. J. Org. Chem 2019, 2019 (39), 6728–6732. 10.1002/ejoc.201901381. [DOI] [Google Scholar]
- (93).Li Z; Wang X; Xia S; Jin J Ligand-Accelerated Iron Photocatalysis Enabling Decarboxylative Alkylation of Heteroarenes. Org. Lett 2019, 21 (11), 4259–4265. 10.1021/acs.orglett.9b01439. [DOI] [PubMed] [Google Scholar]
- (94).Su W; Xu P; Ritter T Decarboxylative Hydroxylation of Benzoic Acids. Angew. Chem. Int. Ed 2021, 60 (45), 24012–24017. 10.1002/anie.202108971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Xu P; López-Rojas P; Ritter T Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination. J. Am. Chem. Soc 2021, 143 (14), 5349–5354. 10.1021/jacs.1c02490. [DOI] [PubMed] [Google Scholar]
- (96).Chen TQ; Pedersen PS; Dow NW; Fayad R; Hauke CE; Rosko MC; Danilov EO; Blakemore DC; Dechert-Schmitt A-M; Knauber T; Castellano FN; MacMillan DWC A Unified Approach to Decarboxylative Halogenation of (Hetero)Aryl Carboxylic Acids. J. Am. Chem. Soc 2022, 144 (18), 8296–8305. 10.1021/jacs.2c02392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Li QY; Gockel SN; Lutovsky GA; DeGlopper KS; Baldwin NJ; Bundesmann MW; Tucker JW; Bagley SW; Yoon TP Decarboxylative Cross-Nucleophile Coupling via Ligand-to-Metal Charge Transfer Photoexcitation of Cu(II) Carboxylates. Nat. Chem 2022, 14 (1), 94–99. 10.1038/s41557-021-00834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Lutovsky GA; Gockel SN; Bundesmann MW; Bagley SW; Yoon TP Iron-Mediated Modular Decarboxylative Cross-Nucleophile Coupling. Chem 2023, 9 (6), 1610–1621. 10.1016/j.chempr.2023.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Kang YC; Treacy SM; Rovis T Iron-Catalyzed Photoinduced LMCT: A 1° C─H Abstraction Enables Skeletal Rearrangements and C(Sp3)─H Alkylation. ACS Catal. 2021, 11 (12), 7442–7449. 10.1021/acscatal.1c02285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Bian K-J; Kao S-C; Nemoto D; Chen X-W; West JG Photochemical Diazidation of Alkenes Enabled by Ligand-to-Metal Charge Transfer and Radical Ligand Transfer. Nat. Commun 2022, 13 (1), 7881. 10.1038/s41467-022-35560-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Bian K-J; Nemoto D; Chen X-W; Kao S-C; Hooson J; West JG, Photocatalytic, Modular Difunctionalization of Alkenes Enabled by Ligand-to-Metal Charge Transfer and Radical Ligand Transfer. Chem. Sci 2024, 15 (1), 124–133. 10.1039/D3SC05231A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Lindner H; Amberg WM; Carreira EM Iron-Mediated Photochemical Anti-Markovnikov Hydroazidation of Unactivated Olefins. J. Am. Chem. Soc 2023, 145 (41), 22347–22353. 10.1021/jacs.3c09122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Yoshimura A; Zhdankin VV Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev 2016, 116 (5), 3328–3435. 10.1021/acs.chemrev.5b00547. [DOI] [PubMed] [Google Scholar]
- (104).Fernández-García S; Chantzakou VO; Juliá-Hernández F Direct Decarboxylation of Trifluoroacetates Enabled by Iron Photocatalysis**. Angew. Chem. Int. Ed n/a (n/a), e202311984. 10.1002/anie.202311984. [DOI] [PubMed] [Google Scholar]
- (105).Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J. Am. Chem. Soc 2014, 136 (4), 1300–1303. 10.1021/ja412342g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Obradors C; Martinez RM; Shenvi RA Ph(i-PrO)SiH2: An Exceptional Reductant for Metal-Catalyzed Hydrogen Atom Transfers. J. Am. Chem. Soc 2016, 138 (14), 4962–4971. 10.1021/jacs.6b02032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).King SM; Ma X; Herzon SB A Method for the Selective Hydrogenation of Alkenyl Halides to Alkyl Halides. J. Am. Chem. Soc 2014, 136 (19), 6884–6887. 10.1021/ja502885c. [DOI] [PubMed] [Google Scholar]
- (108).Ma X; Herzon SB Non-Classical Selectivities in the Reduction of Alkenes by Cobalt-Mediated Hydrogen Atom Transfer. Chem. Sci 2015, 6 (11), 6250–6255. 10.1039/c5sc02476e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Tran HN; West JG Recent Advances in Base Metal-Catalyzed Cooperative Transfer Hydrogenation and Hydrodeuteration of Alkenes. Tetrahedron Lett. 2023, 154404. 10.1016/j.tetlet.2023.154404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Haque MB; Roberts BP Enantioselective Radical-Chain Hydrosilylation of Prochiral Alkenes Using Optically Active Thioi Catalysts. Tetrahedron Lett. 1996, 37, 9123–9126. [Google Scholar]
- (111).Haque MB; Roberts BP; Tocher DA Enantioselective Radical-Chain Hydrosilylation of Alkenes Using Homochiral Thiols as Polarity-Reversal Catalysts. J. Chem. Soc. Perkin 1 1998, No. 17, 2881–2890. 10.1039/a803280g. [DOI] [Google Scholar]
- (112).Shin NY; Ryss JM; Zhang X; Miller SJ; Knowles RR Light-Driven Deracemization Enabled by Excited-State Electron Transfer. Science 2019, 366, 364–369. 10.1021/ba-1991-0228.ch005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Shi Q; Xu M; Chang R; Ramanathan D; Peñin B; Funes-Ardoiz I; Ye J Visible-Light Mediated Catalytic Asymmetric Radical Deuteration at Non-Benzylic Positions. Nat. Commun 2022, 13 (1), 4453. 10.1038/s41467-022-32238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Brill ZG; Grover HK; Maimone TJ Enantioselective Synthesis of an Ophiobolin Sesterterpene via a Programmed Radical Cascade. Science 2016, 352 (6289), 1078–1082. 10.1126/science.aaf6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Barton DHR; Crich D; Motherwell WB New and Improved Methods for the Radical Decarboxylation of Acids. J. Chem. Soc. Chem. Commun 1983, No. 17, 939–941. 10.1039/C39830000939. [DOI] [Google Scholar]
- (116).Buck RC; Franklin J; Berger U; Conder JM; Cousins IT; de Voogt P; Jensen AA; Kannan K; Mabury SA; van Leeuwen SP Perfluoroalkyl and Polyfluoroalkyl Substances in the Environment: Terminology, Classification, and Origins. Integr. Environ. Assess. Manag 2011, 7 (4), 513–541. 10.1002/ieam.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Yerien DE; Barata-Vallejo S; Postigo A Radical Perfluoroalkylation of Aliphatic Substrates. ACS Catal. 2023, 13 (12), 7756–7794. 10.1021/acscatal.3c01922. [DOI] [Google Scholar]
- (118).Barata-Vallejo S; Lantano B; Postigo A Recent Advances in Trifluoromethylation Reactions with Electrophilic Trifluoromethylating Reagents. Chem Eur J 2014, 20 (51), 16806–16829. 10.1002/chem.201404005. [DOI] [PubMed] [Google Scholar]
- (119).De Vleeschouwer F; Jaque P; Geerlings P; Toro-Labbé A; De Proft F Regioselectivity of Radical Additions to Substituted Alkenes: Insight from Conceptual Density Functional Theory. J. Org. Chem 2010, 75 (15), 4964–4974. 10.1021/jo100503e. [DOI] [PubMed] [Google Scholar]
- (120).Fischer H; Radom L Factors Controlling the Addition of Carbon-Centered Radicals to Alkenes. Macromol. Symp 2002, 182, 1–14. . [DOI] [PubMed] [Google Scholar]