

Abstract
Artificial intelligence (AI) and machine learning (ML) models are being deployed in many domains of society and have recently reached the field of drug discovery. Given the increasing prevalence of antimicrobial resistance, as well as the challenges intrinsic to antibiotic development, there is an urgent need to accelerate the design of new antimicrobial therapies. Antimicrobial peptides (AMPs) are therapeutic agents for treating bacterial infections, but their translation into the clinic has been slow owing to toxicity, poor stability, limited cellular penetration and high cost, among other issues. Recent advances in AI and ML have led to breakthroughs in our abilities to predict biomolecular properties and structures and to generate new molecules. The ML-based modelling of peptides may overcome some of the disadvantages associated with traditional drug discovery and aid the rapid development and translation of AMPs. Here, we provide an introduction to this emerging field and survey ML approaches that can be used to address issues currently hindering AMP development. We also outline important limitations that can be addressed for the broader adoption of AMPs in clinical practice, as well as new opportunities in data-driven peptide design.
Introduction
Antimicrobial peptides (AMPs) are short amino acid sequences (typically ranging from 6 to 50 residues in length) that kill various bacteria, viruses and fungi through membrane disruption, specific target binding, immunomodulation, anti-biofilm activity and interference with metabolic processes1–3. Although the discovery of AMPs dates back to the 1940s4 and over 5,000 AMPs have been identified so far5, fewer than 50 AMPs6 have been approved by the US Food and Drug Administration (FDA) or are under clinical investigation6,7. The development of AMPs as therapeutic drugs to treat infectious diseases continues to be hindered by undesirable physicochemical and medicinal chemistry properties (such as high toxicity and poor chemical stability), unspecific or unknown mechanisms of action, the high cost of peptide synthesis, and the generation of more industrial waste than produced by the manufacture of other therapeutic modalities. Yet, antimicrobial resistance remains a major health threat, accounting for more than 35,000 deaths in the United States per year8 and over 1.27 million deaths globally per year9. Because the mechanisms of action of AMPs can differ from those of conventional antibiotics, these peptides remain a promising source of therapeutic agents against antimicrobial resistance and disease-causing pathogens.
Bringing new AMP drugs to the clinic requires computational platforms that can quickly and accurately identify peptides with antimicrobial activity10–17. Such peptides may be mined from nature or from extinct organisms, or generated synthetically. Molecular dynamics (MD) has been used to design AMPs18,19, but it remains a time-consuming and low-throughput approach. In the past few years, machine learning (ML), a subfield of artificial intelligence (AI), has been successfully and widely applied to problems in computational biology, language processing, games and computer vision13,20–34. Some ML models make use of deep learning (DL), which applies a series of complex transformations (implemented using deep neural networks, DNNs) to extract hidden features and make predictions from complex inputs and model data, including images and biomolecules. In computational biology, ML (and especially DL) has been widely used in genomic studies35–38, structural modelling of biomolecules20,39,40, drug discovery and development28,41–44, and medical data analysis45,46. By using and learning knowledge from publicly available peptides, multiple AMPs have been discovered and generated through AI/ML47 (Boxes 1–3), which have been experimentally validated to be effective at targeting bacteria14,15,48–50 (Table 1, Fig. 1). We anticipate that substantial progress will be made on ML-based AMP design in the next few years, and this progress will help to reduce the time and cost associated with AMP discovery and development. In this Review, we survey how ML has been applied to various aspects of AMP design, discuss the limitations of these approaches and suggest future AI/ML-enabled strategies.
Box 1. Experimentation and data generation and curation for peptides.
Antimicrobial activity is typically identified phenotypically by using in vitro screens: that is, antimicrobial agents are determined by their abilities to inhibit the growth of, or kill, whole microbial cells in culture. A common method of quantifying antimicrobial activity is by performing microwell serial dilutions of the agent in growth-permissive media in the presence of an inoculum of microbial culture in each well. After a standard incubation period (for example, overnight for many bacterial cultures), the minimum inhibitory concentration (MIC) is determined as the minimum concentration at which there is no growth of the microbial culture.
To maximize the number of antimicrobial peptides (AMPs) assessed, screening campaigns may screen at a single AMP concentration for growth-inhibitory activity. The appropriate screening concentration can vary depending on the nature of the AMPs screened, the species or strain being targeted, and other experimental conditions such as the growth medium and temperature used. Given the MICs of previously identified AMPs (Table 1), final screening concentrations of 50–100 μM may be appropriate for most initial screens.
For other properties, peptide activity can often be determined analogously; for example, the haemolytic activity of a peptide can be determined by replacing microbial cells with red blood cells and measuring percentage red blood cell lysis (through changes in optical density) instead of microbial growth.
Numerous databases containing AMPs and other peptides are available, including the PDB86 (protein structures), UniProt178 (protein functions), SwissProt140 (expertly curated UniProt), TrEMBL140 (corresponding to translation of all coding sequences in the EMBL database), DBAASP (AMPs)100, CAMP103 (AMPs), LAMP104 (relations between AMPs), APD3106 (AMPs) and Hemolytik123 (haemolysis data).
As experimental conditions and organisms differ between studies and screens, data from the above sources can be variable and non-standardized. The generation of standardized datasets — for example those obtained by running library screens in-house — will be important for proper data quality control, which is paramount for ML model training and benchmarking.
Box 3. Categories of machine learning.
Machine learning (ML) can be divided into supervised, unsupervised and reinforcement learning. In supervised learning, the outputs are labels that are associated with the inputs. The antimicrobial peptide (AMP) activity prediction in Box 2 is an example of supervised learning. In supervised learning, classification means predicting which category the input belongs to. For example, predicting whether a peptide is antimicrobial or not is a binary classification task. Predicting a continuous value (such as the minimum inhibitory concentration (MIC) value) from the input is referred to as regression.
Unsupervised learning involves discerning patterns in the inputs without label information. Typical applications of unsupervised learning include clustering (grouping inputs into several clusters, so that similar inputs are in the same cluster), representation learning (learning features from raw inputs) and generative models (Fig. 3). As some representation learning and generative models use inputs themselves as labels and aim to generate outputs that resemble inputs, these models can also be referred to as self-supervised learning models or self-supervision models.
Reinforcement learning (RL) can be described by scenarios in which an agent in an environment performs some action and receives corresponding continuous feedback (also known as reward) from the environment. Based on the reward, the agent dynamically adjusts its action to maximize its future reward. In the context of AMP sequence generation, the agent may be an ML model that generates an AMP-like sequence for each action. The reward may be the predicted, or experimentally determined, antimicrobial activity of the generated sequence. At the next step, the agent considers the generated sequence and the associated reward as inputs and uses this information to generate another AMP-like sequence. In this process, the ML model becomes trained such that the generated peptide sequences are expected to gradually have more desirable activity values (for example, the generated sequences have decreasing MIC values).
Table 1 |.
Examples of machine learning/artificial intelligence (ML/AI)-based antimicrobial peptide design and discovery
| Ref. | Method | Number of validated AMPs in vitro/in mice | Antimicrobial activity range | Studied bacteria | Toxicity range |
|---|---|---|---|---|---|
| Yoshida et al.164 | GA with ML | 44/N.T. | <4.1 μM (IC50) | Escherichia coli | <1% RBC lysis at antimicrobial IC50 for the most potent peptide |
| Nagarajan et al.174 | Deep generative model (neural language model) | 10/1 | ≤128 μg ml−1 (MIC) | 30 pathogens and commensals (carbapenem-resistant Acinetobacter baumannii for the mouse model) | LD50 ~213–224 μg g−1 in mice, no hepatotoxicity or nephrotoxicity |
| Porto et al.50 | GA | 8/1 | 6.25–100 μg ml−1 (MIC) | E. coli, A. baumannii, Staphylococcus aureus, Pseudomonas aeruginosa, Klebsiella pneumoniae, Streptococcus pyogenes, Listeria ivanovii and Enterococcus faecalis (P. aeruginosa for the mouse model) | Haemolysis HC50 and cytotoxicity CC50 in human erythrocytes and HEK-293 cells both >200 μM for the most potent peptide |
| Dean et al.176 | Deep generative model (VAE) | 6/N.T. | <70 μg ml−1 (IC50) | E. coli, A. baumannii and S. aureus | N.T. |
| Tucs et al.180 | Deep generative model (GAN) | 5/N.T. | 3.1–50 μg ml−1 (MIC) | E. coli | N.T. |
| Das et al.48 | Deep generative model (VAE) with MD | 2/2 (toxicity tests only) | 7.8–128 μg ml−1 (MIC) | E. coli, A. baumannii, S. aureus, P. aeruginosa and K. pneumoniae | 125–500 μg ml−1 (HC50) 158–182 mg kg−1 (LD50 in mice) |
| Boone et al.165 | GA with ML | 1/N.T. | 1.2-cm inhibition zone at 4 mg ml−1 | Staphylococcus epidermidis | N.T. |
| Capecchi et al.126 | Deep generative model (neural language model) | 8/N.T. | ≤64 μg ml−1 (MIC) | E. coli, A. baumannii, S. aureus, P. aeruginosa, Stenotrophomonas maltophilia, Enterobacter cloacae, Burkholderia cenocepacia and S. epidermidis | ≥500 μg ml−1 (minimum haemolytic concentration by visual inspection) |
| Dean et al.177 | Deep generative model (VAE) | 38/N.T. | 0.5–128 μM (MIC) | E. coli, S. aureus and P. aeruginosa | N.T. |
| Torres et al.49 | AMP predictor using physicochemical properties Peptide source: human proteome | 47/2 | ≤64 μM (MIC) | 25 pathogens and commensals (P. aeruginosa and A. baumannii for the mouse models) | No significant weight change in mice |
| Ma et al.14 | DL-based AMP predictors Peptide source: human gut microbiome | 181/3 | 181 peptides had significant reduction in OD600, at 60 μM, 2–200 μM (MIC) for 11 peptides | E. coli, A. baumannii, S. aureus, P. aeruginosa, E. cloacae, K. pneumoniae, S. epidermidis and E. faecalis (K. pneumoniae for the mouse model) | >61 μM (HC50), 22–200 μM (CC50 in HCT116 cells and human erythrocytes) for three effective peptides in mice |
| Huang et al.15 | Simple and DL-based AMP predictorsPeptide source: six to nine amino acids | 54/3 | ≤200 μg ml−1 (MIC) | S. aureus, E. coli, P. aeruginosa, S. haemolyticus and A. baumannii (S. aureus for the mouse model) | >750 μg ml−1 (HC50) >300 μg ml−1 (CC50 in NIH 3T3 cells) for three effective peptides in mice |
| Cao et al.182 | Deep generative model (GAN) with MD | 1/N.T. | 16–256 μg ml−1 (MIC) | Bacillus subtilis, S. maltophilia, P. aeruginosa, Bacillus thuringiensis, S. aureus, E. coli and Lysobacter enzymogenes | N.T. |
| Maasch et al.12 | Simple and DL-based AMP and protein cleavage site predictors Peptide source: Neanderthal and Denisovan proteomes | 6/2 | ≤128 μM (MIC) | E. coli, A. baumannii, S. aureus, P. aeruginosa, K. pneumoniae and S. aureus (P. aeruginosa and A. baumannii for the mouse models) | No significant weight change in mice |
Works using ML/AI techniques to discover or design AMPs, validated using in vitro assays or mouse models. The basic methodology, bacteria studied, antimicrobial and toxicity ranges are provided. AMP, antimicrobial peptide; CC50, half-maximal cytotoxic concentration; DL, deep learning; EC50, half-maximal effective concentration; GA, genetic algorithm; GAN, generative adversarial network; HC50, half-maximal haemolytic concentration; IC50, half-maximal inhibitory concentration; LC50, half-maximal lethal concentration; LD50, half-maximal lethal dose; MD, molecular dynamics; MHC, minimum haemolytic concentration; MIC, minimum inhibitory concentration (MICs were determined by broth microdilution); N.T., not tested; OD600, the optical density of a sample at a wavelength of 600 nm; RBC, red blood cell; VAE, variational autoencoder.
Fig. 1 |. Timelines of major machine learning/artificial intelligence (ML/AI) events and recent studies of ML/AI-driven antimicrobial peptide (AMP) identification and design.

Various ML/AI-driven approaches have been developed to discover AMP-like sequences from available genomic or proteomic data and to design synthetic AMPs. Here, we highlight several studies in which the predictions of ML/AI-driven models were validated in vitro or in mouse models of bacterial infection. Details of highlighted AMP studies can be found in Table 1. DL, deep learning; GAN, generative adversarial network; MD, molecular dynamics; NN, neural network; VAE, variational autoencoder.
Representations of peptides in ML
Properly selecting an input representation (also known as features) for any ML model is a crucial step before model building and training. Input representations should contain information relevant to the properties being modelled; such information helps the ML model to capture the corresponding input–output relationship accurately and improves model quality. Various types of representations, including the ones described below (Fig. 2), can be used to computationally encode peptide information:
Fig. 2 |. Methods of representing peptides as inputs to machine learning models.
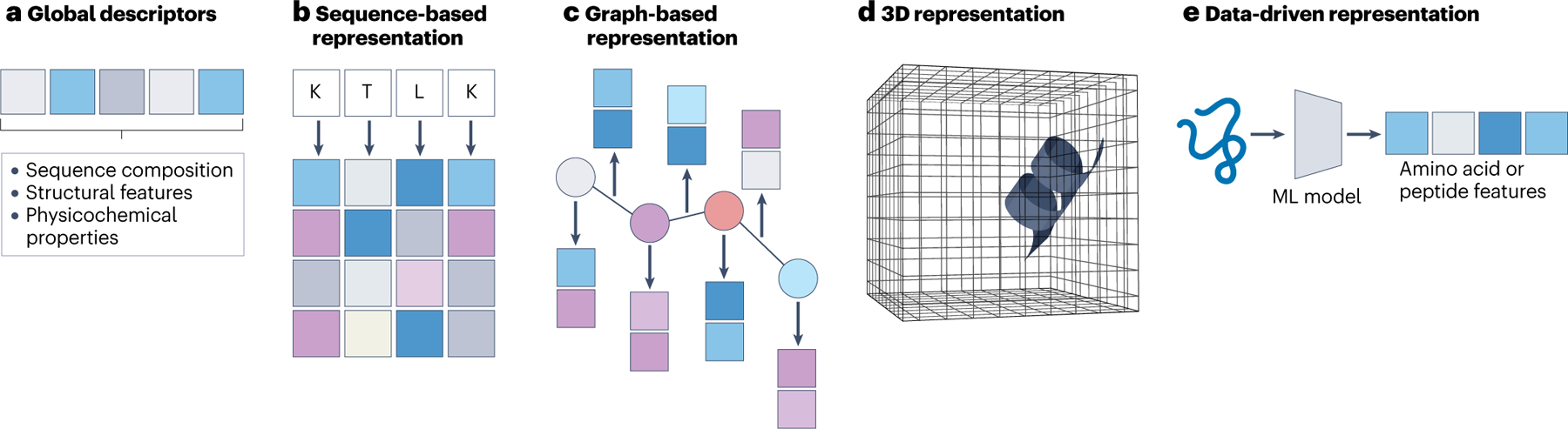
a, For any peptide, global descriptors use fixed-size vectors to encode peptide information such as sequence composition, structural features and physicochemical properties. b, A sequence-based representation encodes a peptide using data from its primary sequence of amino acids. Each type of amino acid is associated with a fixed-size vector encoding the corresponding residue information (such as amino acid type and physicochemical properties). These vectors, or ‘embeddings’, can also be learned from data. c, A graph-based representation consists of nodes and edges. To represent peptides, the nodes can be atoms or residues, whereas the edges can be bonds or geometric distance (given a 3D structure of the peptide) between nodes. Nodes and edges are associated with corresponding vectors representing atom, bond and geometric information. d, When the 3D structure of a peptide is available, the peptide can also be represented by a voxelized (or discretized) form of the structure. Each voxel is represented by a vector, which stores information regarding atom occupancies and atom properties relevant to that voxel. e, Machine learning (ML) models can be used to extract low-dimensional features from peptide inputs from sequence or structure. The extracted features can be used as inputs for other peptide-related tasks.
Global descriptors (0D)
The input representation is typically a fixed-size vector, whose values summarize general (‘global’) properties of the corresponding peptide. These properties might include sequence composition (such as amino acid composition and substring frequency), structural features (such as α-helix and β-sheet arrangements) and physicochemical properties (such as net charge, hydrophobicity and amphiphilicity)51. Although the design of global descriptors for peptides and proteins has been extensively studied52–57, directly using all available descriptors could easily result in high-dimensional vector representations containing irrelevant or redundant information with respect to the properties being modelled. This can increase the models’ complexity, bias them to capture spurious correlations between the input and output, and in turn decrease their generalization ability. To address the issue of properly selecting global descriptors, feature-selection algorithms58 can be used to generate low-dimensional representations that may be better suited for ML models in both supervised and unsupervised approaches. Overall, although constructing global descriptors requires substantial human effort and domain knowledge, this type of representation can be useful for capturing specific information relevant to the property being modelled when only a limited amount of training data is available.
Sequence-based representations (1D)
This input representation type captures primary amino acid sequences. Given a peptide sequence of length L, an n × L matrix is used to store the sequential information of the peptide sequence, where n is the number of features of each letter (that is, amino acid). Here, the information of the ith amino acid in the peptide is encoded by the ith column (an n-dimensional vector) of the matrix; equivalently, this representation can be viewed as encoding an amino acid using a string of length L. A naïve way to generate these n-dimensional vectors so that each amino acid can be uniquely represented is one-hot encoding59. In this encoding scheme, the dimension n reflects the size of the sequence alphabet (that is, possible characters in sequences of interest). For the jth amino acid type in the alphabet, the one-hot encoding representation is a vector containing a value of 1 at the jth position and n − 1 values of 0 at all the other positions. Although the alphabet size n is usually set to 20 when standard amino acids are considered, the alphabet size could be increased to encode non-canonical or chemically modified amino acids. One drawback of using one-hot encoding with the amino acid alphabet is that no additional information about the properties of the amino acids, such as physical or chemical properties, can be encoded or represented. This limitation can be addressed by using computationally or experimentally determined physicochemical, biochemical, and evolutionary features of amino acids60 to replace one-hot encoding. In DL, the n-dimensional vector (or ‘embedding’) for each amino acid can be learned in a data-driven manner61, such that the vector is learned simultaneously as the downstream DNNs perform training operations. Provided that the training data are adequate, the representation can be optimized for the task of interest. Overall, sequence-based representations are suitable inputs for ML models that are designed to handle and process sequential data, such as recurrent neural networks62,63. This type of representation has been widely used for peptide sequence generation64 and property prediction59.
Graph-based representations (2D)
In graph-based representations, the inputs are graphs consisting of nodes and node connectivities (‘edges’). To represent peptides, nodes can be atoms or residues, and edges can be chemical bonds or the geometric distance between atoms or residues. Nodes and edges can be further encoded using one-hot encodings of atom, residue or bond types, geometric features (such as dihedral and torsion angles) and other embeddings. Compared with sequence-based representations, graph-based representations are better inputs for geometry-related ML tasks because they capture connectivity information. Graph neural networks are types of ML models that use graph representations; such models have been applied to various geometry-related tasks, including protein structure prediction20,39,40, structure-based AMP prediction65, molecular conformation generation66 and antibody design67. However, as they capture more connectivity information, graph representations are often more memory-consuming than sequence-based representations, and thus are more computationally costly.
Three-dimensional (3D) representations
In addition to using graph-based representations, peptides with available 3D structures can be represented using voxelization. Specifically, the 3D structure of a peptide can be considered as a 3D image and discretized into fixed-size voxels (for example each with a size of 1 × 1 × 1 Å). For each voxel, a vector storing the occupancies, types and properties of the atoms inside the voxel can be used as features. Three-dimensional convolutional neural networks68,69 can be used to process the voxelized structures and have been applied to protein binding site prediction70, protein–ligand binding affinity prediction71, and other prediction tasks.
Data-driven representations
Feature or representation learning72, which automatically learns features from data, allows for another type of input representation for peptides. State-of-the-art representation learning methods have leveraged the concept of self-supervision — learning ‘supervision signals’, or labels, from the input data itself, which are then used to make sense of the remaining unlabelled data. These methods have demonstrated strong predictive ability in computer vision73, natural language processing21,74,75 and computational biology39,76–79. Specifically, amino acid and protein features learned from protein neural language models39,76,77 trained on large-scale protein sequences have shown strong predictive power on tasks such as predicting protein structure, stability and function after introducing mutations. Peptides and their amino acids can be directly represented using the features extracted from these language models. Moreover, these features and models could serve as initial representations for downstream peptide-related tasks and be fine-tuned based on available peptide data.
ML for antimicrobial peptide design
Structural predictions
Knowledge of protein and peptide secondary and 3D structures can help to elucidate function and guide the design of new proteins and peptides with specified functions and properties. Predicting these structures from the primary sequences of amino acids has been a central and longstanding goal in computational biology and bioinformatics. Although early studies80–82 have aimed to predict peptide secondary structure, more recent studies using ML to predict 3D protein structures have reported exciting advances that can inform AMP development in the near future. In particular, AlphaFold2 (ref. 20) and RoseTTAFold40 are two platforms for sequence-to-3D structure prediction that have demonstrated outstanding accuracy on benchmark sets including CASP14 (ref. 83) and CAMEO84. These platforms use attention network architectures85 (a type of DNN) and co-evolutionary information from multiple sequence alignments (MSAs) to predict 3D structures on the basis of known 3D structures in the Protein Data Bank86 (PDB). Recent platforms, including ESMFold87 and OmegaFold39, have replaced these MSA-based features with features learned from a neural language model (Fig. 3a), achieving prediction accuracy similar to those of AlphaFold and RoseTTAFold.
Fig. 3 |. Schematic illustration of deep generative models for antimicrobial peptides (AMPs).

a, In neural language models, sections of the input (such as certain letters in an input sequence) are missing and the model (often a deep neural network, DNN) is asked to reconstruct the missing parts from the incomplete input. After training, partial inputs are fed into the model to generate new peptides. b, A variational autoencoder (VAE) consists of an encoder and a decoder neural network. The encoder maps the input to a low-dimensional embedding, Z (a vector), which follows some distribution. The decoder then processes the embedding and reconstructs the original input. As the embeddings fall into some probability distribution, new peptides can be generated by decoding embeddings that are sampled from the distribution. c, Normalizing flow models are similar to VAEs, with the exception that the encoder neural network is specified to be dimension-preserving and invertible (hence, the corresponding decoder is the inverse function of the encoder). This makes normalizing flow models capable of inferring exact likelihoods of data. d, A generative adversarial network (GAN) consists of a generator that creates a synthetic peptide from a random vector and a discriminator that aims to identify whether the generated peptide comes from actual data or is synthetic. Because the discriminator and the generator compete with each other, the synthetic peptides produced by the generator will converge to approximate the peptides found in actual data. e, Given any input (X0), diffusion models gradually add Gaussian noise to the input. Sufficient noise addition transforms the input to random Gaussian noise, XT. A DNN is then trained using data to reverse-transform the random noise, XT, back to the original input (from XT to X0). This reverse step specifies the process of how new peptides are generated from random noise.
Although these sequence-to-structure platforms were not specifically designed for short protein sequences (that is, peptides), a recent comparison88 of peptide structure prediction methods showed that these DL-based methods could still achieve the best prediction performance. Especially for α-helical, β-hairpin and disulfide-rich peptides that have increased residue contact and are less solvent-exposed than other peptides (which may result in less conformational flexibility), DL-based methods outperform peptide-specific structure prediction methods such as PEP-FOLD3 (ref. 89) and APPTEST90. Using predicted peptide structures for mechanism of action studies based on MD simulations, or as input features for ML approaches that model peptides (such as AMP prediction), is promising for structure-guided AMP91,92 design. Transfer learning strategies that take models or model-predicted structures and perform fine-tuning (or retraining) based on smaller peptide structures could also improve prediction accuracy for peptides. Extending these sequence-to-structure platforms for chemically modified proteins and peptides remains a challenge to be addressed.
However, accurately identifying the mechanisms of action of small-molecule antimicrobials based on protein structures remains a challenge, and improvements in platforms like molecular docking are needed to better predict protein–ligand interactions93. Using static and rigid protein structures — which are typically produced by sequence-to-structure models — may also be limiting. A single predicted structure (or conformation) may not sufficiently represent a given peptide because of the peptide’s conformational flexibility. In such cases, ML-based predictions could be made more accurate and interpretable by representing the peptide using a set of conformations and allowing models to select the most relevant conformations in a data-driven manner. Generative models66,94–97 and models leveraging reinforcement learning98 have contributed to predicting 3D conformations for small molecules. Here, the 2D or 3D structure of the molecule is provided as inputs, and possible 3D conformations are produced as outputs.
MD simulations have also helped to generate conformational data for larger molecules, such as peptides and proteins; however, MD simulations are time-intensive and computationally expensive. Nonetheless, ML-driven approaches that complement MD simulations have helped to generate useful conformational data. For example, idpGAN99 simulated the conformations of 1,966 and 31 intrinsically disordered proteins for model training and testing, respectively. Based on this modelling, a conditional generative adversarial network (GAN) was developed. This model accepted both a random vector and the amino acid sequence of an intrinsically disordered protein as input, and produced a conformation of the protein as output. Standardized datasets will help to better predict protein and peptide conformations. Of note, DBAASP100, an AMP database containing ~200 peptides for which PDB structures and MD trajectories are available, could be used as a dataset for training and evaluating ML-based models. Accumulating and publicly sharing more data on peptide conformations will better enable us to use ML models to predict AMP conformations and structures.
Property prediction
ML models have been developed to predict properties that inform AMP discovery and development, including antimicrobial activity, toxicity, stability, cell penetration and mechanism of action, as highlighted below.
Antimicrobial activity.
ML-based approaches51,101,102 trained and evaluated on public AMP databases100,103–106 have predicted antimicrobial activity from amino acid sequences, a key step in AMP development. These strategies used simple ML methods (such as random forests and support vector machines)49,107–114, DL-based methods14,115,116, and hybrid methods combining simple and DL-based models15,117. However, these studies differed in the input representations used in each model, how AMPs with and without antimicrobial activity were classified, and how predictions were evaluated, making it difficult to conclude which features and ML models performed best. As feature selection and the classification of training data into positives and negatives greatly affect ML models51,101,102, these tasks should be benchmarked appropriately to make fair comparisons between models. Generating standardized datasets is also important for improving data quality, as public AMP databases often aggregate data from different experimental conditions and organisms (Box 1).
Despite these limitations, some of these models have been applied to mine AMPs from large amino acid sequence spaces, and several predictions have been validated through wet-lab experiments. For example, Deep-AmPEP30 (ref. 115) used convolutional neural networks to discover AMPs from the genome of Candida glabrata, a commensal fungus in humans, and identified a peptide with potent antimicrobial activity in vitro against Bacillus subtilis and Vibrio parahaemolyticus. By training DNNs with different architectures, including recurrent neural networks and attention networks, and by applying the trained DNNs to small open-reading frames in metagenomes from the human gut microbiome, a study validated the activity of 11 peptides against multidrug-resistant Gram-negative pathogens and found that three of these peptides were effective in treating a mouse model of Klebsiella pneumoniae infection14. Moreover, an AMP scoring function that considers net charge, hydrophobicity and sequence length to identify encrypted peptides embedded in the human proteome revealed active encrypted peptides against Pseudomonas aeruginosa and Acinetobacter baumannii infections, which were validated in mouse models49.
These studies demonstrate that genetic and proteomic sequences from various organisms represent a source of naturally occurring AMPs and encrypted peptides. These AMPs may possess useful absorption, distribution, metabolism, excretion and toxicity (ADMET) properties. Thus, mining functional peptides for therapeutic applications from these search spaces is a promising field10,11,16. ML approaches have also been used for molecular de-extinction, whereby the proteomes of our closest relatives, the archaic humans Neanderthals and Denisovans, were mined, and several encrypted peptide antibiotics were resurrected which displayed antimicrobial activity in vitro and in preclinical mouse models12. Moreover, a deep learning algorithm, called APEX, has been recently used to mine the proteomes of all extinct organisms (the ‘extinctome’) as a source of antibiotics, yielding preclinical antibiotic candidates derived from ancient organisms such as the woolly mammoth (for example, the molecule mammuthusin-2), the ancient sea cow, giant sloth and the extinct giant elk16. These studies revealed a new sequence space and opened new avenues for antibiotic discovery through molecular de-extinction.
Typical ML-based AMP prediction models have formulated the problem of AMP prediction as a classification task. Of note, the sequential model ensemble pipeline (SMEP) proposed in ref. 15 formulated this problem according to three criteria (classification, ranking and regression) and virtually screened hundreds of billions of sequences composed of six to nine amino acids to predict peptides with potent antimicrobial activity. The predictions with the highest accuracy were achieved by selecting AMPs according to all three criteria, including the prediction of peptides as active or inactive (classification), the relative antimicrobial activity of peptides (ranking), and the minimum inhibitory concentration (MIC; regression). Of the 55 peptides identified in this way, 54 were found to have antimicrobial activity in vitro, and three were effective in treating disease in a mouse model of bacterial pneumonia15.
Although these studies illustrate how ML-based models have been applied to predict the antimicrobial activities of AMPs, most ML-based models have not yet taken into account bacterial species- or strain-specific information. We expect that extending current ML models to species- or strain-specific predictions of antimicrobial activity will be a focus of future studies, given the large amount of AMP data becoming available for various pathogens and commensals. Such studies are likely to discover peptides that selectively target pathogens without affecting beneficial commensals. Modelling species- or strain-specific antimicrobial activity using aggregated AMP data is challenging, and metadata recording the experimental conditions relevant to the AMPs in the training set may be used as additional inputs for ML models to inform species- or strain-specific biases and make more reliable predictions.
Medicinal chemistry and ADMET.
Toxicity and metabolic instability are major hurdles for the clinical translation of AMPs, including haemotoxicity, cytotoxicity and immunotoxicity, among others118,119. The standard haemolysis assay measures the destruction of red blood cells; because this assay is cost-effective and sensitive, it has been widely used for early toxicity screening119. As limited haemolysis data have been available for peptides, previous ML methods have focused on using simple classifiers119–122 to predict peptide haemotoxicity. Recently, large haemolysis datasets have become available100,123, and DL-based124–127 models predicting haemolysis have been developed. An example of such a model is AMPDeep127, which uses a two-step transfer learning approach for training, whereby a pretrained neural language model for proteins is fine-tuned to classify secreted proteins and haemolytic peptides. By combining the knowledge learned from a related task (classification of secreted proteins) with large sets of training data, AMPDeep obtained state-of-the-art accuracy in predicting haemolysis.
Models using simple ML-driven methods, as well as DL-based methods, have also been developed to predict unspecific cell killing128–132 and anti-inflammatory133–136 or pro-inflammatory137–139 properties of peptides (such as cytotoxicity and immunotoxicity). These models typically curate peptides from various databases, including SwissProt and TrEMBL140. Although these models have good predictive power when validated on splits of the curated data (Matthews correlation coefficients ≥0.9), the extent to which they can help to narrow down AMP candidates found in large search spaces or generated de novo remains unknown. Notably, as with data for antimicrobial activity, the toxicity data underlying these models represent different experimental conditions and cell types, and the generation of standardized datasets will be important for accurately benchmarking these studies and training future models.
To complement predictions of AMP toxicity, the half-life and stability profiles of peptides have also been predicted141–145. AMPs can be particularly susceptible to metabolic or enzyme-catalysed breakdown, resulting in poor bioavailability2. Previous ML models, including those based on support vector machines and linear regression, have predicted AMP stability from sequence and/or structural information144,145. For example, using small datasets reporting the stability properties of ~100 and ~260 peptides revealed that the best-performing models (including support vector machine and K-nearest neighbour) had characteristic values of ~0.7 for accuracy and the Pearson’s correlation coefficient, respectively. The ML-driven approaches used in these studies are promising and can probably be improved by including more data. Moreover, additional ML model architectures beyond the simple ones used in these examples should be explored to improve performance, given these larger datasets.
To summarize, ML-guided predictions of AMP toxicity and chemical stability have generally not been as well studied as those for antimicrobial activity, probably owing to the limited training data available for these tasks. Although additional experimental efforts to generate these data should be undertaken, ML approaches that make better use of the available data can also help to improve model performance. For example, multitask learning (which jointly trains models to perform multiple related tasks) or transfer learning (which fine-tunes ML models pretrained on related tasks) could be used to augment ML models. Aside from haemotoxicity, cytotoxicity and immunotoxicity, AMPs may exhibit other forms of toxicity (such as genotoxicity), and additional data and models are needed to improve our understanding of these important liabilities.
Cell penetration.
Delivery remains a central challenge for AMP development. A bacterial infection may be intracellular, and in such cases AMPs must enter mammalian cells to treat the infection. Thus, in addition to selecting the proper formulation and route of administration, validating that AMPs can enter mammalian cells is important for peptides that target intracellular infections146,147. Cell-penetrating peptides (CPPs), short amino acid sequences that can translocate a variety of biomolecules across cellular membranes, have been used for the delivery of drugs across cellular membranes to reach their targets. Antimicrobial or antineoplastic CPPs may be simultaneously used as both vehicles and drugs, and various studies have aimed to predict CPP activity148–155. For example, a support vector machine to identify CPPs was trained and found four hits predicted to penetrate cells154; this activity was then validated using fluorescence microscopy and quantitative uptake measurements. Similarly, a random forest model was trained to identify CPPs from random peptide sequences to generate and classify CPPs for the delivery of phosphorodiamidate morpholino oligonucleotides (PMOs)151. Another example involved training a neural language model to generate CPP-like sequences and a DL-based model to predict PMO delivery efficacy, and then applying genetic algorithms to optimize the generated CPP-like sequences based on predictions of PMO delivery efficacy148. The predictions made in these two studies were experimentally validated in vitro and in a mouse model, respectively, and offer promising proofs-of-concept for ML-driven models that predict cell penetration by peptides.
Mechanisms of action.
Many AMPs are membrane-active, which may lead to collateral toxicity against human cells and unspecific activity against bacterial cells. MD simulations have helped to identify the mechanism of action of membrane-active AMPs48, but predicting additional mechanisms of action remains a challenge, in part because platforms modelling the binding of these peptides to intracellular targets have low predictive power. For protein targets, platforms that make binding predictions have made use of peptide and target 3D structure156. Combining these approaches with sequence-to-structure prediction pipelines might hold promise, but molecular docking with AlphaFold2-predicted structures cannot accurately identify the protein binding targets of small-molecule antimicrobials, probably owing to limitations in predicting binding pockets and modelling conformational complexity93. Recent models have used ML to predict peptide–protein interactions and peptide–protein binding residues independently of molecular docking156–160. For example, a DL framework, CAMP, was developed to predict binary peptide–protein interactions and identify peptide-binding residues, outperforming molecular-docking-based methods156. Notably, CAMP was benchmarked using several independent datasets, including the ones derived from the PDB and PepBDB, suggesting that the framework might be broadly applied to predict AMP binding targets and guide AMP selection. We anticipate that similar ML- and DL-guided approaches will complement other methods, such as molecular docking, for predicting peptide–protein interactions. Predictive, next-generation platforms that can accurately predict AMP–protein interactions will enable the selection and design of AMPs that have pre-specified targets and favourable mechanisms of action.
Negative data selection for training property predictors.
In a supervised learning framework (Box 3), peptides with and without the property of interest should be provided to train ML models. For classification models, negative data (that is, peptides without the property of interest) should be included in the training set to prevent ML classifiers from minimizing the loss and metric functions (Box 2) by trivially predicting any sample to be positive during and after model training, respectively. Similarly, regression models should be trained to predict different bioactivity values for inactive samples than for active samples. In general, the selection of negative data greatly affects ML model training and the performance of the resulting trained models102. Although researchers have often included unlabelled samples as negative data (for example, previous work14,15 included peptides without certain keywords related to ‘antimicrobial’ from UniProt as negative data), false negatives may exist owing to the lack of experimental validation. These false negatives may mislead ML models that are trained on them.
Box 2. General process of machine learning.
Machine learning (ML) aims to model input–output relationships to perform tasks. For any task to be modelled using ML, it is necessary first to specify the inputs and outputs. The task of predicting a peptide’s antimicrobial activity may, for example, have a peptide sequence as an input and the probability that, at a given concentration, the peptide described by the sequence inhibits the growth of a particular bacterial species as an output.
ML is a data-driven approach — how an ML model maps the input to the output is learned from the training data. Thus, the second step of developing an ML model is to curate task-related data. For example, in the above example of AMP activity prediction, pairs of data points (peptide sequence plus corresponding experimental antimicrobial activity value) curated from public databases or produced by experimental measurements can form a dataset with which the ML model can be trained.
ML model training involves feeding the dataset inputs to the model and obtaining the model’s predicted output values. The predicted outputs are then quantitatively compared with the actual outputs from the training dataset using a loss function. For any pair of predicted and actual outputs, the loss function returns a value measuring how close the predicted output is to the actual output. Larger values correspond to worse predictions. An optimization algorithm is then applied to adjust the parameters of the ML model to minimize the loss value. This training procedure is repeated several times until a stop criterion, for example a particular loss value, is met.
For evaluation, predicted outputs are once again generated by passing inputs from the dataset to the ML model. Metric functions are then used to compare the predicted and actual outputs, returning quantitative values (such as accuracy and Pearson correlation coefficient) that indicate how well the ML model performs. The metric function is not necessarily identical to the loss function.
Although ML models are trained on existing data, a goal of most models is to be able to generalize to new inputs. Therefore, computationally evaluating how well a trained ML model makes predictions requires a separate dataset containing data points that have not been seen by the ML model. In practice, the starting dataset is commonly divided into training, validation and test sets, and only the training dataset is used for model training.
There can be multiple ML models exhibiting good performance, and each model may be parameterized by hyperparameters, which describe model properties (such as depth of a neural network) that should be set before training. In practice, various ML models are evaluated on the validation dataset, and the best-performing ML model and its hyperparameters are selected based on the metric of interest. The performance of this model is then evaluated on the withheld test set and reported.
Several strategies can help to improve the selection of negative data. First, based on the intuition that similar samples may have similar properties of interest, unlabelled samples that are highly similar to positive samples may be filtered out. Similarity can be measured based on feature representation or calculated from sequence or structure alignment. Second, clustering (Box 3) may be performed based on the feature representations of unlabelled samples and subsample data as negative samples from each cluster. This strategy can reduce the redundancy in the negative data and help to balance the training set to include similar numbers of positive and negative samples, which is often useful when facing a highly imbalanced dataset and developing ML models that focus on correctly predicting samples from the minority class. Third, label smoothing has been shown to be effective at regularizing and improving classification models161. Label smoothing assumes that there is a small probability that the label of a given data point is incorrect and prevents classifiers from over-confidently predicting the label by creating ‘soft’ labels for ML models to fit. For example, negative data points whose original class labels are 0 in a binary classification problem may be modified to have higher values (such as between 0 and 0.1 or 0.2) as soft labels, and positive data points whose original class labels are 1 may have lower values (such as between 0.8 or 0.9 and 1) as soft labels. For regression models, learning a statistical distribution of target values can similarly enhance generalization ability162. Fourth, one can make use of positive-unlabelled learning163, in which the unlabelled data are not all considered as negative. Techniques for positive-unlabelled learning, including two-step techniques, may first define reliably negative samples in the unlabelled data based on a criterion (such as dissimilarity to positive samples). In the second step, the positive and inferred-negative samples are used to train ML models, which are then applied to label the rest of the unlabelled samples. The labelled data then make up the final training set for downstream ML models.
AMP generation
As opposed to identifying AMPs among existing amino acid sequences, new peptides can be generated by modifying or optimizing existing peptide sequences and by relying on computational sequence generation de novo. Various ML/AI methods can be used to modify and optimize peptide sequences and graphs (corresponding to 2D or 3D peptide structures), including evolutionary and genetic algorithms, Bayesian optimization and reinforcement learning. Notably, genetic algorithms have been used to optimize AMPs50,164,165; starting from an initial population of template peptide sequences, a genetic algorithm expands the population by creating offspring sequences through crossover (exchange of subsequences) and mutation (modification of subsequences). The antimicrobial potential of each initial and offspring sequence is then assessed based on prior human knowledge or ML-derived rules. Sequences with poor predicted antimicrobial activity are filtered out from the population. By iterating the processes of offspring sequence generation and filtering, the antimicrobial potential of peptides in the population can be optimized. Using such an evolutionary algorithm, AMPs from a glycine-rich guava peptide were designed, revealing that guavanin 2, when synthesized and tested, shows potent antimicrobial activity in vitro and efficacy against P. aeruginosa in a mouse infection model50.
To complement the modification and optimization steps, generative models can model the distribution of the training data and produce data that have properties similar to those of the training dataset. Deep generative models, which combine a generative framework with powerful DNN architectures, include neural language models21,85, variational autoencoder (VAE) models166,167, normalizing flow models168, GANs169 and diffusion models170–172 (Fig. 3), which have been used to generate new AMPs64. Neural language models126,173–175 have largely used recurrent neural networks trained to predict the next amino acid of an AMP sequence given information on the previous amino acids. Although these models were trained using AMP sequences and may be expected to generate AMP-like sequences, most of the outputs produced126,174,175 required additional filters (such as for physicochemical properties, ML-predicted antimicrobial activity and ML-predicted haemolysis) to rank and filter out inactive or weakly active sequences. Using these models, a peptide was generated to be effective for treating carbapenem-resistant A. baumannii in a mouse model of infection174, and eight new, non-haemolytic AMPs were synthesized with activity against multiple-drug-resistant pathogens in vitro126.
As applied to AMPs, VAEs48,176,177 have mapped peptide sequences to low-dimensional embeddings that sample statistical distributions. New peptide sequences can be generated by sampling and decoding embeddings from these distributions. For example, training VAEs on AMP sequences to study the latent space formed by the embeddings revealed that peptides close to each other in the latent space show similar features resulting in the accurate generation of new AMPs176,177. Similarly, using a VAE trained on unlabelled peptides from UniProt178, a sampling strategy was proposed based on predictors of various properties (such as antimicrobial activity and toxicity) to generate AMPs48. MD simulations were then performed to narrow down AMP candidates, and favourable toxicity profiles for two peptides (generated within 48 days) were validated in mice.
Although known for being difficult to train179 (owing, at least in part, to unstable training processes in which either the generator or the discriminator outperforms the other and sensitivity to model hyperparameters), several GANs180–183 have been developed to generate peptides resembling AMPs. Among them, six GAN-derived peptides180,182 have been validated to have antimicrobial activity in vitro. In VAE- and GAN-based approaches, models have often formulated AMP generation in terms of sequence generation while neglecting geometric information from 3D structures and different conformations. Additionally, although flow and diffusion models have been used to generate small molecules184 and proteins185, their use and efficacy for AMP generation remains to be studied.
Notably, introducing peptide structural information as inputs into ML models can help to establish sequence–structure–function relationships by bridging the gap between raw sequence and function. Indeed, although all the information that defines a specific peptide is encoded in the peptide’s sequence, properties such as structure that depend on nonlocal interactions between, and combinatorial subsets of, amino acids may not be sufficiently modelled by simple sequence-to-function frameworks. Thus, we expect that approaches that model and generate AMP sequences using structural and conformational information can improve our understanding of AMP function and mechanisms of action. The peptide sequences generated by any such approach will also be iteratively optimized through rounds of experimental testing and ML model retraining. This optimization will benefit from making use of multi-objective methods that optimize multiple criteria and properties.
Lastly, an important consideration for the generation of AMPs is synthesizability. Peptide synthesis typically occurs through a stepwise elongation process in which certain amino acids are coupled and protecting groups are removed. Although liquid-phase synthesis remains common for large-scale synthesis, most peptides are synthesized using solid-phase technology, in which the first amino acid of a growing chain is linked to a solid resin. Downstream peptide purification commonly relies on techniques such as high-performance liquid chromatography and reverse-phase chromatography, and can result in samples that are >98% pure, a standard often used for in vivo studies and clinical trials. Nevertheless, peptides that form insoluble aggregates, including those containing a high number of amino acids with hydrophobic side chains, remain difficult to synthesize and purify186,187. Independently of such ‘difficult sequences’, longer peptides (for example longer than 50 amino acids) have been more difficult to synthesize and purify than shorter ones owing to the number of sequential reactions that are required to occur; for such peptides, cost and yield are often major bottlenecks. In general, the number of intermediates generated during peptide synthesis, as well as the various hazardous reagents and solvents used (including N,N-dimethylformamide and N-methyl-2-pyrrolidone), makes sustainable peptide synthesis difficult, with estimates of multiple tonnes of waste being generated for each kilogram of produced peptide — as opposed to hundreds of kilograms for small-molecule synthesis188. To help to address these issues, computational approaches such as TANGO and AGGRESCAN may be able to accurately assess the synthetic accessibility of candidate peptide sequences by predicting those that aggregate in solution189,190. Taking into account such predictions during the selection and optimization of candidate peptides will be essential for promoting efficient and cost-effective synthesis.
Moving forward, we anticipate that comprehensive benchmarking will be useful for assessing generative AMP models, allowing us to compare models fairly and to optimize AMP design strategies. This benchmarking may use standardized datasets and metrics. Specifically, to benchmark the performance of generative models fairly, peptide datasets covering diverse amino acid sequence spaces must be compiled. The dataset may be split into a training set (on which all generative models will be trained) and a test set for evaluation. A possible evaluation metric could consist of using the trained generative models to generate a number of peptides, then evaluating how many test peptides share similar properties to those of the generated ones. To evaluate the quality of de novo generated peptides, certain heuristics can be followed, including: (1) lack of redundancy in the generated sequences; (2) novelty with respect to the training set; and (3) diversity of the generated peptides with respect to the general sequence space. Metrics such as the Frechet inception distance191,192 can measure novelty by quantifying the distance between generated peptides and other peptides with respect to a feature representation space. These feature representations are ‘hidden features’ that are derived from ML models trained to perform another task (such as antimicrobial activity prediction).
Current limitations
Explainable and interpretable AI
Simple ML models (such as linear and tree-based models) offer straightforward ways (such as learnt weights and feature importance scores, respectively) to explain important features that contribute to model predictions. Despite generally being more powerful than these simple models, DL models such as neural networks are also more difficult to explain, owing to their black-box nature. Various post-hoc explainable or interpretable methods34,193–198 have been developed to identify important input features for DL models (explainable) or identify the components of DL models that are responsible for certain predictions (interpretable). However, applying these methods to the identification and design of AMPs requires further work. It would be useful for future studies to involve collaborations with experimentalists who can examine whether the important features inferred by explainable approaches (such as physicochemical descriptors, amino acid residues or specific 3D substructures) match expectations. Wet-lab experiments can then comprehensively study peptides with or without the inferred features to validate their importance.
Quantitative modelling of peptide properties
Many peptide property predictors, including those reviewed above, have been used for classification tasks; by definition, this ignores quantitative activity values. Without more quantitative (such as regression) modelling, ML approaches run the risk of selecting for peptides with only weak or moderate property values, such as those for antimicrobial activity. Yet peptides that are more potent and selective than others are more relevant for translation into clinical use, as they can exert their antibacterial effects at lower concentrations without comparable toxicity.
Generalizability and uncertainty estimation
To ensure that ML models make reasonable and reliable predictions, the test set should be similar to the training set. Although domain adaptation techniques199 can be used to address the problem of domain shift, in which the test set differs from the training set, these techniques typically require fine-tuning of ML models, and researchers should aim to assess the generalizability of the ML models they build. Analyses in which dissimilar samples in the training set (where similarity is defined with respect to some property of interest) are withheld from training and then examined as test samples can often provide insightful information with regards to how well a model generalizes in certain contexts. Additionally, property predictors often do not model predictive uncertainty (a surrogate for measuring the reliability or confidence of the predictions), but providing relevant statistics (such as confidence intervals for prediction scores) can better inform situations in which ML models can generalize.
Outlook
ML/AI has been used to tackle key problems relevant to AMP discovery and development, including peptide generation, optimization and property prediction. Multiple AMPs generated or discovered using ML models have been validated to have antibacterial activity in vitro and, more recently, in vivo. This validation supports the feasibility and promise of ML-guided AMP identification and design. We anticipate that, in the coming years, ML will substantially accelerate research and development for AMPs, helping scientists and clinicians to combat infectious diseases. Below, we highlight three areas for future work: data curation and quality; peptide representation and ML model design; and ML model evaluation and selection.
Data curation and quality
ML requires accumulating large amounts of high-quality data, both labelled and unlabelled, as well as positive and negative examples, for model training and evaluation. To predict antimicrobial activity, ML models have mainly been trained on data obtained in vitro (such as MIC values). Developing predictive in vivo models of infection and curating corresponding antimicrobial data for training could accelerate the translation of AMPs identified in silico into the clinic. Publicly available data on medicinal chemistry properties (such as toxicity and stability), although important for AMP development, are scarcer than data on antimicrobial activity. Therefore, future studies should aim to generate and/or curate medicinal chemistry data in addition to data on important functional properties. High-throughput screens that can produce consistent and quantitative data are also needed to enable ML approaches to model activity values quantitatively (for example using regression) for more properties of interest. For example, in addition to using MIC values for regression models that predict antimicrobial activity, measurements of red blood cell half-maximal lethal concentration (LD50) values can inform regression models that predict haemolytic potency.
Public datasets containing data on mechanisms of action can also help to inform AMP discovery. Deciding whether an AMP should be used in combination with other drugs (and whether it may be synergistic) often relies on an understanding of its mechanism of action200. MD19 simulations have been widely used to study AMP mechanisms of action, but these simulations often focus on a few AMPs of interest or on peptide–membrane interactions. The development of ML-based prediction platforms will require more training data containing AMPs with different (non-membrane) targets, as well as improvements to current approaches (such as those using molecular docking) for predicting protein–ligand interactions93. ML approaches can also help to inform combination treatments by directly predicting synergies. Notably, DBAASP is a database that contains information on more than 600 synergistic interactions for AMPs and other antibiotics, representing a useful resource for future ML models to build on100. The generation of high-quality data that examine synergies between AMPs, small-molecule antibiotics and other antimicrobial drugs will continue to enable us to build more robust and accurate models.
Finally, comprehensively leveraging already-available AMP data could be a straightforward way to augment ML-driven models. For example, although MICs are commonly used to define training labels for models predicting antimicrobial activity, additional information (such as time needed for bacterial killing and whether the activity is bacteriostatic or bactericidal) can provide useful knowledge for improving model learning. ML models could use this knowledge as supervision signals during training, improving predictive power. To aid this, the data collected by different groups should be integrated and standardized to ensure quality and consistency. Metadata storing the details of how data were generated should be properly included with each record, as this may help researchers understand biases and limitations of their downstream models. When experimental results are reported, both positive and negative results should be provided, as negative data points can provide useful information to train ML models.
Peptide representation and ML model design
Most ML-based approaches to AMPs have used input peptides containing only the 20 standard amino acids, and peptides with non-canonical (such as d-amino acids) or chemically modified amino acids (such as N- and C-terminal and lysine modifications) have typically been removed for model training and evaluation. As a result, the trained ML models may not generalize to non-standard peptides. Further studies should develop and use peptide representations capable of handling non-standard amino acids and design ML models compatible with this expanded input to improve generality. In addition, 3D structural and conformational information has not yet been widely used for ML-driven modelling of AMP properties. Recent sequence-to-structure platforms, including AlphaFold2 and RoseTTAFold, can help to generate peptide structures, and the resulting structural information can then be used to augment the prediction of peptide properties. Adopting probabilistic approaches to model ML outputs, in addition to providing relevant statistics such as confidence information, can enable ML models to estimate uncertainty. An active area of research uses physics-informed neural networks (PINNs)201–203, which combine neural networks with physics-based models, to quickly and accurately approximate complex and time-consuming calculations for MD simulations. It would be interesting for future work to consider whether these models can be applied to aid in MD-based protein or peptide design204.
ML model evaluation and selection
Benchmarking approaches that use consistent training–validation–test sets and comprehensive evaluation metrics are needed to assess the performance of ML models trained to perform the same task. Although many AMP prediction models have been benchmarked51,101,102, similarly rigorous benchmarking (as described above) should be performed for models trained to predict properties other than antimicrobial activity and for generative models trained to design AMPs. This will aid in selecting appropriate tools to use for in silico AMP optimization and design. Determining suitable hyperparameters for ML models also remains an important problem, and we anticipate that automated ML and neural architecture search tools205–207 can be used to select best-performing hyperparameters in a data-driven manner.
To conclude, we note that AMP development faces challenges beyond those discussed in detail here, including the high cost of peptide synthesis and the generation of industrial waste208. Future work can help to address these challenges by finding ways to better discover and design AMPs with favourable synthesis properties. We expect that ML/AI-guided approaches will continue to accelerate the pace of AMP development, leading to urgently needed treatments for infectious diseases, improvements in human health and exciting discoveries in this nascent field.
Key points.
Machine learning (ML) can aid antimicrobial peptide (AMP) design and discovery. It can be applied to improve drug efficacy, predict medicinal chemistry and reduce the overall time and cost of drug development.
ML can be used for the prediction of therapeutic properties — such as antimicrobial efficacy, and absorption, distribution, metabolism, excretion and toxicity (ADMET) — and macromolecular structures.
Deep generative models are promising approaches to designing new AMPs.
Important limitations in AMP development include lack of selectivity, undesirable physicochemical and medicinal chemistry properties, unspecific or unknown mechanisms of action, high cost of peptide synthesis, and generation of industrial waste. ML can help to overcome these limitations by applying relevant models trained on high-quality datasets.
Acknowledgements
C.d.l.F.-N. holds a Presidential Professorship at the University of Pennsylvania, is a recipient of the Langer Prize by the AIChE Foundation, and acknowledges funding from the IADR Innovation in Oral Care Award, the Procter & Gamble Company, United Therapeutics, a BBRF Young Investigator Grant, the Nemirovsky Prize, Penn Health-Tech Accelerator Award, the Dean’s Innovation Fund from the Perelman School of Medicine at the University of Pennsylvania, the National Institute of General Medical Sciences of the US National Institutes of Health (NIH) under award number R35GM138201, and the Defense Threat Reduction Agency (DTRA; HDTRA11810041, HDTRA1-21-1-0014, and HDTRA1-23-1-0001). We thank K. Pepper for editing the manuscript and de la Fuente Lab members for discussions. F. Wong was supported by the National Institute of Allergy and Infectious Diseases of the NIH under award no. K25AI168451. J.J.C. was supported by the Defense Threat Reduction Agency (grant no. HDTRA12210032), the NIH (grant no. R01-AI146194), and the Broad Institute of MIT and Harvard. This work is part of the Antibiotics-AI Project, which is directed by J.J.C. and supported by the Audacious Project, Flu Lab, LLC, the Sea Grape Foundation, Rosamund Zander and Hansjorg Wyss for the Wyss Foundation, and an anonymous donor.
Footnotes
Competing interests
J.J.C. is scientific co-founder and scientific advisory board chair of EnBiotix, an antibiotic drug discovery company, and Phare Bio, a non-profit venture focused on antibiotic drug development. C.d.l.F.-N. provides consulting services to Invaio Sciences and is a member of the Scientific Advisory Boards of Nowture S.L. and Phare Bio. The remaining authors declare no competing interests.
References
- 1.Fjell CD, Hiss JA, Hancock REW & Schneider G Designing antimicrobial peptides: form follows function. Nat. Rev. Drug. Discov 11, 37–51 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Yan J et al. Recent progress in the discovery and design of antimicrobial peptides using traditional machine learning and deep learning. Antibiotics 11, 1451 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silva ON et al. Repurposing a peptide toxin from wasp venom into antiinfectives with dual antimicrobial and immunomodulatory properties. PNAS 117, 26936–26945 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magana M et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis 20, e216–e230 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Bahar A & Ren D Antimicrobial peptides. Pharmaceuticals 6, 1543–1575 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen CH & Lu TK Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics 9, 24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dijksteel GS, Ulrich MMW, Middelkoop E & Boekema BKHL Review: lessons learned from clinical trials using antimicrobial peptides (AMPs). Front. Microbiol 12, 616979 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention (U.S.); National Center for Emerging Zoonotic and Infectious Diseases (U.S.), Division of Healthcare Quality Promotion, Antibiotic Resistance Coordination and Strategy Unit. Antibiotic Resistance Threats in the United States, 2019 CDC; 10.15620/cdc:82532 (2019). [DOI] [Google Scholar]
- 9.Murray CJ et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santos-Júnior CD et al. Computational exploration of the global microbiome for antibiotic discovery. Preprint at bioRxiv 10.1101/2023.08.31.555663 (2023). [DOI] [Google Scholar]
- 11.Torres MDT et al. Human gut metagenomic mining reveals an untapped source of peptide antibiotics. Preprint at bioRxiv 10.1101/2023.08.31.555711 (2023). [DOI] [Google Scholar]
- 12.Maasch JRMA, Torres MDT, Melo MCR & de la Fuente-Nunez C Molecular de-extinction of ancient antimicrobial peptides enabled by machine learning. Cell Host Microbe 31, 1230–1274.e6 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the use of machine learning (ML) to mine the proteomes of the archaic humans Neanderthals and Denisovans, leading to the discovery of the first antibiotics in extinct organisms (including Neanderthalin-1) and launching the field of molecular de-extinction.
- 13.Wong F, de la Fuente-Nunez C & Collins JJ Leveraging artificial intelligence in the fight against infectious diseases. Science 381, 164–170 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review summarizes state-of-the-art artificial intelligence (AI)/ML approaches to addressing infectious diseases through the lens of biotechnology and medicine.
- 14.Ma Y et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat. Biotechnol 40, 921–931 (2022). [DOI] [PubMed] [Google Scholar]; This study reports the use of multiple language processing neural network models to identify 181 antimicrobial peptides (AMPs) with antimicrobial activity from the human gut microbiome, three of which were validated in vivo in a mouse model of bacterial lung infection.
- 15.Huang J et al. Identification of potent antimicrobial peptides via a machine-learning pipeline that mines the entire space of peptide sequences. Nat. Biomed. Eng 7, 797–810 (2023). [DOI] [PubMed] [Google Scholar]; This study applies a cascading pipeline consisting of multiple ML modules to identify 54 AMPs with antimicrobial activity from combinatorial peptide space.
- 16.Wan F, Torres MDT, Peng J & de la Fuente-Nunez C Molecular de-extinction of antibiotics enabled by deep learning. Preprint at bioRxiv 10.1101/2023.10.01.560353 (2023). [DOI] [Google Scholar]
- 17.Torres MDT & de la Fuente-Nunez C Toward computer-made artificial antibiotics. Curr. Opin. Microbiol 51, 30–38 (2019). [DOI] [PubMed] [Google Scholar]; This review outlines the emerging field of antibiotic discovery enabled by computers.
- 18.Chen CH, Bepler T, Pepper K, Fu D & Lu TK Synthetic molecular evolution of antimicrobial peptides. Curr. Opin. Biotechnol 75, 102718 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Palmer N, Maasch JRMA, Torres MDT & de la Fuente-Nunez C Molecular dynamics for antimicrobial peptide discovery. Infect. Immun 89, e00703–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devlin J, Chang M-W, Lee K & Toutanova K BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Vol. 1 (Long and Short Papers) (eds Burstein J et al.) 4171–4186 (Association for Computational Linguistics, 2019). [Google Scholar]
- 22.Silver D et al. Mastering the game of Go with deep neural networks and tree search. Nature 529, 484–489 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Krizhevsky A, Sutskever I & Hinton GE ImageNet classification with deep convolutional neural networks. Commun. ACM 60, 84–90 (2017). [Google Scholar]
- 24.Valeri JA et al. Sequence-to-function deep learning frameworks for engineered riboregulators. Nat. Commun 11, 5058 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angenent-Mari NM, Garruss AS, Soenksen LR, Church G & Collins JJ A deep learning approach to programmable RNA switches. Nat. Commun 11, 5057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee J et al. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 36, 1234–1240 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu Y et al. Domain-specific language model pretraining for biomedical natural language processing. ACM Trans. Comput. Healthc 3, 1–23 (2022). [Google Scholar]
- 28.Stokes JM et al. A deep learning approach to antibiotic discovery. Cell 180, 688–702. e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin W et al. Deep learning identifies synergistic drug combinations for treating COVID-19. Proc. Natl Acad. Sci. USA 118, e2015070118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong F, Omori S, Donghia NM, Zheng EJ & Collins JJ Discovering small-molecule senolytics with deep neural networks. Nat. Aging 3, 734–750 (2023). [DOI] [PubMed] [Google Scholar]
- 31.Soenksen LR et al. Using deep learning for dermatologist-level detection of suspicious pigmented skin lesions from wide-field images. Sci. Transl. Med 13, eabb3652 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Liu G et al. Deep learning-guided discovery of an antibiotic targeting Acinetobacter baumannii. Nat. Chem. Biol 19, 1342–1350 (2023). [DOI] [PubMed] [Google Scholar]
- 33.Zheng EJ et al. Discovery of antibiotics that selectively kill metabolically dormant bacteria. Cell Chem. Biol 10.1016/j.chembiol.2023.10.026 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong F et al. Discovery of a structural class of antibiotics with explainable deep learning. Nature 626, 177–185 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou J & Troyanskaya OG Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim HK et al. Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol 36, 239–241 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Lopez R, Regier J, Cole MB, Jordan MI & Yosef N Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alipanahi B, Delong A, Weirauch MT & Frey BJ Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol 33, 831–838 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Wu R et al. High-resolution de novo structure prediction from primary sequence. Preprint at bioRxiv 10.1101/2022.07.21.500999 (2022). [DOI] [Google Scholar]
- 40.Baek M et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vamathevan J et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug. Discov 18, 463–477 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li S et al. MONN: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Syst. 10, 308–322.e11 (2020). [Google Scholar]
- 43.Ge Y et al. An integrative drug repositioning framework discovered a potential therapeutic agent targeting COVID-19. Signal. Transduct. Target. Ther 6, 165 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhavoronkov A et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol 37, 1038–1040 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Rajkomar A et al. Scalable and accurate deep learning with electronic health records. NPJ Digit. Med 1, 18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen D, Wu G & Suk H-I Deep learning in medical image analysis. Annu. Rev. Biomed. Eng 19, 221–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melo MCR, Maasch JRMA & de la Fuente-Nunez C Accelerating antibiotic discovery through artificial intelligence. Commun. Biol 4, 1050 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das P et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng 5, 613–623 (2021). [DOI] [PubMed] [Google Scholar]; This study reports the use of a deep generative autoencoder to generate AMPs that were synthesized and tested for antimicrobial activity in vitro and for toxicity in mice.
- 49.Torres MDT et al. Mining for encrypted peptide antibiotics in the human proteome. Nat. Biomed. Eng 6, 67–75 (2022). [DOI] [PubMed] [Google Scholar]; This article reports the exploration of the human proteome as a source of antibiotics, leading to the discovery of thousands of previously unrecognized antimicrobial sequences, and providing a new framework for antibiotic discovery by mining entire proteomes.
- 50.Porto WF et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat. Commun 9, 1490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes an antibiotic molecule designed by a computer, called guavanin 2, which displays anti-infective properties in vivo.
- 51.Xu J et al. Comprehensive assessment of machine learning-based methods for predicting antimicrobial peptides. Brief Bioinform. 22, bbab083 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Osorio D, Rondón-Villarreal P & Torres R Peptides: a package for data mining of antimicrobial peptides. R J. 7, 4–14 (2015). [Google Scholar]
- 53.van Westen GJ et al. Benchmarking of protein descriptor sets in proteochemometric modeling (part 1): comparative study of 13 amino acid descriptor sets. J. Cheminform 5, 41 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Müller AT, Gabernet G, Hiss JA & Schneider G modlAMP: Python for antimicrobial peptides. Bioinformatics 33, 2753–2755 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Romero-Molina S, Ruiz-Blanco YB, Green JR & Sanchez-Garcia E ProtDCal-Suite: a web server for the numerical codification and functional analysis of proteins. Protein Sci. 28, 1734–1743 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barigye SJ, Gómez-Ganau S, Serrano-Candelas E & Gozalbes R PeptiDesCalculator: software for computation of peptide descriptors. Definition, implementation and case studies for 9 bioactivity endpoints. Proteins 89, 174–184 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Chen Z et al. iFeature: a Python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 34, 2499–2502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saeys Y, Inza I & Larranaga P A review of feature selection techniques in bioinformatics. Bioinformatics 23, 2507–2517 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Chen X et al. Sequence-based peptide identification, generation, and property prediction with deep learning: a review. Mol. Syst. Des. Eng 6, 406–428 (2021). [Google Scholar]
- 60.Kawashima S AAindex: amino acid index database. Nucleic Acids Res. 28, 374–374 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.ElAbd H et al. Amino acid encoding for deep learning applications. BMC Bioinform. 21, 235 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hochreiter S & Schmidhuber J Long short-term memory. Neural Comput. 9, 1735–1780 (1997). [DOI] [PubMed] [Google Scholar]
- 63.Chung J, Gülçehre Ç, Cho K & Bengio Y Empirical evaluation of gated recurrent neural networks on sequence modeling. Preprint at arXiv.org/abs/1412.3555 (2014).
- 64.Wan F, Kontogiorgos-Heintz D & de la Fuente-Nunez C Deep generative models for peptide design. Digit. Discov 1, 195–208 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yan K, Lv H, Guo Y, Peng W & Liu B sAMPpred-GAT: prediction of antimicrobial peptide by graph attention network and predicted peptide structure. Bioinformatics 39, btac715 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ganea O et al. GeoMol: torsional geometric generation of molecular 3D conformer ensembles. Adv. Neur. Inf. Process Syst 34, 13757–13769 (2021). [Google Scholar]
- 67.Jin W, Wohlwend J, Barzilay R & Jaakkola TS Iterative refinement graph neural network for antibody sequence-structure co-design. In Proc. 10th International Conference on Learning Representations, ICLR 2022 (OpenReview.net, 2022). [Google Scholar]
- 68.Maturana D & Scherer S VoxNet: a 3D convolutional neural network for real-time object recognition. In Proc. 2015 IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS), 922–928 (IEEE, 2015). [Google Scholar]
- 69.Lecun Y, Bottou L, Bengio Y & Haffner P Gradient-based learning applied to document recognition. Proc. IEEE 86, 2278–2324 (1998). [Google Scholar]
- 70.Jiménez J, Doerr S, Martínez-Rosell G, Rose AS & De Fabritiis G DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 33, 3036–3042 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Jones D et al. Improved protein–ligand binding affinity prediction with structure-based deep fusion inference. J. Chem. Inf. Model 61, 1583–1592 (2021). [DOI] [PubMed] [Google Scholar]
- 72.Bengio Y, Courville A & Vincent P Representation learning: a review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell 35, 1798–1828 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Grill J-B et al. Bootstrap your own latent — a new approach to self-supervised learning. Adv. Neur. Inf. Process Syst 33, 21271–21284 (2020). [Google Scholar]
- 74.Mikolov T, Chen K, Corrado G & Dean J Efficient estimation of word representations in vector space. In Proc. 1st International Conference on Learning Representations, ICLR 2013, Workshop Track (eds Bengio Y & LeCun Y) (OpenReview.net, 2013). [Google Scholar]
- 75.Brown TB et al. Language models are few-shot learners. Preprint at 10.48550/arXiv.2005.14165 (2020). [DOI]
- 76.Madani A et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol 41, 1099–1106 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alley EC, Khimulya G, Biswas S, AlQuraishi M & Church GM Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 16, 1315–1322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rong Y et al. Self-supervised graph transformer on large-scale molecular data. In NIPS’20: Proc. 34th International Conference on Neural Information Processing Systems (eds Larochelle H et al.) 12559–12571 (Curran Assoc., 2020). [Google Scholar]
- 79.Zang X, Zhao X & Tang B Hierarchical molecular graph self-supervised learning for property prediction. Commun. Chem 6, 34 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Geourjon C & Deléage G SOPM: a self-optimized method for protein secondary structure prediction. Protein Eng. 7, 157–164 (1994). [DOI] [PubMed] [Google Scholar]
- 81.Cao X et al. PSSP-MVIRT: peptide secondary structure prediction based on a multi-view deep learning architecture. Brief Bioinform. 22, bbab203 (2021). [DOI] [PubMed] [Google Scholar]
- 82.Peri S, Steen H & Pandey A GPMAW – a software tool for analyzing proteins and peptides. Trends Biochem. Sci 26, 687–689 (2001). [DOI] [PubMed] [Google Scholar]
- 83.Pereira J et al. High‐accuracy protein structure prediction in CASP14. Protein 89, 1687–1699 (2021). [DOI] [PubMed] [Google Scholar]
- 84.Robin X et al. Continuous Automated Model EvaluatiOn (CAMEO) — perspectives on the future of fully automated evaluation of structure prediction methods. Proteins 89, 1977–1986 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vaswani A et al. Attention is all you need. In NIPS’17: Proc. 31st International Conference on Neural Information Processing Systems (eds Guyon I et al.) 6000–6010 (Curran Assoc., 2017). [Google Scholar]
- 86.Berman HM The protein data bank. Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin Z et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023). [DOI] [PubMed] [Google Scholar]
- 88.McDonald EF, Jones T, Plate L, Meiler J & Gulsevin A Benchmarking AlphaFold2 on peptide structure prediction. Structure 31, 111–119.e2 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lamiable A et al. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 44, W449–W454 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Timmons PB & Hewage CM APPTEST is a novel protocol for the automatic prediction of peptide tertiary structures. Brief Bioinform. 22, bbab308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boaro A et al. Structure-function-guided design of synthetic peptides with anti-infective activity derived from wasp venom. Cell Rep. Phys. Sci 4, 101459 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Torres MDT et al. Structure-function-guided exploration of the antimicrobial peptide polybia-CP identifies activity determinants and generates synthetic therapeutic candidates. Commun. Biol 1, 221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wong F et al. Benchmarking-enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol 18, e11081 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luo S, Shi C, Xu M & Tang J Predicting molecular conformation via dynamic graph score matching. Adv. Neur. Inf. Process Syst 34, 19784–19795 (2021). [Google Scholar]
- 95.Hoogeboom E et al. Equivariant diffusion for molecule generation in 3D. Proc. Mach. Learn. Res 162, 8867–8887 (PMLR, 2022). [Google Scholar]
- 96.Xu M et al. GeoDiff: a geometric diffusion model for molecular conformation generation. In Proc. 10th International Conference on Learning Representations, ICLR 2022 (OpenReview.net, 2022). [Google Scholar]
- 97.Mansimov E, Mahmood O, Kang S & Cho K Molecular geometry prediction using a deep generative graph neural network. Sci. Rep 9, 20381 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gogineni T et al. TorsionNet: a reinforcement learning approach to sequential conformer search. In NIPS’20: Proc. 34th International Conference on Neural Information Processing Systems (eds Larochelle H et al.) 20142–20153 (ACM, 2020). [Google Scholar]
- 99.Janson G, Valdes-Garcia G, Heo L & Feig M Direct generation of protein conformational ensembles via machine learning. Nat. Commun 14, 774 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pirtskhalava M et al. DBAASP v3: database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 49, D288–D297 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.García-Jacas CR, Pinacho-Castellanos SA, García-González LA & Brizuela CA Do deep learning models make a difference in the identification of antimicrobial peptides? Brief Bioinform. 23, bbac094 (2022). [DOI] [PubMed] [Google Scholar]
- 102.Sidorczuk K et al. Benchmarks in antimicrobial peptide prediction are biased due to the selection of negative data. Brief Bioinform. 23, bbac343 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Waghu FH, Barai RS, Gurung P & Idicula-Thomas S CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 44, D1094–D1097 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao X, Wu H, Lu H, Li G & Huang Q LAMP: a database linking antimicrobial peptides. PLoS ONE 8, e66557 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Witten J & Witten Z Deep learning regression model for antimicrobial peptide design. Preprint at bioRxiv 10.1101/692681 (2019). [DOI] [Google Scholar]
- 106.Wang G, Li X & Wang Z APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meher PK, Sahu TK, Saini V & Rao AR Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep 7, 42362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiao X, Wang P, Lin W-Z, Jia J-H & Chou K-C iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem 436, 168–177 (2013). [DOI] [PubMed] [Google Scholar]
- 109.Fingerhut LCHW, Miller DJ, Strugnell JM, Daly NL & Cooke IR ampir: an R package for fast genome-wide prediction of antimicrobial peptides. Bioinformatics 36, 5262–5263 (2021). [DOI] [PubMed] [Google Scholar]
- 110.Santos-Júnior CD, Pan S, Zhao X-M & Coelho LP Macrel: antimicrobial peptide screening in genomes and metagenomes. PeerJ 8, e10555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Burdukiewicz M et al. Proteomic screening for prediction and design of antimicrobial peptides with AmpGram. Int. J. Mol. Sci 21, 4310 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lawrence TJ et al. amPEPpy 1.0: a portable and accurate antimicrobial peptide prediction tool. Bioinformatics 37, 2058–2060 (2021). [DOI] [PubMed] [Google Scholar]
- 113.Bhadra P, Yan J, Li J, Fong S & Siu SWI AmPEP: sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep 8, 1697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pane K et al. Antimicrobial potency of cationic antimicrobial peptides can be predicted from their amino acid composition: application to the detection of ‘cryptic’ antimicrobial peptides. J. Theor. Biol 419, 254–265 (2017). [DOI] [PubMed] [Google Scholar]
- 115.Yan J et al. Deep-AmPEP30: improve short antimicrobial peptides prediction with deep learning. Mol. Ther. Nucleic Acids 20, 882–894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Veltri D, Kamath U & Shehu A Deep learning improves antimicrobial peptide recognition. Bioinformatics 34, 2740–2747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xiao X, Shao Y-T, Cheng X & Stamatovic B iAMP-CA2L: a new CNN-BiLSTM-SVM classifier based on cellular automata image for identifying antimicrobial peptides and their functional types. Brief. Bioinform 22, bbab209 (2021). [DOI] [PubMed] [Google Scholar]
- 118.Robles-Loaiza AA et al. Traditional and computational screening of non-toxic peptides and approaches to improving selectivity. Pharmaceuticals 15, 323 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Plisson F, Ramírez-Sánchez O & Martínez-Hernández C Machine learning-guided discovery and design of non-hemolytic peptides. Sci. Rep 10, 16581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chaudhary K et al. A web server and mobile app for computing hemolytic potency of peptides. Sci. Rep 6, 22843 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Win TS et al. HemoPred: a web server for predicting the hemolytic activity of peptides. Future Med. Chem 9, 275–291 (2017). [DOI] [PubMed] [Google Scholar]
- 122.Hasan MM et al. HLPpred-Fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 36, 3350–3356 (2020). [DOI] [PubMed] [Google Scholar]
- 123.Gautam A et al. Hemolytik: a database of experimentally determined hemolytic and non-hemolytic peptides. Nucleic Acids Res. 42, D444–D449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zakharova E, Orsi M, Capecchi A & Reymond J Machine learning guided discovery of non-hemolytic membrane disruptive anticancer peptides. ChemMedChem 17, e202200291 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Timmons PB & Hewage CM HAPPENN is a novel tool for hemolytic activity prediction for therapeutic peptides which employs neural networks. Sci. Rep 10, 10869 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Capecchi A et al. Machine learning designs non-hemolytic antimicrobial peptides. Chem. Sci 12, 9221–9232 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Salem M, Keshavarzi Arshadi A & Yuan JS AMPDeep: hemolytic activity prediction of antimicrobial peptides using transfer learning. BMC Bioinform. 23, 389 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gupta S et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8, e73957 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sharma N, Naorem LD, Jain S & Raghava GPS ToxinPred2: an improved method for predicting toxicity of proteins. Brief. Bioinform 23, bbac174 (2022). [DOI] [PubMed] [Google Scholar]
- 130.Naamati G, Askenazi M & Linial M ClanTox: a classifier of short animal toxins. Nucleic Acids Res. 37, W363–W368 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wei L, Ye X, Sakurai T, Mu Z & Wei L ToxIBTL: prediction of peptide toxicity based on information bottleneck and transfer learning. Bioinformatics 38, 1514–1524 (2022). [DOI] [PubMed] [Google Scholar]
- 132.Wei L, Ye X, Xue Y, Sakurai T & Wei L ATSE: a peptide toxicity predictor by exploiting structural and evolutionary information based on graph neural network and attention mechanism. Brief. Bioinform 22, bbab041 (2021). [DOI] [PubMed] [Google Scholar]
- 133.Zhang J, Zhang Z, Pu L, Tang J & Guo F AIEpred: an ensemble predictive model of classifier chain to identify anti-inflammatory peptides. IEEE/ACM Trans. Comput. Biol. Bioinform 18, 1831–1840 (2021). [DOI] [PubMed] [Google Scholar]
- 134.Khatun MS, Hasan MM & Kurata H PreAIP: computational prediction of anti-inflammatory peptides by integrating multiple complementary features. Front. Genet 10, 219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Manavalan B, Shin TH, Kim MO & Lee G AIPpred: sequence-based prediction of anti-inflammatory peptides using random forest. Front. Pharmacol 9, 276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gupta S, Sharma AK, Shastri V, Madhu MK & Sharma VK Prediction of anti-inflammatory proteins/peptides: an insilico approach. J. Transl. Med 15, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gupta S, Madhu MK, Sharma AK & Sharma VK ProInflam: a webserver for the prediction of proinflammatory antigenicity of peptides and proteins. J. Transl. Med 14, 178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Manavalan B, Shin TH, Kim MO & Lee G PIP-EL: a new ensemble learning method for improved proinflammatory peptide predictions. Front. Immunol 9, 1783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Khatun MS, Hasan MM, Shoombuatong W & Kurata H ProIn-Fuse: improved and robust prediction of proinflammatory peptides by fusing of multiple feature representations. J. Comput. Aided Mol. Des 34, 1229–1236 (2020). [DOI] [PubMed] [Google Scholar]
- 140.Boeckmann B The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sharma A, Singla D, Rashid M & Raghava GPS Designing of peptides with desired half-life in intestine-like environment. BMC Bioinform. 15, 282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yin S, Ding F & Dokholyan NV Eris: an automated estimator of protein stability. Nat. Methods 4, 466–467 (2007). [DOI] [PubMed] [Google Scholar]
- 143.Persikov AV, Ramshaw JAM & Brodsky B Prediction of collagen stability from amino acid sequence. J. Biol. Chem 280, 19343–19349 (2005). [DOI] [PubMed] [Google Scholar]
- 144.Wang F et al. Advancing oral delivery of biologics: machine learning predicts peptide stability in the gastrointestinal tract. Int. J. Pharm 634, 122643 (2023). [DOI] [PubMed] [Google Scholar]
- 145.Mathur D, Singh S, Mehta A, Agrawal P & Raghava GPS In silico approaches for predicting the half-life of natural and modified peptides in blood. PLoS ONE 13, e0196829 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cardoso MH et al. Non-lytic antibacterial peptides that translocate through bacterial membranes to act on intracellular targets. Int. J. Mol. Sci 20, 4877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ho Y-H, Shah P, Chen Y-W & Chen C-S Systematic analysis of intracellular-targeting antimicrobial peptides, bactenecin 7, hybrid of pleurocidin and dermaseptin, proline–arginine-rich peptide, and lactoferricin b, by using Escherichia coli proteome microarrays. Mol. Cell. Proteom 15, 1837–1847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schissel CK et al. Deep learning to design nuclear-targeting abiotic miniproteins. Nat. Chem 13, 992–1000 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fu X, Cai L, Zeng X & Zou Q StackCPPred: a stacking and pairwise energy content-based prediction of cell-penetrating peptides and their uptake efficiency. Bioinformatics 36, 3028–3034 (2020). [DOI] [PubMed] [Google Scholar]
- 150.Nasiri F, Atanaki FF, Behrouzi S, Kavousi K & Bagheri M CpACpP: in silico cell-penetrating anticancer peptide prediction using a novel bioinformatics framework. ACS Omega 6, 19846–19859 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wolfe JM et al. Machine learning to predict cell-penetrating peptides for antisense delivery. ACS Cent. Sci 4, 512–520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kumar V et al. Prediction of cell-penetrating potential of modified peptides containing natural and chemically modified residues. Front. Microbiol 9, 725 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Manavalan B, Subramaniyam S, Shin TH, Kim MO & Lee G Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res 17, 2715–2726 (2018). [DOI] [PubMed] [Google Scholar]
- 154.Sanders WS, Johnston CI, Bridges SM, Burgess SC & Willeford KO Prediction of cell penetrating peptides by support vector machines. PLoS Comput. Biol 7, e1002101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Qiang X et al. CPPred-FL: a sequence-based predictor for large-scale identification of cell-penetrating peptides by feature representation learning. Brief. Bioinform 21, 11–23 (2018). [DOI] [PubMed] [Google Scholar]
- 156.Lei Y et al. A deep-learning framework for multi-level peptide–protein interaction prediction. Nat. Commun 12, 5465 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cunningham JM, Koytiger G, Sorger PK & AlQuraishi M Biophysical prediction of protein–peptide interactions and signaling networks using machine learning. Nat. Methods 17, 175–183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li Z, Miao Q, Yan F, Meng Y & Zhou P Machine learning in quantitative protein–peptide affinity prediction: implications for therapeutic peptide design. Curr. Drug. Metab 20, 170–176 (2019). [DOI] [PubMed] [Google Scholar]
- 159.Trisciuzzi D, Siragusa L, Baroni M, Cruciani G & Nicolotti O An integrated machine learning model to spot peptide binding pockets in 3D protein screening. J. Chem. Inf. Model 62, 6812–6824 (2022). [DOI] [PubMed] [Google Scholar]
- 160.Wang R, Jin J, Zou Q, Nakai K & Wei L Predicting protein–peptide binding residues via interpretable deep learning. Bioinformatics 38, 3351–3360 (2022). [DOI] [PubMed] [Google Scholar]
- 161.Müller R, Kornblith S & Hinton G When does label smoothing help? In Proc. 33rd International Conference on Neural Information Processing Systems (eds Wallach HM et al.) 4694–4703 (ACM, 2019). [Google Scholar]
- 162.Imani E & White M Improving regression performance with distributional losses. In Proc. 35th International Conference on Machine Learning, Vol. 80 (eds Dy JG & Krause A) 2162–2171 (PMLR, 2018). [Google Scholar]
- 163.Bekker J & Davis J Learning from positive and unlabeled data: a survey. Mach. Learn 109, 719–760 (2020). [Google Scholar]
- 164.Yoshida M et al. Using evolutionary algorithms and machine learning to explore sequence space for the discovery of antimicrobial peptides. Chem 4, 533–543 (2018). [Google Scholar]
- 165.Boone K, Wisdom C, Camarda K, Spencer P & Tamerler C Combining genetic algorithm with machine learning strategies for designing potent antimicrobial peptides. BMC Bioinforma. 22, 239 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rezende DJ, Mohamed S & Wierstra D Stochastic backpropagation and approximate inference in deep generative models. In Proc. 31st International Conference on Machine Learning, Vol. 32 (eds Xing EP & Jebara T) 1278–1286 (PMLR, 2014). [Google Scholar]
- 167.Kingma DP & Welling M Auto-encoding variational Bayes. In Proc. 2nd International Conference on Learning Representations, ICLR 2014, Conference Track (eds Bengio Y & LeCun Y) (OpenReview.net, 2014). [Google Scholar]
- 168.Rezende D & Mohamed S Variational inference with normalizing flows. In Proc. 32nd International Conference on Machine Learning, Vol. 37 (eds Bach F & Blei D) 1530–1538 (PMLR, 2015). [Google Scholar]
- 169.Goodfellow I et al. Generative adversarial networks. Commun. ACM 63, 139–144 (2020). [Google Scholar]
- 170.Song Y et al. Score-based generative modeling through stochastic differential equations. In 9th International Conference on Learning Representations, ICLR 2021 (OpenReview.net, 2021). [Google Scholar]
- 171.Ho J, Jain A & Abbeel P Denoising diffusion probabilistic models. In Proc. 34th Conference on Neural Information Processing Systems, Advances in Neural Information Processing Systems 33 (eds Larochelle H et al.) (NeurIPS, 2020). [Google Scholar]
- 172.Sohl-Dickstein J, Weiss E, Maheswaranathan N & Ganguli S Deep unsupervised learning using nonequilibrium thermodynamics. In Proc. 32nd International Conference on Machine Learning Vol. 37 (eds Bach F & Blei D) 2256–2265 (PMLR, 2015). [Google Scholar]
- 173.Müller AT, Hiss JA & Schneider G Recurrent neural network model for constructive peptide design. J. Chem. Inf. Model 58, 472–479 (2018). [DOI] [PubMed] [Google Scholar]
- 174.Nagarajan D et al. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem 293, 3492–3509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang C, Garlick S & Zloh M Deep learning for novel antimicrobial peptide design. Biomolecules 11, 471 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dean SN & Walper SA Variational autoencoder for generation of antimicrobial peptides. ACS Omega 5, 20746–20754 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Dean SN, Alvarez JAE, Zabetakis D, Walper SA & Malanoski AP PepVAE: variational autoencoder framework for antimicrobial peptide generation and activity prediction. Front. Microbiol 10.3389/fmicb.2021.725727 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.UniProt Consrtioum. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Arjovsky M & Bottou L Towards principled methods for training generative adversarial networks. In Proc. 5th International Conference on Learning Representations, ICLR 2017, Conference Track (OpenReview.net, 2017). [Google Scholar]
- 180.Tucs A et al. Generating ampicillin-level antimicrobial peptides with activity-aware generative adversarial networks. ACS Omega 5, 22847–22851 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Van Oort CM, Ferrell JB, Remington JM, Wshah S & Li J AMPGAN v2: machine learning-guided design of antimicrobial peptides. J. Chem. Inf. Model 61, 2198–2207 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cao Q et al. Designing antimicrobial peptides using deep learning and molecular dynamic simulations. Brief. Bioinform 24, bbad058 (2023). [DOI] [PubMed] [Google Scholar]
- 183.Ferrell JB et al. A generative approach toward precision antimicrobial peptide design. Preprint at bioRxiv 10.1101/2020.10.02.324087 (2021). [DOI] [Google Scholar]
- 184.Shi C et al. GraphAF: a flow-based autoregressive model for molecular graph generation. In Proc. 8th International Conference on Learning Representations, ICLR 2020 (OpenReview.net, 2020). [Google Scholar]
- 185.Anand N & Achim T Protein structure and sequence generation with equivariant denoising diffusion probabilistic models. Preprint at 10.48550/arXiv.2205.15019 (2022). [DOI]
- 186.Coin I, Beyermann M & Bienert M Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nat. Protoc 2, 3247–3256 (2007). [DOI] [PubMed] [Google Scholar]
- 187.Mueller LK, Baumruck AC, Zhdanova H & Tietze AA Challenges and perspectives in chemical synthesis of highly hydrophobic peptides. Front Bioeng. Biotechnol 8, 162 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Isidro-Llobet A et al. Sustainability challenges in peptide synthesis and purification: from r&d to production. J. Org. Chem 84, 4615–4628 (2019). [DOI] [PubMed] [Google Scholar]
- 189.Conchillo-Solé O et al. AGGRESCAN: a server for the prediction and evaluation of ‘hot spots’ of aggregation in polypeptides. BMC Bioinform. 8, 65 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J & Serrano L Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol 22, 1302–1306 (2004). [DOI] [PubMed] [Google Scholar]
- 191.Heusel M, Ramsauer H, Unterthiner T, Nessler B & Hochreiter S GANs trained by a two time-scale update rule converge to a local nash equilibrium. In Proc. 31st International Conference on Neural Information Processing Systems, 6629–6640 (Curran Assoc., 2017). [Google Scholar]
- 192.Preuer K, Renz P, Unterthiner T, Hochreiter S & Klambauer G Fréchet chemnet distance: a metric for generative models for molecules in drug discovery. J. Chem. Inf. Model 58, 1736–1741 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Ribeiro MT, Singh S & Guestrin C ‘Why should I trust you?’ explaining the predictions of any classifier. In Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 1135–1144 (ACM, 2016). [Google Scholar]
- 194.Shrikumar A, Greenside P & Kundaje A Learning important features through propagating activation differences. In Proc. 34th International Conference on Machine Learning, Vol. 70, 3145–3153 (JMLR.org, 2017). [Google Scholar]
- 195.Sundararajan M, Taly A & Yan Q Axiomatic attribution for deep networks. In Proc. 34th International Conference on Machine Learning, Vol. 70, 3319–3328 (JMLR.org, 2017). [Google Scholar]
- 196.Jiménez-Luna J, Grisoni F & Schneider G Drug discovery with explainable artificial intelligence. Nat. Mach. Intell 2, 573–584 (2020). [Google Scholar]
- 197.Lundberg SM & Lee S-I A unified approach to interpreting model predictions. In Proc. 31st International Conference on Neural Information Processing Systems, 4768–4777 (Curran Assoc., 2017). [Google Scholar]
- 198.Yuan H, Yu H, Gui S & Ji S Explainability in graph neural networks: a taxonomic survey. IEEE Trans. Pattern Anal. Mach. Intell 45, 5782–5799 (2023). [DOI] [PubMed] [Google Scholar]
- 199.Farahani A, Voghoei S, Rasheed K & Arabnia HR In Advances in Data Science and Information Engineering. Transactions on Computational Science and Computational Intelligence (eds Stahlbock R et al.) 10.1007/978-3-030-71704-9_65 (Springer, 2021). [DOI] [Google Scholar]
- 200.Reffuveille F, de la Fuente-Núñez C, Mansour S & Hancock REW A broad-spectrum antibiofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrob. Agents Chemother 58, 5363–5371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Karniadakis GE et al. Physics-informed machine learning. Nat. Rev. Phys 3, 422–440 (2021). [Google Scholar]
- 202.Doerr S et al. TorchMD: a deep learning framework for molecular simulations. J. Chem. Theory Comput 17, 2355–2363 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Husic BE et al. Coarse graining molecular dynamics with graph neural networks. J. Chem. Phys 153, 194101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Omar SI, Keasar C, Ben-Sasson AJ & Haber E Protein design using physics informed neural networks. Biomolecules 13, 457 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ren P et al. A comprehensive survey of neural architecture search. ACM Comput. Surv 54, 1–34 (2022). [Google Scholar]
- 206.He X, Zhao K & Chu X AutoML: a survey of the state-of-the-art. Knowl. Based Syst 212, 106622 (2021). [Google Scholar]
- 207.Valeri JA et al. BioAutoMATED: an end-to-end automated machine learning tool for explanation and design of biological sequences. Cell Syst. 14, 525–542 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ferrazzano L et al. Sustainability in peptide chemistry: current synthesis and purification technologies and future challenges. Green. Chem 24, 975–1020 (2022). [Google Scholar]