

Abstract
Antibody generation requires the use of one or more time-consuming methods, namely animal immunization, and in vitro display technologies. However, the recent availability of large amounts of antibody sequence and structural data in the public domain along with the advent of generative deep learning algorithms raises the possibility of computationally generating novel antibody sequences with desirable developability attributes. Here, we describe a deep learning model for computationally generating libraries of highly human antibody variable regions whose intrinsic physicochemical properties resemble those of the variable regions of the marketed antibody-based biotherapeutics (medicine-likeness). We generated 100000 variable region sequences of antigen-agnostic human antibodies belonging to the IGHV3-IGKV1 germline pair using a training dataset of 31416 human antibodies that satisfied our computational developability criteria. The in-silico generated antibodies recapitulate intrinsic sequence, structural, and physicochemical properties of the training antibodies, and compare favorably with the experimentally measured biophysical attributes of 100 variable regions of marketed and clinical stage antibody-based biotherapeutics. A sample of 51 highly diverse in-silico generated antibodies with >90th percentile medicine-likeness and > 90% humanness was evaluated by two independent experimental laboratories. Our data show the in-silico generated sequences exhibit high expression, monomer content, and thermal stability along with low hydrophobicity, self-association, and non-specific binding when produced as full-length monoclonal antibodies. The ability to computationally generate developable human antibody libraries is a first step towards enabling in-silico discovery of antibody-based biotherapeutics. These findings are expected to accelerate in-silico discovery of antibody-based biotherapeutics and expand the druggable antigen space to include targets refractory to conventional antibody discovery methods requiring in vitro antigen production.
Keywords: antibody, biotherapeutics, machine learning, developability, drug discovery
Graphical Abstract
Graphical Abstract.

Introduction
Antibody generation is the first in a long series of steps needed for discovery and development of therapeutic antibodies. It begins with production and qualification of the target antigen, which can sometimes take considerable time and effort itself. The antibody generation campaign is then commonly initiated to obtain tools or reagent antibodies that can help provide initial exploration and validation of the therapeutic concept. Once the initial experiments show promise, larger antibody generation campaigns are then devoted to obtaining higher quality antibodies to be used for therapeutic purposes. Irrespective of whether it is a tool or a therapeutic antibody generation campaign, a few major pathways, all experimental in nature, have been developed over several decades to generate antibodies against a given target antigen. These are summarized in Fig. 1 along with their advantages and disadvantages, and more details can be found in Gray et al. 2020 [1]. Monoclonal antibodies can be generated from immunized animals via B-cell cloning or by using animal-free systems such as phage or yeast display of natural repertoires or rationally designed libraries.
Figure 1.

Antibody generation is the first in a long series of steps needed for discovery and development of biotherapeutics. It begins as soon as a novel therapeutic concept has been formed, antigen to be targeted has been identified, and initial experimental material for the antigen has been produced in the laboratory. An antibody generation campaign is now initiated to obtain tool antibodies that can help provide initial exploration and validation of the therapeutic concept. Once the initial experiments show promise, larger antibody generation campaigns are then devoted to obtaining higher quality antibodies to be used for therapeutic purposes. Irrespective of whether it’s a tool or therapeutic antibody generation campaign, a few major pathways all experimental in nature have been developed over the years. These are summarized here, and more details can be found at Gray et al. 2020 [1]. Abs stands for antibodies; PolyAbs stands for polyclonal antibodies; and mAbs stands for monoclonal antibodies.
Numerous technological advances in immunology, molecular biology, and next generation sequencing have collectively made a very large number of antibody sequences available in the public domain in recent years. Publicly accessible databases such as Observed Antibody Space (OAS) [2, 3] and Adaptive Immune Receptor Repertoire (AIRR) [4–6] now contain billions of antibody sequences from both naïve and antigen experienced repertoires derived from various species. Availability of antibody sequences on this scale opens new opportunities for training machine learning algorithms with desired antibody sequence–structural descriptors to generate new sequences in-silico. Furthermore, if we can take advantage of this approach, we may accelerate antibody discovery by generating sequences with good developability profiles [7–10] upfront, before screening for potential binders to a given target by computational and/or experimental means [11]. Together, these developments have led us to develop an innovative conceptual roadmap for discovery of antibodies in-silico (DAbI, [11]). Figure 2 shows our roadmap to enable de novo antibody-based drug discovery. Contrary to prevailing emphasis on finding the antigen-binding antibodies, the first step in our roadmap is in-silico generation of antigen-agnostic antibody libraries with good developability attributes. This strategic pivot allows us to mitigate the risk of costly failures during biotherapeutic drug product and clinical development by discovering the candidates that are already developable. Once fully enabled, DAbI shall help expand the antigen space potentially druggable by antibodies in addition to improving both efficiency and speed of biotherapeutic drug discovery and development cycles. This complements the efforts already underway to find novel drug targets and make them available in public repositories [12–15].
Figure 2.

Our conceptual roadmap to enable DAbI. The workflow drawn in this figure is focused at in-silico generation of highly developable antibody libraries that can be further interrogated for antigen binding using various in-silico tools such as protein modeling, molecular dynamics simulations, paratope - epitope predictions, virtual screening, molecular docking and affinity maturation. Note that the focus of this roadmap is to pre-pay for developability at the early stages of antibody library construction to minimize the risk of late-stage hurdles and failures during the drug product development, clinical trials, and regulatory approval. This report describes a proof-of-concept study for in-silico generation of antigen agnostic antibodies with desirable developability attributes.
In this work, we trained a Wasserstein Generative Adversarial Network with Gradient Penalty (WGAN+GP, [16, 17]) model on 31416 IGHV3-IGKV1 antibody variable region sequences that were pre-screened for high percent humanness, low chemical liabilities in the CDRs, and high medicine-likeness [10, 11, 16–18]. Here, the term ‘medicine-likeness’ implies similarity with the intrinsic physicochemical descriptors computed using sequences and structural models of the variable regions of the marketed antibody-based biotherapeutics. It does not have any implications for antigen binding (function) and should not be used to make inferences about binding specific targets. The in-silico generated sequences recapitulated all the desirable intrinsic features of the training sequences (high percent humanness, low incidence of physicochemical liabilities in their CDRs, and high medicine-likeness) but were found to have minimal duplicates of those in the training dataset or among themselves. A small number of highly diverse and medicine-like in-silico generated sequences were experimentally tested in two independent laboratories for developability in the spirit of the Turing test [19]. The results from both labs confirmed that these sequences possess desirable developability attributes. This work confirms the first foundational step in our conceptual roadmap to enable DAbI (Fig. 2) by building an initial antigen-agnostic, highly developable human antibody library through machine-learning (ML).
Results
Antibody sequence collection and WGAN+GP model development
The training data was obtained from internal and external sources (see methods section supporting information for details) to form an initial pool of 400000 VH:VL paired human Fv sequences, including those from the OAS [2, 3]. The sequences were grouped by pairs of germline loci. IGHV3-IGKV1, which comprises of IGHV3-* heavy chains paired with IGKV1-* light chains, was found to be the most common germline pair among the collected sequences and in the OAS collection (Figures S1A and S1B in Supporting Information). In this study, we therefore chose to train with, and in-silico generate sequences belonging to this highly represented germline pair as a proof of concept. The training sequence data belonging to this pair was further processed to ensure all sequences were highly human, contained no non-canonical Cys residues, no N-linked glycosylation motifs, low incidence of physicochemical liabilities in the CDRs, and qualify in-house developed high medicine-likeness metrics with a hypothesis that our deep learning model trained with these high-quality sequences might in turn generate new high-quality sequences. A total of 31416 IGHV3-IGKV1 paired antibody sequences belong to this training dataset. In addition to the training dataset, a control dataset containing 71283 paired antibody sequences belonging to the same germline loci pair and with ≥80% humanness, but without any additional filters, was also created. The training dataset is a subset of this control dataset. A WGAN+GP [16, 17] was used to train on both datasets, and to in-silico generate 100000 paired antibody sequences in the ScFv format (see materials and methods) in each case. The in-silico sequences generated using the control dataset are only used as ‘reference’ wherever necessary, while the focus of our analyses is on the ‘main’ sequence generated using the training dataset.
While several deep learning algorithms have been developed for in-silico generation of antibody sequences and structures [20–28], we chose to use Generative Adversarial Network (GAN) because the adversarial relationship between the generator and the discriminator neural networks intuitively resembles the feedback loop mechanism ubiquitous in cellular and physiological processes and in natural evolution. We consider this important because algorithms that imbibe natural processes and easily learn characteristics of natural antibodies without requiring a large number of sequences in their training datasets or a large number of machine learning features, are of greater practical use within the biopharmaceutical industry. They may also have a better chance of de novo producing experimentally verifiable antibody sequences with desired properties. Furthermore, we chose to use Wasserstein GAN over simple GAN because the use of Wasserstein distance rather than binary feedback from the discriminator allows for more stable model training and generation of diverse antibody sequences. However, this diversity needed to be contained with the boundary conditions imposed by the specific germline pair and medicine-likeness profile. Coupling WGAN with Gradient Penalty allowed us to achieve this and keep the generated sequences realistic. To our knowledge this is the first attempt to produce antigen-agnostic but highly developable antibodies via deep learning. Earlier attempts have focused on generating and optimizing antigen-specific antibodies [20, 25, 27, 28]. Therefore, a direct benchmarking of different methods for in-silico antibody generation methods with our protocol is infeasible. Furthermore, our work focuses on obtaining experimental validation of the in-silico generated antibodies, thereby, creating a viable pathway for antibody discovery in addition to animal immunizations, hybridomas, and display libraries (see Figs. 1 and 2).
Figure 3(A-F) describes the salient features of our procedure, and the details are provided in the materials and methods section. Briefly, the amino acid sequence of the variable region (Fv) of each antibody in the training dataset was converted into single chain variable fragments (ScFvs) by combining the sequences of VH and VL in VH-(GGGGS)4-VL format to preserve their VH and VL pairings and the integrity of the conserved sequence motifs found at the end of framework 4 (β-strand G) in VH and at the beginning of Framework 1 (β-strand A) in VL. Note that a vast majority of publicly available repositories of antibody sequence data do not contain the chain pairing information and therefore most available language models, although trained on hundreds of millions of antibody sequences (>> > 31 416 sequences available to us), generate VH and VL sequences separately and do not provide the pairing information [20, 24, 26]. All the ScFvs were multiply aligned and the individual multiple sequence alignment positions were numbered using ANARCI [29] as described in the material and methods section. Use of the multiple sequence alignment (MSA) of all sequences in the training set allowed our algorithm to learn features associated with the antibody structure–function such as mapping of the framework and complementarity determining regions, motifs found at the junctions of these regions, and positions of canonical Cysteine residues in the V-genes along with the junctions between V, D (heavy chain only), and J genes of the heavy and light chains [30], without the need to introduce additional sequence-structural features or embeddings required for methods such as LSTM [28]. The creation of the MSA for the training sequence data set was followed by one-hot encoding [31], which allowed us to convert the amino acid sequence of each ScFv into an image (see Fig. 3A–C). This yielded 31 416 images which were then used to train the WGAN+GP model over 1000 epochs. Conversion of the sequences into images via one hot encoding allowed the generator and discriminator neural networks to learn the covariances among sequentially adjacent as well as distant residues and the sequence - structural relationships inherent to the antibodies. The competition between the generator and the Wasserstein critic along with use of the gradient penalty limits the ability of the model to produce unrealistic outputs. Figure 3D plots convergence of the loss functions of the generator and the Wasserstein critic over the learning epochs. Figure 3E and 3F show emergence of clearer images and greater overlaps between the training and the generated data as a function of the number of epochs. In this work 100 000 images were generated computationally and translated back into 100 000 in-silico generated antibody sequences in the same ScFv format. Protein MPNN, Variational Autoencoder (VAE) and diffusion models [21, 25] could have been also used at this step. However, these methods lack a feedback mechanism and were therefore at greater risk producing spurious results than the WGAN+GP combination. The number of sequences to be generated was arbitrarily decided, keeping in mind computational resources available to us. The number of sequences that can be generated using this model is expected to be astronomically large. However, there may be increased risk of redundancy among the generated sequences as their number increases, although this relationship was not explored here. Nonetheless, sequence—structural characteristics of the 100 000 in-silico generated sequences show minimal redundancy (see below). Based on these results, one could speculate that future computationally generated antibody repertoires could potentially match or even exceed in sequence diversities than those feasible from B-cell repertoires [32, 33] and display technologies [34–36]. This possibility was not tested here, however, the observation that 100 000 generated antibodies did not fully exhaust CDR diversity learnt by our model implies this potential (see below).
Figure 3.

Key aspects of the algorithm used in this work for in-silico generation of IGHV1-IGKV3 antibodies. (A) Antibody sequences in the training set were set in ScFv format and multiply aligned. (B) One-hot encoding was used to convert the multiple sequence alignment into a matrix. (C) the matrix was used to convert each antibody sequence into an image. These images were used to train a WGAN+GP model. (D) Plot showing the convergence of the generator and the Wasserstein critic loss functions versus the number of training epochs. (E) Samples of the images generated by the WGAN+GP model at different epochs. These images were converted back into the antibody sequences using the one-hot matrix derived from the multiple sequence alignment of the training sequences. (F) Dimensionality reduction by the tSNE analyses show the overlaps between the training and in-silico generated sequences over the number of epochs.
Computational analyses of in-silico generated antibody sequences
Amino acid composition of the in-silico generated antibodies is same those in the training dataset
Figure S2 in the supporting information shows that antibodies generated in-silico have the same amino acid composition as those in the training set. This conclusion is confirmed by the ꭓ2 - test that accepts the null hypothesis that antibodies belonging to training and in-silico generated dataset have the same amino acid composition (ꭓ2 - value = 0.00085 (and 0.00079 for reference sequences), p-value =1 or 100% level of confidence for 19 degrees of freedom). This observation shows that our model preserves the fundamental residue composition of the IGHV3-IGKV1 germline pairs in both the control and the training datasets, and therefore generation of de novo sequences arises from the changes in sequence ordering.
In-silico generated antibodies show low redundancy
Among the 100 000 antibody sequences generated in-silico by using the training dataset, 10 (0.01%) were found to be duplicates (Table 1a). Individually assessing the VH and VL portions showed that in-silico generated sequences contained 95541 (95.5%) unique VH and 84183 (84.1%) unique VL sequences. The remainder of the unique 99990 ScFvs generated in this work arise from the unique VH and VL pairings. Overall, these observations suggest that in-silico generated sequences have low redundancy, thereby demonstrating that our algorithm is effective at generating nonidentical paired antibody sequences. This conclusion is further supported by the observation that the 100 000 reference antibody sequences generated in-silico by using the control dataset showed only one duplicate (Table 1a). Note that the control dataset contains more than twice as many sequences as the training dataset and therefore the WGAN+GP model had larger latent space to explore.
Table 1a.
Quantitative analysis of sequence redundancies among the in-silico sequences generated using the control and training datasets.
| Measurement | Value | Value |
|---|---|---|
| Number of training sequences in IGHV3_IGKV1 germline pair loci | 71 283 (Control dataset) | 31 416 (Training dataset) |
| Number of in-silico generated sequences | 100 000 | 100 000 |
| Number of duplicates within the set of in-silico generated sequences | 1 (0.001%) | 10 (0.01%) |
| Number of duplicates in-silico generated VH regions | 3121 (3.1%) | 4459 (4.5%) |
| Number of duplicates in-silico generated VL regions | 3960 (3.9%) | 15 827 (15.8%) |
| Number of duplicates between the generated sequences and the training sequences | 0 | 9 |
Levenshtein’s distance shows antibodies in the in-silico generated set are different from those in the training set
To understand how different the antibodies are in the training and generated sets, we used Levenshtein distance [37]. This distance was calculated for each of the 100 000 in-silico generated antibody sequences by comparing them with their respective closest sequences in the training dataset. Figure S3 in the supporting Information shows a histogram of Levenshtein distance values for the in-silico generated sequences. On average, an in-silico generated antibody sequence is 23 ± 6 edits away from its closest sequence in the training data set with the maximum number of edits being 46 (range = 0–46) (Table 1b). Among the 100 000 reference sequences generated using the control dataset, the average Levenshtein distance is similar at 25 ± 5 (range 4–46), suggesting that our WGAN+GP model performs consistently for both the datasets (Table 1b).
Table 1b.
Levenshtein distances of in-silico generated antibody sequences from their closest training antibody sequences in the control and training datasets.
| Dataset | L-dista | ScFv | VH | VL | HCDR1 | HCDR2 | HCDR3 | LCDR1 | LCDR2 | LCDR3 |
|---|---|---|---|---|---|---|---|---|---|---|
| 71 283 sequences in control dataset | # of 0 L-dist | 0 | 231 | 0 | 78 851 | 56 156 | 9031 | 79 854 | 96 170 | 67 842 |
| Mean ± std | 24.8 ± 5.1 | 11.7 ± 4.5 | 7.0 ± 2.1 | 0.3 ± 0.6 | 0.9 ± 1.2 | 4.1 ± 2.4 | 0.2 ± 0.5 | 0.1 ± 0.2 | 0.4 ± 0.6 | |
| Range (min - max) | 4–46 | 0–31 | 1–16 | 0–4 | 0–8 | 0–13 | 0–3 | 0–2 | 0–3 | |
| 31 416 sequences in training dataset | # of 0 L-dist | 9 | 1184 | 1464 | 74 609 | 48 517 | 11 169 | 78 326 | 91 163 | 68 081 |
| Mean ± std | 22.7 ± 6.2 | 11.1 ± 5.0 | 5.4 ± 2.3 | 0.3 ± 0.6 | 0.9 ± 1.1 | 4.1 ± 2.6 | 0.2 ± 0.5 | 0.1 ± 0.3 | 0.4 ± 0.6 | |
| Range (min - max) | 0–46 | 0–31 | 0–15 | 0–4 | 0–6 | 0–14 | 0–3 | 0–4 | 0–3 |
L-dist stands for Levenshtein distance.
Nine out of the 100 000 (0.009%) ScFv sequences generated in-silico using the training dataset have Levenshtein distance of 0, indicating they are exact copies of the training sequences. Looking deeper into individual chains, VH and VL showed 1184 (~1.2%) and 1464 (~1.5%) instances, respectively, with the Levenshtein distance of 0. The overall Levenshtein distance distribution ranges from 0 to 15 (Average = 5 ± 2) for the light chains (VL) and 0 to 31 (Average = 11 ± 5) for the heavy chains (VH). The individual CDRs in the light and heavy chains of the in-silico generated antibodies also show different degrees of variation with average Levenshtein distance for the HCDR3s being 4 ± 3 (range 0–14, Table 1b), suggesting that in-silico generated antibodies may be functionally diverse. Therefore, more than 98% of generated VH and VL sequences are novel, individually, and the remainder (~1%) of the 99 991 in-silico generated ScFv sequences involve novel VH and VL pairings. In summary, our method is effective at generating paired antibody sequences.
Germline pair distribution of the in-silico generated antibodies
All the in-silico generated antibody sequences belong to the IGHV3-IGKV1 germline loci pair, same as that of the training set. Within these, sequences with IGHV3–23 and IGKV1–39 pairing are observed in the largest proportions in both training and generated sequences. Overall, the in-silico generated sequences closely mimic the germline pair distribution within the training set as shown in Fig. 4 using the germline pairing heat maps. Looking deeper into individual germline subclusters within the IGHV3 and IGVK1 loci, we found that the antibody sequences in the training set belong to 664 unique germline pairs whereas the in-silico generated antibody sequences span across 556 unique germline pairs. Of these, 404 germline pairs are common in both the training and in-silico generated antibody sequences. Approximately 87% (87 675 of 100 000) of the in-silico generated sequences and ~ 83% (26 694 of 31 416) of the training sequences belonged to this common set of 404 unique germline pairs. 12 325 (12%) of the 100 000 generated sequences belong to 152 unique germline pairs that were not found in the training set, whereas 4722 (15%) of 31 416 training sequences belong to 240 unique germline pairs that were not present among the in-silico generated antibody sequences (Fig. 4 and supplementary data S9). These observations show that our algorithm can explore further the germline pairing in the latent space available within IGHV3-IGKV1 loci. Simultaneously, it has not yet fully explored all the germline pairs made available by the training dataset. This suggests that there is room for greater sampling.
Figure 4.

Germline pair heatmap for (A) 31 416 antibodies in the training dataset and for (B) 100 000 in-silico generated sequences. This figure shows that the in-silico generated antibody sequences, in general, retain most of the germ-line pairings seen among the training sequences while exploring new ones within the IGHV3-IGKV1 pair of germline loci.
In addition to germline pairing, we asked if interfaces between the light and the heavy chains of the in-silico generated antibodies resemble those of the real antibodies. Towards this goal, we computed the buried surface area between VH and VL (BSAVH:VL) as well as the molecular mechanics based interaction energies between VH and VL domains (Eint VH:VL) for all the 31 416 antibodies in the training dataset, the 100 000 antibodies in-silico generated dataset, and 468 antibodies from a non-redundant set of antigen: antibody crystal structures available in the Protein Data Bank (PDB, as of Feb 2022 [38], our unpublished work). Figure 5A and 5B show that VH:VL interfaces of the in-silico generated antibodies are like those in antibodies of the training dataset and in the crystal structures (p-values >0.05).
Figure 5.

Computational analyses show that the in-silico generated antibodies recapitulate the sequence, structural and physicochemical characteristics of the training ones. (A) Box plots comparing the surface area buried at the VH: VL interfaces (BSAVH:VL) of the antibody sequences in the training dataset, in-silico generated dataset, and in the crystal structures of 468 antibody: Antigen complexes. (B) Box plots comparing the interaction energies (Eint) between VH and VL domains of the antibody sequences in the training dataset, in-silico generated dataset, and in the crystal structures of 468 antibody: Antigen complex. (C) Box plots showing the percent humanness of the VH and VL domains of the antibody sequences in the training and in-silico generated datasets. (D) CDR diversity among the training and in-silico generated antibody sequences was measured in terms of variations in CDR lengths as well as sequences. (i) Boxplots showing distributions of CDR lengths among the training and in-silico generated antibody sequences. (ii) bar plots showing Shannon entropies, used as measure of sequence variations, among the CDRs of the training and in-silico generated antibody sequences. (E) Violin plot comparing the medicine-likeness percentile scores for the antibody sequences in the training and in-silico generated datasets. The medicine-likeness percentile scores were computed using sequence- and structure-based calculations of a set of five non-redundant physicochemical descriptors, along with percent humanness, physicochemical liabilities in the CDRs, and TANGO aggregation scores of 113 marketed Fv structures. See materials and methods for details.
Percent humanness of the in-silico generated antibodies
In-silico generated sequences show a high human germline content. Figure 5C shows that 99.9% (99991) of the light chain variable regions (VL) are more than 80% human while 99.3% (99298) of the heavy chain variable regions (VH) are more than 80% human, when compared with their respective closest human germlines, identified using IgBlast [39]. In total, 99103 out of 100 000 in-silico generated sequences are more than 80% human for both the VL and VH domains. Note that all the sequences in our training dataset were 80% or more human. This observation shows that our model can generate highly human antibody sequences. Approximately 99.7% of the reference in-silico sequences (99699 out of 100000) generated using the control dataset also show high percent humanness.
CDR diversity of the in-silico generated antibodies
Binding to a given antigen was not part of our selection criteria for the antibody sequences to be included in the training dataset. Therefore, the in-silico generated sequences are expected to be antigen-agnostic, with diverse CDRs capable of recognizing various antigens. No antigen binding assays were performed in this work. Instead, CDR diversity was used as a computational measure to quantify antigen agnosticism of the in-silico generated antibodies. The CDR diversity was evaluated based on variations in CDR lengths and Shannon entropy [40] of the CDR sequences. Figure 5D-(i) and Table 2a compare length distributions of all light chain (LCDRs 1 to 3) and heavy chain CDRs (HCDRs 1 to 3). LCDR1, LCDR2 and LCDR3, HCDR1 and HCDR2 of the training as well as generated sets show almost identical distributions and the average values for CDR lengths are the same in both datasets. However, the range of CDR lengths tends to be smaller for in-silico generated sequences. For example, lengths of the HCDR3s in the training set ranges from 3 to 24 amino acid residues, while it is five to 22 residues among the generated ones. Similarly, LCDR3s among the training sequences are five to 12 residues long, while in the generated sequences LCDR3s are seven to 10 residues long (Table 2a).
Table 2a.
Comparison of CDR length diversity in the training dataset and the 100 000 main in-silico generated sequences.
| CDR name | CDR lengths of 31 416 sequences in the training dataset | CDR lengths of 100 000 sequences generated In-silico using the training dataset | ||
|---|---|---|---|---|
| (Mean ± Std | Range | Mean ± Std | Range | |
| LCDR1 | 11.00 ± 0.05 | 9–14 | 11.00 ± 0.03 | 10–11 |
| LCDR2 | 7.00 ± 0.06 | 6–10 | 7.00 ± 0.01 | 6–7 |
| LCDR3 | 9.00 ± 0.47 | 5–12 | 8.95 ± 0.40 | 7–10 |
| HCDR1 | 10.17 ± 0.50 | 6–12 | 10.14 ± 0.43 | 9–12 |
| HCDR2 | 17.11 ± 0.81 | 14–19 | 17.06 ± 0.73 | 15–19 |
| HCDR3 | 13.11 ± 2.96 | 3–24 | 12.82 ± 2.78 | 5–22 |
Shannon entropy (S) [40], as described by Equation 3 in the materials and methods section, was used to measure diversity at each multiple alignment position of the antibody sequences in the training and in-silico generated datasets (Fig. 6). For a given CDR, its sequence diversity was then measured by summing the Shannon entropies over the CDR length (Equation 4). Figure 5D-(ii) and Table 2b show CDR sequence diversities for each of the six CDRs present in the sequences in both generated and training sets. Expectedly, the HCDR3s show the greatest sequence diversity in both training and in-silico generated sequences. Like the smaller CDR length diversity, the CDRs of the in-silico generated sequences also show generally smaller Shannon entropies (Table 2b). Interestingly, the under sampling of the CDR diversity by the generated sequences in comparison to the training sequences is more pronounced for the CDRs in the heavy chains (ΔSHCDRs = SHCDRs (in-silico generated sequences) - SHCDRs (training sequences) = −16.1 bits) than the ones in the light chains (ΔSLCDRs = −10.9 bits) (Table 2b). These observations suggest that generative sampling of 100 000 sequences didn’t fully explore the diversity learnt by the model and therefore there is scope for constructing larger antibody libraries, in-silico. This inference is consistent with the observation that the in-silico generated sequences are highly non-redundant (Table 1a).
Figure 6.

Shannon entropy quantifies sequence diversity at each position in the multiple sequence alignments of the light and heavy chains in the 31 416 training and 100 000 in-silico generated antibody sequences. The shaded regions indicate the CDR positions. In general, the in-silico generated antibody sequences recapitulate the sequence diversity observed for the training ones.
Table 2b.
Comparison of CDR sequence diversity in the training dataset and the 100 000 main in-silico generated sequences.
| CDR name | 31 416 sequences in the training dataset | 100 000 sequences generated in-silico using the training dataset | |
|---|---|---|---|
| Shannon Entropy (S, bits) | Shannon Entropy (S, bits) | Difference (ΔS (bits))a | |
| LCDR1 | 13.7 | 9.7 | −4.0 |
| LCDR2 | 10.3 | 7.3 | −3.0 |
| LCDR3 | 17.6 | 13.7 | −3.9 |
| All LCDRs | 41.6 | 30.7 | −10.9 |
| HCDR1 | 16.2 | 11.4 | −4.8 |
| HCDR2 | 25.0 | 19.2 | −5.8 |
| HCDR3 | 53.2 | 47.7 | −5.2 |
| All HCDRs | 94.4 | 78.3 | −16.1 |
| All CDRs | 136.0 | 109.0 | −27.0 |
ΔSCDRs = SCDRs (in-silico generated sequences) - SCDRs (training sequences)
Incidence of physicochemical liability motifs among the in-silico generated antibodies
Incidence of potential N-linked glycosylation sites and non-canonical unpaired cysteines in the variable portions of the antibody drug candidates is undesirable from the perspective of drug development [41]. Therefore, antibody sequences in the training set did not contain any non-canonical cysteines or potential glycosylation motifs in their CDRs. However, a total of 7816 (7.8%) of the in-silico generated sequences were found to contain N-linked glycosylation motifs in their CDR regions (Table 3). Furthermore, 3775 (~3.8%) VH CDR regions and 4197 (~4.2%) VL CDR regions of the in-silico generated antibody sequences contained at least one N-linked glycosylation motif, and 156 (<0.2%) contained these motifs in both VH and VL. In addition to the N-linked glycosylation sites, 480 (~0.5%) in-silico generated sequences contained one or more non-canonical unpaired cysteines in their CDRs. A majority of these were observed in the heavy chain CDRs (438 out of 480; and 410 in HCDR3). The light chain CDRs contained non-canonical cysteines in 42 instances (Table 3). Only eleven (0.01%) in-silico generated sequences contained non-canonical cysteines in their framework regions. While low, this incidence of non-canonical cysteines among the in-silico generated sequences was unexpected since the sequences in the training set did not contain any of them. Taken together, these observations show our model’s high fidelity in reproducing the quality attributes of the training sequences.
Table 3.
Incidence of non-canonical Cys residues and N-linked glycosylation motifs among the CDRs of the 100 000 in-silico generated antibody sequences generated using the control and training datasets.a
| CDR name | Number of reference sequences with non-canonical cysteine residues | Number of main sequences with non-canonical cysteine residues | Number of reference sequences with N-linked glycosylation motifs | Number of main sequences with N-linked glycosylation motifs |
|---|---|---|---|---|
| HCDR1 | 56 (0.05%) | 19 (0.02%) | 91 (0.09%) | 490 (0.49%) |
| HCDR2 | 110 (0.11%) | 9 (0.01%) | 1279 (1.28%) | 2567 (2.6%) |
| HCDR3 | 1344 (1.34%) | 410 (0.41%) | 1340 (1.34%) | 744 (0.74%) |
| All HCDRs | 1508 (1.5%) | 438 (0.44%) | 2694 (2.69%) | 3775 (3.77%) |
| LCDR1 | 82 (0.08%) | 9 (0.01%) | 749 (0.75%) | 3706 (3.71%) |
| LCDR2 | 387 (0.39%) | 12 (0.01%) | 25 (0.02%) | 13 (0.01%) |
| LCDR3 | 12 (0.01%) | 21 (0.02%) | 3246 (3.24%) | 478 (0.48%) |
| All LCDRs | 481 (0.48%) | 42 (0.04%) | 3967 (3.96%) | 4197 (4.20%) |
| All CDRs | 1980 (1.98%) | 480 (0.48%) | 6556 (6.54%) | 7816 (7.81%) |
The 100 000 antibody sequences generated in-silico using the control dataset are called as reference sequences in the second and fourth columns of this table. The 100 000 antibody sequences generated in-silico using the training dataset are called as main sequences in the third and fifth columns of this table. Note that the number of paired antibody sequences containing unpaired Cys and N-linked glycosylation motifs in All HCDRs, All LCDRs and All CDRs may not be the sum of individual CDRs. This is because a given sequence may contain these liabilities in multiple CDRs.
Table S2 in the Supporting Information compares the incidence of the chemical liability motifs in the 100 000 reference sequences with those in the 100 000 main in-silico generated sequences. 1980 (~1.9%) of the reference in-silico generated sequences contain non-canonical unpaired cysteines in at least one of their CDRs. This is ~ 4-fold greater incidence when compared with 480 among the CDRs of the main in-silico generated sequences. Moreover, 1344 (~1.3%) of the reference sequences have cysteines in their HCDR3, which is about thrice the number of non-canonical Cys residues containing HCDR3s among the main ones. On the other hand, the incidence of N-linked glycosylation motifs in the CDR regions of the reference in-silico generated sequences is lower (~6.5%, 6556) than that (~7.8%, 7816) for main sequences generated using the training data.
The reference sequences also contain significantly more (14.6% greater incidence) Asn-deamidation motifs in their CDRs, while those for oxidation and asp-isomerization remain similar (Table S2). The overall chemical liability score for the main sequences is 459 ± 645, lower than that (521 ± 790) for the reference sequences. The large standard deviation values over the mean chemical liabilities scores imply large scatter in the data. Taken together, these results justify additional criteria, besides percent humanness, used to make the training set for generating medicine-like antibody sequences, since maximizing the developability of in-silico generated sequences is our goal.
Medicine-likeness of the in-silico generated antibodies
Next, we checked if the antibodies generated in-silico using the training data resemble the antibodies which have already undergone product and clinical development, granted regulatory approvals, and are available in the market as biotherapeutic medicines, in terms of their intrinsic physicochemical properties. Figure 5E shows distributions of percentile scores for medicine-likeness for the training and generated datasets. The medicine-likeness percentiles were computed using sequence and structure-based physicochemical descriptors for the 100 000 in-silico generated sequences in three environmental conditions, namely, pH 3.5 and no added salt (viral inactivation step during manufacturing), pH 6.0 and no added salt (formulation condition), and pH 7.4 with 137 mM salt (physiological condition). Figure S4 in Supporting Information shows statistical distributions of the five non-redundant physicochemical descriptors, calculated using the Fv region molecular models, for the 100 000 in-silico generated antibodies and 31 416 antibodies in the training dataset, in reference to 113 Fvs from the marketed antibodies. These descriptors were identified by Ahmed et al. [10] for constructing a medicine-likeness profile using the intrinsic physicochemical parameters of the marketed antibodies. The details of these calculations are provided in the Materials and Methods section of the supporting Information. More information on our profile-based methodology can be found elsewhere [10, 18]. More than two-thirds of (69.7%) of the in-silico generated sequences have medicine-likeness scores of >50th percentile. However, this is lower than the proportion (78.8%) of the training sequences that have medicine-likeness scores of >50 percentile (Table S1, Fig. 5E). Furthermore, to assess how variations in the calculated structural descriptors that constitute the medicine-likeness profile may reflect diversity in the 3D structures of the antibodies, we selected structural models of the antibodies showing the extreme (minimum and maximum) and median values for each of the five descriptors (BSAVH:VL, HI, DM/HM, RP, and pI3D, more details in the methods section). A total of 30 antibodies, 15 in each of the training and generated datasets, were selected for this analysis and their 3D structures were superimposed using MOE [42]. Overall structural alignment for the selected antibodies in the training dataset shows the mean HCDR3 RMSD (root mean square deviation) of 4.9 Å (RMSD range = 0.8 Å - 8.7 Å). The corresponding statistic is 5.1 Å (RMSD range = 1.0 Å, max: 10.7 Å) for the in-silico generated antibodies (Fig. S5). Taken together, these observations suggest that in-silico generated sequences have recapitulated physicochemical characteristics of the training sequences and there may be scope to further sample this physicochemical space by generating more de novo antibody sequences. Furthermore, we assessed a randomly chosen sample of 500 reference in-silico sequences generated using the control dataset for their medicine-likeness. The results are summarized in Table S3 and Fig. S7 in the Supporting Information. The median medicine-likeness of the reference sequences is 40 (mean = 47 ± 30). This is significantly lower than the median value of 70 for the main in-silico sequences generated using the training dataset (mean = 63 ± 29). This result further reinforces the importance of physicochemical quality of the sequences chosen for training the WGAN+GP model for generating medicine-like antibody sequences.
In summary, computational analyses described above show that the antibody sequences generated by the WGAN+GP model using the 31 416 sequences in the training dataset are likely to possess good developability attributes. The next section provides experimental evidence for this inference.
Experimental results
A sample of 51 high-quality in-silico generated antibody sequences was sent to two laboratories for independent assessments of their developability attributes such as expression levels, purity, thermal stability, hydrophobicity, self-association, and poly-specificity. The choice of 51 sequences was based on several considerations described below. First, each one of these 51 sequences is highly medicine-like (≥90th percentile medicine likeness) and possesses ≥90% humanness. There are no unpaired Cys or N-linked glycosylation motifs in these sequences as well as no chemical liability (oxidation, Asn - deamidation, Asp - isomerization, and fragmentation) in the CDRs of any of them. These 51 selected sequences represent 42 387 (42%) of the sequences in the generated dataset, spanning across 38 unique germline pairs. An artificial phylogenetic tree constructed using HCDR3 loops of these antibodies is shown in Fig. S6. The HCDR3 sequences are distributed over a wide spectrum of smaller clusters, indicating potential functional diversity among the experimentally tested sample. In addition to the above-mentioned scientific considerations, experimental resources available to us were also a significant consideration in determining the sample size.
As stated above, the 51 in-silico generated antibody sequences were passed on to two independent laboratories for experimental validation. The first experimental laboratory (referred to as Lab I) is within Biotherapeutics Discovery at Boehringer Ingelheim located in Ridgefield, CT. The second experimental laboratory (referred to as Lab II) is in the Biointerfaces Institute in the University of Michigan located at Ann Arbor, MI. There was no exchange of material among the experimental labs and the approaches taken by them to analyze the performance of the in-silico generated sequences were also independent of each other. In Lab I, experimental results obtained from the in-silico generated antibodies (referred to as GAN set) were compared with a set of 100 marketed or clinical stage antibodies (referred to as EXT set). In Lab II, our sample of 51 in-silico generated antibodies was subjected to additional criteria internal to Lab II. These criteria are described in the materials and methods section. Only 11 of the 51 in-silico generated antibodies passed these additional criteria and were experimentally produced. Performance of these 11 selected in-silico generated antibodies were compared to approved antibodies known to show desirable and poor developability attributes.
All in-silico generated antibody sequences expressed well in the mammalian cells and could be purified in sufficient quantities needed for the experimental work, as described in the materials and methods section. This observation demonstrates that our algorithm is effective at generating experimentally verifiable antibodies. Furthermore, all experiments either included control molecules to compare with historical values collected over the previous years (Lab I) or were conducted multiple independent times (Lab II), both following well-established protocols and employing automation whenever feasible to minimize the risk of random and human error. The reproducibility and the reliability of the findings have been further confirmed by the consistent results obtained by the both laboratories.
Experimental results from Lab I
The variable region (Fv) sequences of all EXT (100 clinical and marketed) and GAN (51 in-silico generated) antibodies were cloned into an IgG1KO(LALA) backbone, regardless of the published isotype, to minimize any differences associated with the constant regions. Small-scale transient transfection, purification via Protein A affinity resin, and biophysical characterization were all conducted on automated platforms to minimize variance associated with manual operations. Quantitative analytics were collected to assess how similar GAN generated antibodies perform relative to existing therapeutic antibodies. The distributions of titer, purity, thermal stability, and hydrophobicity are shown in Fig. 7A. Comparing GAN to EXT sets, the production metrics (titer and purity) are statistically different with greater titer and slightly higher purity for the GAN set (Table 4a). Expression of the EXT set was temporally distinct from the GAN set; given the variability of transient production, the difference in mean titer is within normal operational range. The difference in average purity was less significant and appeared to be driven by a bias towards high purity antibodies in the GAN set coupled with four particularly low purity antibodies in the EXT set. In contrast to the production metrics, both thermal stability and hydrophobicity were highly similar between the two sets of molecules (Table 4a). Indeed, the distributions of Fab thermal stability were nearly identical (p-value: 0.983), demonstrating the effectiveness of our GAN method in producing high-quality antibodies. Consistent with the computational results, the ranges of these experimentally measured developability attributes are smaller for the antibodies in the GAN dataset than those in the EXT dataset. This could be due to the smaller number of antibodies in the GAN dataset (51 sequences) versus those (100) in the EXT dataset. Alternatively, there could also be room for generating more antibodies via our WGAN+GP model to fully sample the biophysical property space observed for the antibodies in the EXT dataset. This is consistent with our computational results described in section 2 of this manuscript.
Figure 7.

Biophysical analyses show that in-silico generated antibodies possess desirable developability attributes. A. (Lab I) quantitative experimental analyses of (i) titer, (ii) purity, (iii) thermal stability (Fab Tm), and (iv) hydrophobicity (analytical hydrophobic interaction chromatography [aHIC] retention time), comparing 51 in-silico generated antibodies (GAN dataset) with 100 marketed and clinical stage biotherapeutics (EXT set). B. (lab II) experimental analyses of 11 in-silico generated antibodies. (i) the yield of antibodies after purification via protein a chromatography. (ii) percentage monomeric antibody after protein a purification, as judged by size exclusion chromatography. (iii) apparent melting temperatures (Fab Tm) of IgGs, as measured using differential scanning fluorimetry. (iv) non-specific binding of IgGs, as evaluated using the PolySpecificity Particle (PSP) assay. The control antibodies were IgG1s with the variable regions of elotuzumab (negative control) and emibetuzumab (positive control). (v) Self-association of IgGs, as measured using the charge-stabilized self-interaction nanoparticle spectroscopy (CS-SINS) assay. The control antibodies were NISTmAb (negative control) and an IgG1 containing the variable regions of omalizumab (positive control). Tras.; trastuzumab. Elot.; elotuzumab. Emi.; emibetuzumab. Oma.; omalizumab. RFU; median fluorescence intensity.
Table 4a.
Statistical summary of quantitative analytics of antibodies in the GAN and the EXT datasets for titer, purity, thermal stability (Fab Tm) and hydrophobicity (aHIC retention time)a.
| Developability attribute |
EXT dataset mean ± std (range) |
GAN dataset Mean ± std (range) |
p-value |
|---|---|---|---|
| Titer (mg/L) | 96.8 ± 41.0 (20–245) | 127.9 ± 33.5 (62–210) | <0.0001 |
| Purity (% Main Peak) | 96.7 ± 2.5 (86.4–100) | 97.9 ± 2.0 (91.4–100) | 0.0033 |
| Thermal Stability (Fab, ° C) | 75.5 ± 6.6 (55.5–91.7) | 75.4 ± 6.6 (56.1–89.4) | 0.9830 |
| Hydrophobicity (aHIC RT, min) | 4.5 ± 3.7 (0.1–16.6) | 4.7 ± 3.4 (0.6–13.7) | 0.7706 |
These experiments were performed in Lab I. The GAN dataset consists of 51 in-silico generated antibodies using the WGAN+GP method; The EXT dataset consists of 100 approved or clinical stage antibodies. Note that all antibodies were produced in IgG1KO(LALA) format. See materials and methods for details in the supporting information for details.
Experimental results from Lab II
The performance of 11 in-silico generated antibodies was assessed in reference to five control antibodies (trastuzumab, omalizumab, elotuzumab, emibetuzumab, and NISTmab) as described in the materials and methods. Trastuzumab was selected as the primary control due to its well-characterized properties including high expression yield, robust thermal stability, and low non-specific binding and self-association [43]. As a clinically approved and well-behaved IgG1K antibody, trastuzumab serves as a reliable benchmark for evaluating in-silico generated antibodies. The other control antibodies include NISTmab and three well-characterized biotherapeutics (omalizumab, elotuzumab, and emibetuzumab) showing a range of biophysical attributes that are relevant to their developability (Table 4b), as previously described [44, 45]. All the 15 antibodies that were expressed and NISTmab [46] that was purchased commercially were IgG1K monoclonal antibodies. The results are shown in Fig. 7B and Table 4b. Notably, all the antibodies expressed at generally similar levels as trastuzumab, ranging from 27% to 116%. The in-silico generated IgGs were also largely monomeric after Protein A purification, ranging from 91%–99% monomer relative to 98% monomer for trastuzumab, and highly stable (melting temperatures of 62–90°C relative to ~83°C for trastuzumab). Moreover, the in-silico generated antibodies displayed low levels of non-specific binding and self-association, which were similar or even lower than those for trastuzumab.
Table 4b.
Experimental characterization of 11 in-silico generated antibody sequences in lab IIa.
| Antibodies | Yield (mg/L) | Monomer (%) after 1-step purification | Tm (Fab, °C) | PSP (RFU) | CS-SINS score |
|---|---|---|---|---|---|
| trastuzumab | 28.3 ± 6.1 | 97.9 ± 1.4 | 82.8 ± 0.1 | 50.2 ± 10.2 | 0.10 ± 0.04 |
| M4 | 12.2 ± 8.5 | 95.6 ± 4.4 | 77.2 ± 0.1 | 50.6 ± 7.4 | 0.07 ± 0.02 |
| M10 | 19.9 ± 10.6 | 97.5 ± 0.0 | 72.5 ± 0.2 | 59.9 ± 5.7 | 0.44 ± 0.06 |
| M20 | 19.5 ± 2.4 | 97.6 ± 0.1 | 90.4 ± 0.4 | 49.2 ± 6.3 | 0.07 ± 0.06 |
| M23 | 26.3 ± 8.3 | 96.4 ± 1.3 | 80.1 ± 0.1 | 49.0 ± 11.8 | 0.13 ± 0.03 |
| M25 | 16.2 ± 3.0 | 97.7 ± 0.3 | 69.8 ± 0.1 | 59.2 ± 6.2 | 0.07 ± 0.04 |
| M30 | 32.7 ± 6.8 | 97.7 ± 0.8 | 82.8 ± 0.0 | 50.3 ± 6.1 | 0.06 0.03 0.03 |
| M33 | 23.5 ± 5.8 | 98.0 ± 0.8 | 82.7 ± 0.1 | 47.4 ± 7.0 | 0.18 0.06 0.06 |
| M36 | 25.5 ± 7.5 | 91.4 ± 5.1 | 79.3 ± 0.1 | 48.1 ± 9.8 | 0.10 0.05 0.05 |
| M37 | 14.3 ± 10.2 | 98.6 ± 0.6 | 71.8 ± 0.1 | 51.8 ± 6.9 | 0.10 0.05 0.05 |
| M41 | 32.0 ± 8.2 | 97.2 ± 2.4 | 74.3 ± 0.1 | 80.8 ± 13.1 | 0.08 ± 0.09 |
| M45 | 7.5 ± 4.1 | 98.2 ± 0.9 | 61.6 ± 0.1 | 92.9 ± 7.0 | 0.14 ± 0.07 |
| NISTmAb | 0.03 ± 0.02 | ||||
| omalizumab | 1.00 ± 0.02 | ||||
| elotuzumab | 56.3 ± 7.3 | ||||
| emibetuzumab | 6547.7 ± 1304.3 |
Lab II used additional criteria on our initial sample of 51 in-silico generated antibodies to further qualify 11 in-silico generated antibodies for experimental testing. The numbers M4, M10, and so on indicate the in-silico generated antibodies. Note that trastuzumab was used as a control antibody for all experimental assays. Furthermore, elotuzumab and emibetuzumab were used as additional controls for the poly-specificity (PSP); and NIST mAb and omalizumab were used as additional controls for the self-association (CS-SINS). Monomer content values reported in this table are after Protein A purification step. After the 2-step purification, all antibodies were 98.3%–100% monomer. All antibodies were produced as IgG1K monoclonal antibodies. See materials and methods in the supporting information for details.
Discussion
In-silico generation of antibodies has been hailed as the third revolution in antibody discovery after animal immunizations and display technologies [47]. There can be several advantages to generating antibodies in-silico, both in academic and industrial research. For example, the earliest stages of antibody discovery projects involve generation of reagent antibodies to interrogate a given target. However, high-quality tool antibodies that bind targets of interest are not always commercially available. This necessitates in-house generation of reagent-quality monoclonal antibodies, which can be both expensive and time-consuming even for well-behaved targets. In the case of targets that are not well-behaved, the costs and time required can escalate quickly, creating a major project bottleneck. In the realm of therapeutic antibodies, the opportunity is much greater. Here, in-silico antibody discovery can lead to a greater number of therapeutic products in the clinic by expanding the druggable antigen-space to include difficult targets as well as novel ones by enabling the discovery process initiation even before the availability of sufficient antigen material needed to start the experiments. This is in addition to accelerating discovery and development of therapeutic drug candidates. Such an ability not only saves cost, time, and use of animals in drug discovery, but also significantly increases the opportunities for the biopharmaceutical industry to serve an increased population of patients with unmet medical needs.
To enable DAbI, we have developed an innovative conceptual roadmap to discover developable antibody-based biotherapeutics [11]. Figure 2 describes it schematically, and the results presented in this work test the feasibility of the first step in this roadmap. This first step is in-silico generation of an antigen-agnostic human antibody library with good developability attributes. The next steps involve screening of this library to find potential antigen-specific antibody binders and further refine them for binding affinity and cross-species reactivity to enable pre-clinical testing in animal models. These steps are out of scope for the current work. Literature reports show that researchers are using different learning theory-based methods for designing antigen-specific antibodies [48] and for humanization [49]. Generation of antigen or epitope specific antibodies via machine learning is an attractive proposition because it holds promise for early success in a discovery project by addressing a specific therapeutic concept. Therefore, initial in-silico antibody generation algorithms have focused on finding antigen or epitope-specific antibodies [25, 27, 47]. This is analogous to finding the antibody binders via animal immunizations or panning of display libraries against a given antigen. However, this approach suffers from the key disadvantage that one needs to start from ground zero every time a new biologic drug discovery program is initiated. Therefore, in this work, we focused on generating antigen-agnostic antibody libraries with favorable developability features included. Such libraries can be used to simultaneously facilitate multiple drug discovery projects. These libraries are analogous to naïve B-cell repertoires, e.g. obtained from humanized mice prior to their immunization with specific antigens. Another example of such libraries shall be the phage or yeast display libraries that can be panned for binding specific antigens. Indeed, it is attractive to combine in-silico generated antigen-independent but highly developable antibody libraries with the display technologies to obtain functional antibody binders, rapidly, without requiring animal immunizations [1]. True value of such computationally generated antigen-agnostic libraries is that they need to be constructed only once or a small number of instances in a biologic drug discovery organization and then screened to pre-compute potential antigen-specific libraries for all the targets of interest to the organization. One can only imagine the potential impact of such pre-computed antibody binder libraries on accelerating biologic drug discovery projects and towards expanding druggable antigen space to include targets that are difficult to express and purify in the lab, such as integral membrane proteins, GPCRs, and tight junction proteins. Availability of computationally generated highly developable but antigen-agnostic antibody libraries can also be used to start the discovery process as soon as potential novel drug targets become available in the databases [12–15], prior to their in vitro production and start of immunization experiments. In summary, DAbI can improve the biotherapeutic drug discovery paradigm and set the discovery and development project cycles for faster, resource saving, and more efficient pathways.
In recent years several generative AI models have been developed to generate novel protein and antibody sequences [25, 27, 50–56]. As stated in the Results section, a direct benchmarking of all available antibody generation methods is infeasible because the goal of this study (generation of antigen-agnostic antibodies with desirable developability features) differs from most of the earlier reports that have focused on generating antigen-specific antibodies. For example, we note that most generative language models train on the antibody sequences and generate new ones by iteratively predicting the next element in a sequence. Such sequence in-filing algorithms have been successful at predicting potential antibodies to specific antigens [24, 26, 51] since they rely on local attributes such as CDRs. In contrast, our goal requires prediction of the entire sequences de novo since the developability attributes can be traced to both local and global features of antibody sequences and structures [41]. Therefore, we chose to approach this problem by building a protocol that addresses the challenges unique to our task. We preferred WGAN+GP over other algorithms because of its ability to mimic the natural feedback loop mechanism ubiquitous in biochemistry and natural evolution. Additionally, our protocol has other novel features. First, we developed a physicochemical profile (medicine-likeness profile) using the intrinsic biophysical properties of the variable regions of the marketed antibody based biotherapeutics. Next, we queried publicly available as well as internal sequence repositories for paired antibody sequences whose variable regions belong to the most successful pair germline loci (IGHV3-IGKV1) and do not contain too many physicochemical liabilities to construct a training dataset of 31 416 paired antibody sequences. The sequences in the training set were converted to ScFv format, multiply aligned, and converted into images via one-hot encoding. Treating the training dataset sequence as images and using convolutional layers to identify features and representations inherent to antibody variable region (Fvs), allowed us to capture the spatial context of each element in the sequence and learn framework constraints faster. WGAN+GP [16, 17] was chosen for the purpose of this experiment over plain GAN [57] or WGAN [16] because WGAN+GP [17] mitigates issues of gradient collapse and vanishing gradients with the gradient penalty term. This ensures the model remains trainable and the training loss converges consistently. It also avoids overfitting of the model to the point where it starts generating unrealistic data [58]. Numerous research studies have successfully employed GANs for generating in-silico data across several fields, including the biological sciences [59–67]. Compared to other models like VAE, WGAN+GP can generate more representative in-silico data due to its adversarial training approach [63]. Diffusion models typically generate new samples by reversing a diffusion process that adds noise to real data. While this can produce diverse outputs, it could also prove challenging to control the noise levels and ‘artifacts’ in the generated samples [68]. In our case, it was crucial that we keep the noise levels low, so that the germline-pairing as well as physicochemical attributes of the in-silico generated antibodies are like those of the training set sequences. Another limitation of Diffusion Models is their slow sampling rate, and it takes a huge number of model evaluations to generate good quality samples [69]. Diffusion Models also require significantly more computational resources to train, and this made WGANs a more suitable choice for us given the tradeoff between resources and quality. Moreover, in several scenarios WGANs may outperform Diffusion Models on smaller datasets, making it the more practical choice for real world applications [70, 71].
To ensure that in-silico generated antibodies are developable, we focused on sequence-structural properties of the marketed antibodies because they have overcome all the hurdles in their technical as well as clinical development, won regulatory approval, and are available in the market to serve patients with unmet medical needs. Distributions of non-redundant physicochemical descriptors computed using the variable regions of the marketed biotherapeutics [10] were used for creating the training dataset of antibody sequences as well as for assessing developability of the in-silico generated ones. These distributions measure medicine-likeness of the antibodies by assessing similarity of their variable regions’ physicochemical characteristics with those of the variable regions of the marketed antibodies. In the context of our work, the term ‘medicine-likeness’ does not imply any functional attributes and therefore should not be used to infer antigen binding. A sample of 51 in-silico generated antibodies with high medicine-likeness percentiles was sent for testing to two independent labs, one in industry and the other in academia. The protocols followed by the two labs are different and the two labs neither collaborated nor exchanged any material related to this study. The results from both laboratories show that in-silico generated antibodies possess good developability attributes. The sample size of 51 antibodies was determined based on the cost and resource availability for performing the experiments. However, within this boundary condition, the antibodies in the experimentally tested sample are highly diverse in terms of their sequences and HCDR3 loops as described in the results section. Furthermore, note that our method was effective at not only generating unique paired antibody sequences, but also experimentally verifiable ones. This is significant because in an unrelated study, most of the antibodies generated using a language model and a variational autoencoder model either failed to express at all or produced insufficient material for biophysical characterization routinely used in biopharmaceutical industry for pre-clinical developability assessments (our unpublished results).
The choice of antibody sequences used in the training dataset and algorithm(s) used for training can influence the quality of the computationally generated antibody sequences. In our case, the antibody sequences included in the training set were not randomly chosen, since the objective of our study was to develop an experimentally verifiable tool for computational generation of highly developable antigen agnostic antibodies. On the contrary, the training dataset is intentionally biased towards the following attributes. First, the sequences used in the training data set of this study are paired VH:VL antibody sequences. This limited the data availability because most of the sequences in the public antibody sequence repositories are unpaired. This limitation meant that we could not make a language model for the purpose of our study, even if it were to be useful for our goal. Second, all the antibody sequences in the training dataset belong to IGHV3-IGKV1 loci of germline pairs, which is the most productive loci among the marketed antibodies. Furthermore, as described in the materials and methods, all the sequences are ≥80% human, non-redundant, and possess good developability features such as low incidence of chemical liabilities, no non-canonical Cys residues and N-linked glycosylation sites. We also used a control dataset of 71 283 paired antibody sequences that belong to the same germline pair loci and are ≥80% human. No additional developability filters were applied to the control dataset. This control dataset was used to generate another set of 100 000 reference sequences using the same WGAN+GP model as the one used for the training dataset. As shown in the results section, the WGAN+GP model was effective in generating highly non-redundant sequences with a low number of duplicates again. However, the reference antibodies generated using the control dataset were of lower physicochemical quality than those generated using the training dataset. Therefore, use of additional physicochemical quality filters assured that we are training our generative algorithm on high quality data.
Along with the training dataset, the choice of algorithm was also not random as discussed earlier. Similarly, the in-silico generated antibodies selected for experimental testing are also biased towards high medicine-likeness and humanness. There is one aspect where our training data is not biased. Binding to specific antigens was not used as a selection criterion for antibodies in our training and generated datasets and analysis of sequence-structural properties described in the results section does suggest that antibodies in both training and generated datasets may be functionally diverse. However, this was not confirmed via experiments in this report. A control study with randomized data and benchmarking multiple models, though desirable, was not undertaken at this stage because of both computational and experimental resource limitations and our goal to demonstrate practical utility of DAbI.
This study is novel on several counts. Unlike the traditional methods of antibody generation against a specific target, our computationally generated library is antigen-agnostic and pre-pays for developability. That is, antibody binders discovered using this library are likely to present minimal, if any, developability issues as they progress through drug discovery and development cycles. This changes the current biotherapeutic drug discovery paradigm of function first to developability first. This is novel because most computational antibody generation algorithms are focused on building antigen-specific antibody libraries and do not address developability. Therefore, we followed a novel protocol, and a novel deep learning algorithm as described earlier. Experimental validation is generally lacking in this field. In this report, two independent experimental validation tests were performed to see if our AI generated antibody library met its design objectives. This adaptation of the ‘Turing test’ within the context of this work was an important element of this study. Results show that the in-silico generated antibodies are medicine-like and behave in the same way as the clinical stage and marketed antibodies, thereby completing the Turing imitation game [19]. As far as we know this is the first report where AI generated antibodies have been experimentally validated, not in one but two laboratories. Overall, we have been able to demonstrate the feasibility of the first part of our roadmap (Fig. 2) to enable DAbI [11]. Insights gained from this work shall not only help pivot the biological drug discovery paradigm towards greater use of computation to discover developable biotherapeutic drug candidates but also be useful towards making well defined diagnostic and reagent antibodies with good reproducibility.
Materials and methods
Computational methods
Sequence data collection
A diverse set of 400 000 VH-VL paired human Fv sequences were obtained from multiple sources and compiled together to form the primary pool of training sequences including the sequences from Observed Antibody Space (OAS) [2, 3], deep sequencing of Yeast display libraries [72], published literature [3, 10] as well as our internal sequencing data.
Grouping of sequences by germline pair families
The sequences were first grouped by genetic loci pairs of germlines, such as IGHV3-IGKV1 group comprising of all antibody sequences with VH-VL pairing between IGHV3-* heavy chain germlines and IGKV1-* light chain germlines, and so on. The total number of sequences in each germline pair group is shown in supporting information Fig. S1. To group the sequences by germline pair family, the closest human VH and VL germline sequences for each Fv sequence in the training dataset were determined using IgBlast tool [39], with the IMGT database [73] as a reference of human germline sequences. The germline data was then parsed using in-house python script to build a dictionary with each germline pair as dictionary- ‘key’ and sequences belonging to them as dictionary- ‘items’.
Control and training datasets
After sorting by germline pair families and eliminating sequences with low percent humanness, the dataset of interest consisted of 71 283 paired antibody sequences that are ≥80% human and belong to IGHV3-IGKV1 germline loci pair. This dataset is referred to as the control dataset. This control dataset was further filtered to retain 31 416 paired antibody sequences with high physicochemical quality (medicine-likeness, described below). This dataset is called the training set in this work. Both reference and training datasets were used to train our deep learning model (see below) and generate 100 000 new paired antibody sequences in-silico in each case. The 100 000 antibody sequences generated by using the control set are used as ‘reference’ sequences, while those generated by using the training set are the ‘main’ sequences analyzed in this work.
Multiple sequence alignment
The sequences in each pair of light and heavy chain germline loci were aligned using an in-house protocol that employs a positional numbering-based alignment. Each residue in each sequence was assigned its positional number according to the Martin numbering scheme as implemented by ANARCI python-based tool [29]. These positional numbers were treated as guides for referring to each residue according to its respective Martin position number using python scripts written in-house.
From the grouped set of sequences, the groups with germline pairs that are highly represented in the natural antibody space (obtained from analyzing OAS paired human sequences) [2, 3] and market-stage antibody-based biotherapeutics [10], were chosen for training a generative deep learning model described later in this section. The sequences within each of the selected germline pair groups were analyzed to eliminate redundancies and poor-quality sequences exhibiting high risk chemical liabilities, and to ensure high human germline identity as described below in detail. The germline pair of IGHV3 and IGKV1 was selected for further method development and calculations, based on its high prevalence in OAS natural paired antibodies distribution (Fig. S1). Similarly, a dominant population of IGHV3-IGKV1 germline loci pair was also found in our training set.
Elimination of redundant sequences from the training set
The sequences with at least one unique CDR were retained and treated as unique or non-redundant sequences. The CDR positions were defined for each sequence in the dataset using MOE CCG numbering that implements Martin numbering scheme-based CDR definitions via svl script [42, 74]. All the six CDR sequences for every antibody in the dataset were sorted to obtain unique sets of six CDR sequences using in-house python script, and the sequences presenting duplicates of all six CDRs were subsequently eliminated from further analysis.
Input data filtering based on non-canonical cysteine and N-linked glycosylation liabilities
The training set sequences were ensured to contain exactly four cysteines at the canonical positions within antibody variable regions, by discarding the ones that were found to be containing more than four cysteines or lacking one or more of the canonical cysteines. The presence of canonical cysteines was assessed by collecting and examining the positional number of cysteines in the multiple sequence alignments. Sequence-based motifs—‘NXS’ and ‘NXT’ where X is any amino acid residue except Proline [75]—were used to detect potential Asn-linked glycosylation motifs in the antibody variable regions. The sequences containing these motifs were subsequently eliminated from the input training dataset.
Percent humanness
The percent identity of training sequences with the closest human germline was extracted from the output obtained from running the IgBLAST tool [39]. The sequences with at least 80% identity with a human germline sequence V-region were retained as part of the training set.
Antibody sequence formatting
The final set of training sequences that meet the above criteria for IGHV3 - IGVK1 germline cluster pair is 31 416. These sequences were written as ScFvs in text file format with VH sequence followed by a (GGGGS)4 linker and then its pairing VL sequence as shown below:
 |
All these sequences were then multiply aligned and the gaps in the multiple sequence alignment were replaced with ‘X’ or ‘Z’ depending on their position on the Fv region. Gaps in the CDRs were replaced with ‘X’ while those in the framework regions (FR) were replaced with ‘Z’. Different notations for gaps were implemented to enable the learning algorithm to distinguish the variations in hypervariable regions from the rest of the sequence.
Antibody sequence embedding
Each sequence was encoded into a 2D image using one-hot encoding technique [31], where each multiple sequence alignment column represents an alignment position, and each row represents an amino acid sequence as shown in Fig. 3. This encoding of 1-D amino acid sequence into a 2-D matrix enables us to represent each sequence as a grayscale image, suitable for machine learning via convolutional neural networks (CNN, [76, 77]).
Training the deep learning model
Generative adversarial network (GAN) model generation and training
GANs are deep learning neural networks that can be utilized to generate new data after being trained on given input data [57]. A GAN consists of two competing convolutional neural networks, a generator and a discriminator. The generator model in a GAN is thought to capture the semantic features of real data, which enables it to generate new real-like data that did not exist previously. The discriminator tries to distinguish between the generated and the real data as accurately as possible, while the generator tries to generate samples closely resembling real data and beat the discriminator [57]. Both networks perform adversarial learning to optimize their goals based on their loss function. Training GANs is a minimax optimization problem shown below as equation (1):
 |
(1) |
Wasserstein GANs
The GAN models used in this work are Wasserstein GANs. Wasserstein Generative Adversarial Network, or Wasserstein GAN, is an extension to GAN that both improves the stability when training the model and provides a new loss function that correlates with the quality of generated images known as Wasserstein Loss [16]. Instead of using a discriminator to classify or predict the probability of generated images as being real or artificial, the WGAN changes or replaces the discriminator model with a criterion that scores the realness or artificialness of a given image. The original Wasserstein GAN leverages the Wasserstein distance [16] to produce a value function that has better theoretical properties than the value function used in the original GAN paper.
 |
(2) |
In this work, we developed a WGAN model to generate antibody sequences using Keras and Tensorflow packages in python [78, 79]. The WGAN architecture with gradient penalty was used as described above. A latent vector of size 128 was fed into the generator model, which was then passed to multiple up-sampling layers and 2D convolutional transpose layers followed by a SoftMax activation layer at the end. The output was then trimmed using a cropping layer to match the length of the sequences being trained. The resulting 2D array (in this case of size 264 x 22) corresponds to the aligned ScFv chain length (264) one-hot encoded over the second dimension of size 22 for 20 amino acids and Xs and Zs for the gaps found in multiple sequence alignments of the antibody sequences (Xs for the CDRs, and Zs for the framework regions) as described above. The model was trained on IGHV3-IGKV1 sequences for a maximum of 1000 epochs, with an early stopping function to stop the training after the model converges and there is no significant improvement in generator loss over 100 consecutive epochs.
Dimensionality reduction to assess training convergence
T-distributed Stochastic Neighbor embedding (T-SNE) is a non-linear dimensionality reduction technique used to visualize high-dimensional data [80]. T-SNE technique was used to visually represent the convergence of generated sequence space with that of the training sequences. This method was implemented in python using sklearn package [81]. The performance of the model in generating real-like images was assessed using dimensionality reduction methods. The training data and the generated data were reduced to 2-dimensional embeddings using T-SNE algorithm. The performance was calculated by the Euclidean distance between the embeddings of the generated data and the training data embeddings.
Computational assessments of in-silico generated sequences
The converged GAN model was used to generate 100 000 sequences. The sequence redundancy was measured based on CDR uniqueness as described above, both within the generated sequences and across training sequences to determine the sequence duplication from the training set.
Amino acid composition
Each of the training and generated sequences was treated as a text string and the total number of occurrences of each of the 20 amino acids was computed using in-house python script and stored as a python dictionary. The (G4S)4 linker was removed before counting the number of amino acids in the sequences. The frequencies of amino acids in the amino acid count dictionaries for the training and generated sets were normalized and analyzed using the ꭓ2 analyses.
Levenshtein distance analysis
Levenshtein distance [37] was calculated using the python package called Levenshtein (https://rapidfuzz.github.io/Levenshtein/). Each generated sequence was compared with each of the training sequences represented as a text string with no gaps. The smallest Levenshtein distance corresponding to each generated sequence was the number of edits that its closest training sequence needs to undergo to result in that sequence. Similar method was applied to VH, VL and individual CDR portions of the sequence to compute their respective Levenshtein distances.
Shannon entropy
Diversity in generated sequences versus training sequences was determined by comparing the Shannon entropy for each aligned sequence position computed using the equation below.
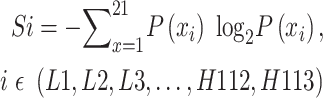 |
(3) |
where Si is the Shannon entropy at position i in the antibody sequence, P(xi) is probably the occurrence of amino acid residue x or a gap at the position i. VL positions are numbered L1, L2, L3, …, while VH positions are numbered H1, H2, H3, …, according to the Martin numbering scheme. Note that the summation runs from 1 to 21 to account for 20 amino acid residues and a gap.
CDR diversity among the generated sequences was compared with the training sequence CDR diversity using CDR length distributions and CDR sequence variations. The number of residues in each CDR was calculated to determine the CDR length diversity. For CDR sequence variations, the Shannon entropy values for positions belonging to the CDR regions were individually summed to give the Shannon entropy for each CDR.
 |
(4) |
Where SCDR(j) is the Shannon entropy of CDR (j). Nj is the length of the CDR (j) and Sij is the Shannon entropy of the position i within the CDR (j).
Variable region structural diversity
We identified the sequences exhibiting extreme (highest and lowest) and median structure-based physicochemical properties considered in this work [10] including average hydrophobic imbalance (HI), surface area buried at VH:VL interface (BSAVH:VL), ratio of dipole moment to hydrophobic moment (DM/HM), ratio of surface areas covered by the charged and hydrophobic patches (RP), and structure based pI of the Fv region (pI3D_Fv). Three structures per physicochemical property resulted in a total of 15 structures for training and 15 for generated sequences. These structures were sequence and structure aligned and superposed in MOE. The superposed structures were used to obtain pairwise RMSD of CDRs in MOE.
Germline pair frequency comparison
In-house python script was written to parse the IgBlast output to obtain the germline of each sequence. The heavy chain and light chain germlines were treated as a 2D matrix, and each cell value was updated with the total number of sequence observations belonging to each corresponding germline pair.
Chemical liabilities
CDR located chemical degradation sequence motifs suggesting susceptibility to loss of function due to oxidation, deamidation, Asp-isomerization, etc, were identified using an in-house script. Incidence of Asn-linked Glycosylation motifs and non-canonical cysteines were also noted using the same script. The extent of risk was determined based on which motif it is, which CDR it is in, and location in the CDR, i.e. stem versus middle of the loop by using different arbitrarily chosen weights.
High-throughput Fv modeling
The training Fv sequences were homology modeled with MOE Antibody modeler implemented in high throughput using in-house scripts. The models were built in Amber10: EHT. Generalized Born implicit solvent force field model was used for modeling with internal and external dielectric values set to 4 and 80, respectively. The modeled structures were prepared using default ‘QuickPrep’ function in MOE and energy minimized with a root mean squared gradient threshold of 10−6 kCal.mol−1.Å−2. The models were protonated and energy minimized in three different conditions, namely, pH 3.5 and no added salt, pH 6 and no added salt, pH 7.4 and 0.137 M salt, to mimic the conditions faced by antibodies during manufacturing (the viral inactivation step), formulation, and in vivo (physiological condition), respectively.
Medicine-likeness calculations
A comparative analysis of sequence and structure-based physicochemical descriptors was performed between the in-silico generated sequences against a benchmark set of 113 Marketed antibody Fv sequences/structures [82] for assessing their medicine likeness. Sequence-based quantities such as % human germline identity, TANGO aggregation scores, chemical liability scores (glycosylation, unpaired cysteines, isomerization prediction) were combined with five non-correlated structure-based descriptors described by Ahmed et al. (2021) [10], namely, average hydrophobic imbalance (HI), surface area buried at VH:VL interface (BSAVH:VL), ratio of dipole moment to hydrophobic moment (DM/HM), ratio of surface areas covered by the charged and hydrophobic patches (RP), and structure based pI of the Fv region (pI3D_Fv). These descriptors were computed at three different solution conditions via protonation of the Fv models at pH 7.4, ionic strength 137 mM (physiological condition), at pH 6.0 with zero ionic strength (formulation condition) and at pH 3.5 with zero ionic strength (low pH hold during biomanufacturing). The reader is referred to Licari et al. 2022 [18] for detailed descriptions of these conditions. The data generated allowed us to make three different profiles for the variable regions of 113 marketed antibodies. In each profile, the distributions for each descriptor were normalized to range between −1 to 1 based on their physicochemical meaning. The normalized descriptor scores were summed to present a total score over each profile and then all the three profiles for each sequence in the benchmark set. This procedure gives us a reference distribution of scores that were then used to assign a percentile value to the sequence to suggest its medicine-likeness in reference to the 113 marketed biotherapeutics. For evaluation of 100 000 in-silico generated antibody sequences, the same set of sequence and structure-based descriptors were computed. These descriptors were again scaled for normalization as described above and summed up for a total score. The total score for each in-silico generated sequence was then evaluated by identifying the respective percentile value of the generated sequence’s total score on the reference distribution of 113 approved Fvs.
Antibody: Antigen complex crystal structures
818 unique antibody: antigen (Ab: Ag) co-crystal structures were searched (filters: Species – Human, Resolution – 3 Å, Antigen – protein) and download as of February 2022 from structural antibody database (sAbDab, http://opig.stats.ox.ac.uk/webapps/newsabdab/sabdab/) [38, 83]. From these, structures with non-redundant CDR sequences, high % identity with human germlines (>70%) and complexes with antigens larger than 50 amino acids were retained, which resulted in 468 final set of complexes which were prepared in MOE using QuickPrep and energy minimized [42]. The dataset was used to compute VH:VL affinity and buried surface areas (BSA) in MOE, for comparison with the modeled Fv regions of training and generated sequences.
Selection of antibody sequences for experimental characterization
A sample of fifty-one in-silico generated antibody sequences with more than 90% human germline content, >90 percentile medicine-likeness, high CDR diversity, no unpaired Cys residues, no N-linked glycosylation sites, and chemical liabilities in the CDRs was taken from these 100 000 in-silico generated sequences and sent to two independent experimental laboratories for their experimental analyses. The experiments consisted of expression, purification, and biophysical characterization of these antibodies.
Experimental methods
Experimental methods at Lab I
Antibody expression and titer determination
CHO-3E7 (CHO-E) cells from the National Research Council, Canada (NRC) stably expressing truncated Epstein–Barr virus Nuclear Antigen-1 were maintained in an actively dividing state in FreeStyle CHO (FS-CHO, Thermo Scientific) medium supplemented with 8 mM Glutamax (Gibco). For antibody expression, ~ 26 ml of CHO-E cells (4 x 106 cells/ml in Irvine media supplemented with 4 mM Glutamax) were aliquoted into 50 ml TubeSpin Bioreactor tubes (Thermo Scientific) and transfected with 10.5 μg light chain and 5.25 μg heavy chain plasmid DNA supplemented with 36.75 μg non-coding filler DNA and complexed with 52.5 μl of TransIT Pro (Mirus Bio LLC) transfection reagent in 1 ml OptiPro SFM [64]. Cultures were incubated at 37°C, 5% CO2 and 300 rpm. Twenty-four hours after transfection, the temperature was reduced to 30°C. Cultures were maintained for 7 days and harvested by centrifugation at 4815 g at 4°C for 25 minutes. Aliquots (100 μl) of clarified cell culture supernatants were transferred to flat bottom polypropylene microplates (Greiner Bio-One) for titer measurements using an Octet Red 96 with Protein A quantitation biosensors (Sartorius). The biosensors were equilibrated in conditioned medium (media conditioned by mock-transfected CHO-E cells), and the sample plate was incubated at 30°C for 10 minutes prior to data acquisition. Measurements were performed at 1000 rpm with data acquired for 2 minutes. The raw data was analyzed with manufacturer’s software and plotted against a standard curve of matching IgG isotype, conditioned medium, and shake speed.
Antibody purification
Automated affinity capture via Protein A chromatography was performed using a Hamilton Vantage liquid handler. Briefly, ~ 22 ml of each clarified cell culture supernatant was loaded onto separate ProPlus PhyTips (1 ml pipet tip containing 80 μl resin; Biotage) at a flow rate of 0.16 ml/min. The resin was sequentially washed with 1 ml of DPBS, pH 7.2 (Invitrogen), DPBS plus 1 M NaCl, and DPBS at the flow rate of 0.5 ml/min. Captured antibodies were eluted thrice, each with 0.35 ml of 30 mM sodium acetate, pH 3.6, at the flow rate of 0.5 ml/min. The three elution pools were combined, and pH adjusted to pH 5 with addition of 0.03 ml of 1 M sodium acetate, pH ~9. Sample concentrations were measured using UV spectroscopy on a Lunatic Plate Reader (Unchained Labs) with calculated extinction coefficients.
Analytical size exclusion chromatography
Analytical size exclusion chromatography was performed on an Agilent 1290 Infinity II LC UPLC system with a Waters BEH200 SEC column (200 Å, 1.7 μm, 4.6 mm X 150 mm); 10 μg injections with 50 mM sodium phosphate, pH 6.8, 200 mM arginine, and 0.05% sodium azide as the mobile phase and a 0.5 ml/min constant flow rate. Chromatograms were analyzed within Empower 3 Pro software (Waters).
Buffer exchange and dynamic light scattering
Prior to further characterization, all samples were exchanged into 10 mM histidine, pH 6.0, 20 mM NaCl using the Freeslate Jr. automated buffer exchange system (Unchained Labs). After five rounds, the cumulative volumetric exchange percentage was determined to exceed 95% for all samples. Samples (20 ml) were then transferred to a 384-well glass bottom plate (Griener) for dynamic light scattering analysis (DynaPro II, Wyatt Technology) to verify the buffer exchange process did not significantly alter the sample quality.
Thermal stability analysis
Thermal stability profiles were acquired using a QuantStudio 5 Flex real-time PCR system (Applied Biosystems) with SYPRO Orange (Invitrogen) as the extrinsic fluorophore. Briefly, each sample was diluted to ~ 0.4 g/L in 10 mM histidine, pH 6.0 with 20 mM sodium chloride and 5X (final) SYPRO Orange. Melt curves were generated using a thermal ramp from 25 to 95°C at a rate of 2°C/min, with data collected approximately every 0.4°C through Ex1 (l = 470 ± 15 nm) and Em3 (l = 623 ± 10 nm) excitation and emission filters, respectively. Raw fluorescence data were transformed and analyzed with a custom Python script to detect inflection points in the first derivative of fluorescent data (Tm). The plots were then visually inspected to confirm which reported value was specific to the respective Fab domain.
Analytical hydrophobic interaction chromatography
Samples were prepared by diluting 1:1 with 2 M ammonium sulfate and analyzed using a Waters Acquity UPLC H-class system with a Sepax Proteomic HIC butyl-NP 1.7 column, 10 μg per injection. Chromatograms were generated from UV absorbance (l = 220 nm) as a function of decreasing ammonium sulfate concentration using a 16-minute single gradient elution from 1 M to 0 M ammonium sulfate in 0.1 M sodium phosphate, pH 6.0 with a 0.8 ml/minute flow rate. Data were analyzed for retention times using Empower 3 software (Waters).
Experimental methods at lab II
Selection of in-silico generated antibodies for experimental analysis
The down-selection process for the 51 in silico-generated antibodies to the final 11 antibodies for experimental evaluation involved the following five steps. First, a sequence-based machine learning model was used to assess the polyreactivity of the antibodies. One of the 51 antibodies was predicted to be polyreactive and was subsequently removed. Second, 20 of the remaining 50 antibodies were removed because they were predicted to display either high self-association or non-specific binding by a second set of models reported previously [44]. Third, eight of the remaining 30 antibodies were removed because their pairwise Fv sequence similarity was >90%. Fourth, seven of the remaining 22 antibodies were removed because the VH and/or VL domains were too similar (>90% sequence similarity) to those found in a Lab II internal database of pre-existing antibodies. Finally, four of the 15 antibodies were eliminated because they contained consecutive aromatic residues in their CDRs, which resulted in the final set of 11 antibodies.
Production of soluble antibodies
Cloning of plasmids, antibody expression, and purification were performed as previously described [45]. Briefly, variable light (VL) and heavy (VH) chain domains of each antibody were purchased as geneblocks (Integrated DNA Technologies). Next, the PCR-amplified fragments and pTT5 expression vectors containing common IgG1 heavy and light chain (kappa) frameworks were digested using desired restriction enzymes (EcoRI-HF and BsiWI-HF for VL; EcoRI-HF and NheI-HF for VH; New England Biolabs). The digested DNA fragments were directly purified (Qiagen, 28104), whereas the digested vectors were first treated with calf alkaline phosphatase (New England Biolabs, M0525L) and then purified by 1% agarose gel electrophoresis and DNA extraction (Qiagen, 28704). Finally, digested fragments and vectors were ligated with T4 DNA ligase (New England Biolabs, M0202L) and transformed into DH5α component cells. Antibody sequences were confirmed by Sanger sequencing.
Soluble antibody expression was performed as previously described [84]. Briefly, the mixture of the light chain and heavy chain expression plasmids (7.5 μg for each) and polyethylenimine (45 μg) (PEI MAX, 247651, Polysciences Inc.) in 3 ml of F17 media (Invitrogen, A1383502) was used for transient transfection of 25 ml HEK293-6E (L-11565, National Research Council Canada) cell culture grown in F17 media supplemented with L-glutamine (Gibco, 25030081), Kolliphor (Fisher, NC0917244), and G418 antibiotics (Gibco, 10131035) under the conditions of 37°C, 5.0% CO2, and 250 rpm agitation. The day after transfection (24–36 h), protein expression was enhanced by adding 750 μl of 20% yeastolate (BD Sciences, 292804). After four additional expression days, the supernatant was harvested and purified using Protein A agarose resin (20334, Pierce; Thermo Fisher Scientific). After purification, antibodies were buffer exchanged into 20 mM acetate (pH 5.0) using Zeba desalting columns (Thermo Fisher Scientific, 89890).
The purity of the antibody samples after the Protein A purification was evaluated by size-exclusion chromatography (SEC) using a Shimadzu Prominence HPLC System with an LC-20AT pump, SIL-20 AC autosampler, and FRC-10A fraction collector. For analytical SEC, 100 μl of antibody sample (0.1 mg/ml) was loaded onto an SEC column (Superdex 200 Increase 10/300 GL column; GE, 28990944) and analyzed at 0.75 ml/min using 100 mM sodium acetate running buffer supplemented with 200 mM arginine (pH 5). Absorbance was monitored at 280 nm. The percentage of antibody monomers was evaluated by analyzing the peak areas between the void volume and buffer elution times. After evaluation of the monomeric content, the antibodies were further purified via the same SEC system. The collected sample fractions were buffer exchanged into 20 mM acetate (pH 5), filtered, snap-frozen, aliquoted, and stored at -80C.
Polyspecificity analysis (PSP assay)
Polyspecificity analysis was performed as previously described [44]. Briefly, Protein A Dynabeads (Invitrogen, 10002D) were washed three times with cold PBSB and diluted to 54 μg/ml in cold PBSB. The beads (30 μl) were then incubated with antibodies (85 μl, 15 μg/ml) overnight at 4°C with mild agitation. The next day, the coated beads were washed twice with cold PBSB by centrifugation (2500 xg for 5 min). The washed beads were then resuspended in 200 μl of 0.1 mg/ml biotinylated SMP reagent (prepared as previously described [44], and incubated at 4°C for 20 min. Next, the beads were washed once with cold PBSB and incubated with secondary reagent, 0.001x streptavidin Alexa Flour 647 (Invitrogen, S32357) and 0.001x goat anti-human Fc F(ab’)2 Alexa Flour 488 (Invitrogen, H10120) in cold PBSB, on ice for 4 min. Finally, the beads were washed once more, resuspended in cold PBSB, and analyzed via flow cytometry using a Bio-Rad ZE5 flow cytometer to measure their relative fluorescence unit (RFU) values for 5000 events per sample. IgG1s with the variable domains of elotuzumab (negative control) and emibetuzumab (positive control) were analyzed in each experiment as control antibodies.
Melting temperature (Tm) analysis
The melting temperatures of the soluble antibodies were determined using differential scanning fluorimetry (DSF), as previously described [84]. Briefly, antibodies (0.12 mg/ml; in 1x PBS buffer, pH 7.4) were mixed with Protein Thermal Shift Dye (Applied Biosystems, 4461146) at a volume ratio of 7:1 (protein: dye) so that the final concentration of dye equals 1x. The samples were then added to individual wells of a clear 384-well plate. The plates were submitted to the University of Michigan Advanced Genomics Core for analysis. The plates were centrifuged at 1000–2000 rpm for 1 min and then inserted into a QuantStudio 12 K Flex Real-Time PCR System (Applied Biosystems). The thermal cycle conditions were set to examine fluorescence signals at increasing temperatures between 25–98°C. The fluorescence signals were monitored by the ROX channel. Frequency distribution curves for each sample using the negative first derivative of fluorescence signals were generated using the Gaussian distribution model. In general, a three-peak distribution model was observed, wherein the CH2 and CH3 domains exhibited nearly identical inflection points for most antibodies, whereas the Fab fragment displayed variation, with its inflection point being designated as the Tm value. In some samples, however, a broad peak was observed at the same point where the peaks of the Fab and CH3 domains, or even the three peaks (CH2/CH3/Fab), overlapped. The Tm values of the overlapping peaks, such as CH3/Fab or CH2/CH3/Fab, were denoted in these samples as Fab.
Self-association analysis (CS-SINS)
The self-association features of antibodies were evaluated by the Charge-Stabilized Self-Interaction Nanoparticle Spectroscopy (CS-SINS) as previously described [45]. To prepare immunogold-conjugates, first, 20 nm gold nanoparticles (Ted Pella Inc., 15705) were ~ 24-fold concentrated (e.g. 2.4 ml to 100 μl) by centrifugation (21 300 xg, 6 min). Separately, a capture antibody/polylysine mixture (0.10 poly-lysine/IgG fraction) was prepared by mixing 3.05 μl of polylysine reagent (Sigma, P1274) (2.67 mg/ml) and 97 μl of anti-human Fc-specific antibody (Jackson ImmunoResearch, 109–005-008) (0.8 mg/ml, pH 4.3, 20 mM acetate buffer). Finally, the concentrated gold nanoparticles and the capture antibody/polylysine mixture were mixed at a 1:1 volume ratio and incubated overnight at room temperature.
To analyze the self-association of a given antibody, 5 μl of immunogold-conjugate and 45 μl of antibody sample (11.1 μg/ml) in 10 mM histidine buffer (pH 6) were mixed in a well of a 384-well plate, incubated at room temperature for 4 h, and the absorbance spectrum (450–650 nm, 1 nm increments) was measured.
The self-association scores were determined by using the following data processing procedure. The plasmon wavelength was determined by fitting ~40 points around the maximum absorbance value using a quadratic equation and setting the first derivative to zero (ref for Github: https://github.com/Tessier-Lab-UMich/Methods_data_analysis). Calibration test #1 was performed by evaluating plasmon wavelengths for human polyclonal antibody (Jackson ImmunoResearch Laboratories, 009–000-003) and NISTmAb (Sigma, NIST8671). If plasmon wavelengths are below pre-set limits (<534 nm for human polyclonal antibody and < 533 nm for NISTmAb), then calibration test #2 was performed. Calibration test #2 was performed by evaluating the CS-SINS scores for calibration panel #1: tocilizumab, cetuximab, evolocumab, denosumab, pembrolizumab, and omalizumab. For these six monoclonal antibodies, the CS-SINS scores were calculated using historical data for tocilizumab (parameter #1) and omalizumab (parameter #2) CS-SINS plasmon wavelengths. Parameter #1 is the plasmon wavelength of low self-association antibody which is 531.01 for tocilizumab and parameter #2 is the plasmon wavelength of a high self-association antibody which is 532.97 for omalizumab. CS-SINS scores were calculated using the following formula:
 |
(5) |
For the two mAbs (tocilizumab and omalizumab), the parameters in the CS-SINS score equation were fitted to maximize the agreement of the linear fit between the new and historical measurements by minimizing the following term: (1-slope)2 + (intercept)2. The historical measurements of the calibration panel #1 are: 0 for tocilizumab; 0.085 for denosumab; 0.22 for pembrolizumab; 0.36 for evolocumab; 0.47 for cetuximab; and 1 for omalizumab).
For test #2, the following requirements for the degree of linear fit performance between historical reference and the new data set were evaluated.
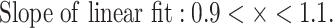 |
 |
 |
If the measurements pass both calibrations, then the final step is to evaluate the CS-SINS scores for additional antibodies, which were scored using parameters #1 and #2 that are fit instead of using the plasmon wavelengths of tocilizumab and omalizumab for these parameters.
Key Points
This report explores the following question, is it feasible to computationally generate highly developable antibody libraries for de novo biologic drug discovery?
Paired IGHV3-IGKV1 Fv sequences were collected and filtered into a training set based on the similarity of their intrinsic physicochemical attributes with those of the Fv regions from marketed antibodies (Medicine-likeness).
A deep learning model developed using the training set was able to generate highly medicine-like novel Fv sequences in silico that are different from those in the training set.
A sample of in silico generated sequences was sent to two laboratories for experimental testing.
Experimental results from both laboratories show that in silico generated antibodies are developable, thereby answering the question affirmatively.
Supplementary Material
Acknowledgements
The authors acknowledge discussions with numerous colleagues in Boehringer Ingelheim and external collaborators on the topic of in-silico generation of antibodies. The authors acknowledge helpful discussions with Stephen Comeau, Niksa Kastrapeli, Noah Pefour, Alexander Jung, and Jan Kriegl on the topic of in-silico drug discovery. Gina Moretti is thanked for designing the plasmid vectors and ordering them. Javier Rivera, Zhong-Fu Huang, and Bryce Klair are thanked for expression and purification of the antibodies. Giuseppe Licari, Joschka Bauer and Lucky Ahmed are acknowledged for their contributions to Medicine-likeness profile, which played a crucial part in analyses of the in-silico generated antibodies. Dr. Prabakaran Ponraj is thanked for the critical reading of this manuscript. The authors also acknowledge contributions of three anonymous reviewers for their constructive criticism of this work. Their feedback helped improve this work significantly.
Contributor Information
Nandhini Rajagopal, Biotherapeutics Molecule Discovery, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Udit Choudhary, Global Computational Biology and Digital Sciences, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Kenny Tsang, Biotherapeutics Molecule Discovery, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Kyle P Martin, Biotherapeutics Molecule Discovery, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Murat Karadag, Departments of Chemical Engineering, Pharmaceutical Sciences and Biomedical Engineering, Biointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48105, United States.
Hsin-Ting Chen, Departments of Chemical Engineering, Pharmaceutical Sciences and Biomedical Engineering, Biointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48105, United States.
Na-Young Kwon, Departments of Chemical Engineering, Pharmaceutical Sciences and Biomedical Engineering, Biointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48105, United States.
Joseph Mozdzierz, Biotherapeutics Molecule Discovery, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Alexander M Horspool, Biotherapeutics Molecule Discovery, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Li Li, Global Computational Biology and Digital Sciences, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Peter M Tessier, Departments of Chemical Engineering, Pharmaceutical Sciences and Biomedical Engineering, Biointerfaces Institute, University of Michigan, 2800 Plymouth Road, Ann Arbor, MI 48105, United States.
Michael S Marlow, Biotherapeutics Molecule Discovery, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Andrew E Nixon, Biotherapeutics Molecule Discovery, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Sandeep Kumar, Biotherapeutics Molecule Discovery, Boehringer Ingelheim Pharmaceutical Inc., 900 Ridgebury Road, Ridgefield, CT 06877, United States.
Conflict of interest: NR, KT, UC, KPM, JM, AMH, LL, MSM, AEN, and SK worked for Boehringer Ingelheim Pharmaceutical Inc. at the time this research was performed. PMT is a member of the scientific advisory boards for Nabla Bio, Aureka Biotechnologies, and Dualitas Therapeutics.
Funding
National Institutes of Health (R35GM136300 to PMT) and the Albert M. Mattocks Chair (to PMT).
Data availability
All the data used and created in this study has been disclosed in SI datasets.
Author contributions
Sandeep Kumar, Li Li, Michael S. Marlow, Peter M. Tessier, and Andrew E. Nixon conceived the research problem and directed the work reported in this work. Nandhini Rajagopal collected the antibody sequences and Udit Choudhary trained the WGAN+GP model. Nandhini Rajagopal and Kenny Tsang analyzed the input antibody sequences as well as the in-silico generated antibody sequences and wrote most of the in-house developed scripts using this work. Kyle P. Martin collected the marketed antibody sequences and contributed towards the development of medicine-likeness profiles. Murat Karadag, Hsin-Ting Chen, Na-Young Kwon, and Joseph Mozdzierz performed experimental studies. Alexander M. Horspool contributed Fig. 1. All authors contributed towards manuscript writing, revision, and agree to the content of this report.
References
- 1. Gray AC, Bradbury ARM, Knappik A. et al. Animal-derived-antibody generation faces strict reform in accordance with European Union policy on animal use. Nat Methods 2020;17:755–6. 10.1038/s41592-020-0906-9. [DOI] [PubMed] [Google Scholar]
- 2. Kovaltsuk A, Leem J, Kelm S. et al. Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires. The Journal of Immunology 2018;201:2502–9. 10.4049/jimmunol.1800708. [DOI] [PubMed] [Google Scholar]
- 3. Olsen TH, Boyles F, Deane CM. Observed antibody space: a diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci 2021:141–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Breden F, Luning Prak ET, Peters B. et al. Reproducibility and reuse of adaptive immune receptor repertoire data. Front Immunol 2017;8:1418. 10.3389/fimmu.2017.01418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vander, Heiden JA, Marquez S, Marthandan N. et al. AIRR community standardized representations for annotated immune repertoires. Front Immunol 2018;9:2206. 10.3389/fimmu.2018.02206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collins AM, Ohlin M, Corcoran M. et al. AIRR-C IG reference sets: curated sets of immunoglobulin heavy and light chain germline genes. Front Immunol 2024;14:1330153. 10.3389/fimmu.2023.1330153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lauer TM, Agrawal NJ, Chennamsetty N. et al. Developability index: a rapid In Silico tool for the screening of antibody aggregation propensity. J Pharm Sci 2012;101:102–15. 10.1002/jps.22758. [DOI] [PubMed] [Google Scholar]
- 8. Raybould MIJ, Deane CM. The therapeutic antibody profiler for computational Developability assessment. Methods Mol Biol 2022;2313:115–25. 10.1007/978-1-0716-1450-1_5. [DOI] [PubMed] [Google Scholar]
- 9. Raybould MIJ, Marks C, Krawczyk K. et al. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci 2019;116:4025–30. 10.1073/pnas.1810576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahmed L, Gupta P, Martin KP. et al. Intrinsic physicochemical profile of marketed antibody-based biotherapeutics. PNAS 2021;118. 10.1073/pnas.2020577118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bauer J, Rajagopal N, Gupta P. et al. How can we discover developable antibody-based biotherapeutics? Front Mol Biosci 2023;10:1221626. 10.3389/fmolb.2023.1221626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ochoa D, Hercules A, Carmona M. et al. Open targets platform: supporting systematic drug-target identification and prioritisation. Nucleic Acids Res 2021;49:D1302–10. 10.1093/nar/gkaa1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou Y, Zhang Y, Zhao D. et al. TTD: therapeutic target database describing target druggability information. Nucleic Acids Res 2024;52:D1465–77. 10.1093/nar/gkad751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mendez D, Gaulton A, Bento AP. et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 2019;47:D930–40. 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harding SD, Armstrong JF, Faccenda E. et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2022: curating pharmacology for COVID-19, malaria and antibacterials. Nucleic Acids Res 2022;50:D1282–94. 10.1093/nar/gkab1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arjovsky M, Chintala S, Bottou L. Wasserstein GAN [Preprint]. arXiv:1701.07875, 2017. Available at: http://arxiv.org/abs/1701.07875 [Accessed 12 November 2023].
- 17. Gulrajani I, Ahmed F, Arjovsky M. et al. Improved training of Wasserstein GANs [Preprint]. arXiv:1704.00028, 2017. Available at: http://arxiv.org/abs/1704.00028 [Accessed 25 February 2024].
- 18. Licari G, Martin KP, Crames M. et al. Embedding dynamics in intrinsic physicochemical profiles of market-stage antibody-based biotherapeutics. Mol Pharm 2023;20:1096–111. 10.1021/acs.molpharmaceut.2c00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turing AM. Computing machinery and intelligence. Mind 1950;LIX:433–60. 10.1093/mind/LIX.236.433. [DOI] [Google Scholar]
- 20. Ruffolo JA, Gray JJ, Sulam J. Deciphering antibody affinity maturation with language models and weakly supervised learning [Preprint]. arXiv:2112.07782, 2021. Available at: http://arxiv.org/abs/2112.07782 [Accessed 30 October 2024].
- 21. Dauparas J, Anishchenko I, Bennett N. et al. Robust deep learning-based protein sequence design using ProteinMPNN. Science 2022;378:49–56. 10.1126/science.add2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng L, Shi S, Lu M. et al. AnnoPRO: a strategy for protein function annotation based on multi-scale protein representation and a hybrid deep learning of dual-path encoding. Genome Biol 2024;25:41. 10.1186/s13059-024-03166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rives A, Meier J, Sercu T. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci U S A 2021;118:e2016239118. 10.1073/pnas.2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shuai RW, Ruffolo JA, Gray JJ. IgLM: infilling language modeling for antibody sequence design. cels 2023;14:979–989.e4. 10.1016/j.cels.2023.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eguchi RR, Choe CA, Huang P-S. Ig-VAE: generative modeling of protein structure by direct 3D coordinate generation. PLoS Comput Biol 2022;18:e1010271. 10.1371/journal.pcbi.1010271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olsen TH, Moal IH, Deane CM. AbLang: an antibody language model for completing antibody sequences. Bioinformatics. Advances 2022;2:vbac046. 10.1093/bioadv/vbac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akbar R, Robert PA, Weber CR. et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale. MAbs 2022;14:2031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saka K, Kakuzaki T, Metsugi S. et al. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci Rep 2021;11:5852. 10.1038/s41598-021-85274-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dunbar J, Deane CM. ANARCI: antigen receptor numbering and receptor classification. Bioinformatics 2016;32:298–300. 10.1093/bioinformatics/btv552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Branden CI, Tooze J. Introduction to Protein Structure 2nd edn. NY, USA: Garland Science, 1998. [Google Scholar]
- 31. Harris D, Harris S. Digital Design and Computer Architecture 2nd edn. Morgan Kaufman Publishers, Burlington, Massachusetts: Elsevier Inc., 2012. [Google Scholar]
- 32. Brüggemann M, Caskey HM, Teale C. et al. A repertoire of monoclonal antibodies with human heavy chains from transgenic mice. Proc Natl Acad Sci 1989;86:6709–13. 10.1073/pnas.86.17.6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee E-C, Liang Q, Ali H. et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat Biotechnol 2014;32:356–63. 10.1038/nbt.2825. [DOI] [PubMed] [Google Scholar]
- 34. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985;228:1315–7. 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 35. McCafferty J, Griffiths AD, Winter G. et al. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 1990;348:552–4. 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 36. Bradbury ARM, Sidhu S, Dübel S. et al. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol 2011;29:245–54. 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levenshtein VI. Binary codes capable of correcting deletions, insertions and reversals. Soviet Physics Doklady 1966;10:707. [Google Scholar]
- 38. Berman HM, Westbrook J, Feng Z. et al. The protein data Bank. Nucleic Acids Res 2000;28:235–42. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ye J, Ma N, Madden TL. et al. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 2013;41:W34–40. 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shannon CE. A mathematical theory of communication. Bell Syst Tech J 1948;27:379–423. 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- 41. Kumar S, Kumar Singh S. (Eds.). Developability of Biotherapeutics: Computational Approaches (1st ed.). Boca Raton, FL, USA: CRC Press, 2015. [Google Scholar]
- 42. Molecular Operating Environment (MOE) , 2022.02. (2023). Deposited 2023.
- 43. Jain T, Sun T, Durand S. et al. Biophysical properties of the clinical-stage antibody landscape. PNAS 2017;114:944–9. 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Makowski EK, Wu L, Desai AA. et al. Highly sensitive detection of antibody nonspecific interactions using flow cytometry. MAbs 2021;13:1951426. 10.1080/19420862.2021.1951426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Starr CG, Makowski EK, Wu L. et al. Ultradilute measurements of self-Association for the Identification of antibodies with Favorable high-concentration solution properties. Mol Pharm 2021;18:2744–53. 10.1021/acs.molpharmaceut.1c00280. [DOI] [PubMed] [Google Scholar]
- 46. Bergonzo C, Gallagher DT. Atomic model structure of the NIST monoclonal antibody (NISTmAb) reference material. J Res Natl Inst Stand Technol 2021;126:126012. 10.6028/jres.126.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sormanni P, Aprile FA, Vendruscolo M. Third generation antibody discovery methods: in silico rational design. Chem Soc Rev 2018;47:9137–57. 10.1039/C8CS00523K. [DOI] [PubMed] [Google Scholar]
- 48. Gao K, Wu L, Zhu J. et al. Pre-training antibody language models for antigen-specific computational antibody design. In: Proceedings of the 29th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, KDD ‘23, pp. 506–17. New York, NY, USA: Association for Computing Machinery, 2023. [Google Scholar]
- 49. Vashchenko D, Nguyen S, Goncalves A. et al. AbBERT: learning antibody humanness via masked language Modeling [Preprint]. bioRxiv 2022.08.02.502236, 2022. Available at: 10.1101/2022.08.02.502236v1 [Accessed 25 February 2024]. [DOI]
- 50. Amimeur T, Shaver JM, Ketchem RR. et al. Designing feature-controlled humanoid antibody discovery libraries using generative adversarial networks. bioRxiv 2020.04.12.024844 2020.
- 51. Xu X, Xu T, Zhou J. et al. AB-gen: antibody library design with generative pre-trained transformer and deep reinforcement learning. Genomics Proteomics Bioinformatics 2023;21:1043–53. 10.1016/j.gpb.2023.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Strokach A, Kim PM. Deep generative modeling for protein design. Curr Opin Struct Biol 2022;72:226–36. 10.1016/j.sbi.2021.11.008. [DOI] [PubMed] [Google Scholar]
- 53. Ingraham JB, Baranov M, Costello Z. et al. Illuminating protein space with a programmable generative model. Nature 2023;623:1070–8. 10.1038/s41586-023-06728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chu AE, Kim J, Cheng L. et al. An all-atom protein generative model. Proc Natl Acad Sci 2024;121:e2311500121. 10.1073/pnas.2311500121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen Y, Wang Z, Wang L. et al. Deep generative model for drug design from protein target sequence. J Chem 2023;15:38. 10.1186/s13321-023-00702-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu Z, Johnston KE, Arnold FH. et al. Protein sequence design with deep generative models. Curr Opin Chem Biol 2021;65:18–27. 10.1016/j.cbpa.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 57. Goodfellow IJ, Pouget-Abadie J, Mirza M. et al. Generative adversarial networks [Preprint]. arXiv:1406.2661, 2014. Available at: http://arxiv.org/abs/1406.2661 [Accessed 12 November 2023].
- 58. Thanh-Tung H, Tran T. On catastrophic forgetting and mode collapse in generative adversarial networks [Preprint]. arXiv:1807.04015, 2020. Available at: http://arxiv.org/abs/1807.04015 [Accessed 30 October 2024].
- 59. Nair A, Deshmukh J, Sonare A. et al. Image Outpainting using Wasserstein generative adversarial network with gradient penalty. In: 2022 6th International Conference on Computing Methodologies and Communication (ICCMC), pp. 1248–55, 2022.
- 60. Liu L, Xia Y, Tang L. An overview of biological data generation using generative adversarial networks. In: 2020 IEEE Conference on Telecommunications, Optics and Computer Science (TOCS), pp. 141–4, 2020.
- 61. Park J, Kim H, Kim J. et al. A practical application of generative adversarial networks for RNA-seq analysis to predict the molecular progress of Alzheimer’s disease. PLoS Comput Biol 2020;16:e1008099. 10.1371/journal.pcbi.1008099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marouf M, Machart P, Bansal V. et al. Realistic in silico generation and augmentation of single-cell RNA-seq data using generative adversarial networks. Nat Commun 2020;11:166. 10.1038/s41467-019-14018-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. El-Kaddoury M, Mahmoudi A, Himmi MM. Deep generative models for image generation: A practical comparison between Variational autoencoders and generative adversarial networks. In: Renault É, Boumerdassi S, Leghris C. et al. (eds.), Mobile, Secure, and Programmable Networking, pp. 1–8. New York, NY, USA: Springer International Publishing, 2019. [Google Scholar]
- 64. Lan L, You L, Zhang Z. et al. Generative adversarial networks and its applications in biomedical informatics. Front Public Health 2020;8:164. 10.3389/fpubh.2020.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Osokin A, Chessel A, Salas REC. et al. GANs for biological image synthesis. arxiv preprint arXiv:1708.04692, 2017. Available at: http://arxiv.org/abs/1708.04692.
- 66. Riley R, Mathieson I, Mathieson S. Interpreting generative adversarial networks to infer natural selection from genetic data. Genetics 2024;226:iyae024. 10.1093/genetics/iyae024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mascolini A, Cardamone D, Ponzio F. et al. Exploiting generative self-supervised learning for the assessment of biological images with lack of annotations. BMC Bioinformatics 2022;23:295. 10.1186/s12859-022-04845-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Szeliga A. A comparative study of deep generative models for image generation. Hochschule Hannover 2023. [Google Scholar]
- 69. Salimans T, Ho J. Progressive distillation for fast sampling of diffusion models. arxiv preprint arXiv:2202.00512, 2022. Available at: http://arxiv.org/abs/2202.00512 [Accessed 30 October 2024].
- 70. Does Diffusion Beat GAN in Image Super Resolution? arxiv preprint arXiv:2405.17261. Available at: https://arxiv.org/html/2405.17261v1.
- 71. Cherednichenko O, Poptsova M. Generative models for prediction of non-B DNA structures [Preprint]. bioRxiv 2024.03.23.586408 2024. Available at: 10.1101/2024.03.23.586408v1 [Accessed 30 October 2024]. [DOI]
- 72. Makowski EK, Chen H, Lambert M. et al. Reduction of therapeutic antibody self-association using yeast-display selections and machine learning. MAbs 2022;14:2146629. 10.1080/19420862.2022.2146629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lefranc M-P. IMGT, the international ImMunoGeneTics database®. Nucleic Acids Res 2003;31:307–10. 10.1093/nar/gkg085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abhinandan KR, Martin ACR. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol Immunol 2008;45:3832–9. 10.1016/j.molimm.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 75. Bause E, Hettkamp H. Primary structural requirements for N-glycosylation of peptides in rat liver. FEBS Lett 1979;108:341–4. 10.1016/0014-5793(79)80559-1. [DOI] [PubMed] [Google Scholar]
- 76. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521:436–44. 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 77. Schmidhuber J. Deep learning in neural networks: An overview. Neural Netw 2015;61:85–117. 10.1016/j.neunet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 78. Chollet F., et al. , Keras. Deposited 2015 2015.
- 79. Abadi M, Agarwal A, Barham P. et al. TensorFlow: large-scale machine learning on heterogeneous distributed systems. arxiv preprint arXiv:1603.04467, 2016. 10.48550/arXiv.1603.04467. [DOI]
- 80. van der Maaten L, Hinton G. Visualizing data using t-SNE. Journal of Machine Learning Research 2008;9:2579–605. [Google Scholar]
- 81. Pedregosa F. et al. Scikit-learn: machine learning in python. Journal of Machine Learning Research 2011;12:2825–30. [Google Scholar]
- 82. Martin KP, Grimaldi C, Grempler R. et al. Trends in industrialization of biotherapeutics: a survey of product characteristics of 89 antibody-based biotherapeutics. MAbs 15:2191301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dunbar J, Krawczyk K, Leem J. et al. SAbDab: the structural antibody database. Nucleic Acids Res 2014;42:D1140–6. 10.1093/nar/gkt1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zupancic JM, Desai AA, Schardt JS. et al. Directed evolution of potent neutralizing nanobodies against SARS-CoV-2 using CDR-swapping mutagenesis. Cell Chemical Biology 2021;28:1379–1388.e7. 10.1016/j.chembiol.2021.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data used and created in this study has been disclosed in SI datasets.