

Abstract
L-type amino acid transporter 1 (LAT1, SLC7A5), overexpressed in various cancers, mediates the uptake of essential amino acids crucial for tumor growth. It has emerged as a promising target for cancer therapy. Nanvuranlat (JPH203/KYT-0353), a LAT1 inhibitor, has shown antitumor activity in preclinical studies and efficacy in biliary tract cancer during clinical trials. This study provides a comprehensive pharmacological characterization of nanvuranlat and its N-acetyl metabolite, including structural insights into their LAT1 interactions. Both compounds demonstrated high selectivity for LAT1 over LAT2 and other amino acid transporters. Nanvuranlat acts as a competitive, non-transportable LAT1 inhibitor (Ki = 38.7 nM), while its N-acetyl metabolite retains selectivity but with reduced affinity (Ki = 1.68 µM). Nanvuranlat exhibited a sustained inhibitory effect on LAT1 even after its removal, indicating the potential for prolonged therapeutic effects. Both compounds showed comparable dissociation rates, suggesting that N-acetylation does not affect the interaction responsible for slow dissociation. The U-shaped conformation adopted by nanvuranlat when bound to LAT1 likely contributes to its high affinity, selectivity, sustained inhibitory effect, and non-transportable nature observed in this study. These insights into nanvuranlat’s mechanism and metabolic impact provide essential information for understanding its clinical efficacy and advancing LAT1-targeted cancer therapies.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-87522-6.
Keywords: Amino acid transporter, LAT1 inhibitor, Pharmacological selectivity, Drug metabolism & efficacy, Cancer therapy
Subject terms: Pharmacology, Drug development
Introduction
Amino acid transporters are crucial in cellular metabolism, growth, and proliferation1,2. L-type amino acid transporter 1 (LAT1, SLC7A5) has attracted significant attention due to its overexpression in various cancer types2,3. As a solute carrier family 7 member, LAT1 forms a heterodimeric complex with CD98hc (4F2hc, SLC3A2) for its functional expression in the plasma membrane3–6. LAT1 mediates the Na+-independent transport of large neutral amino acids, including branched-chain and aromatic amino acids, many of which are essential amino acids3,4,7. While LAT1 is expressed at moderate levels in the blood-brain barrier and is normally found in the placenta, it is notably overexpressed in various human cancers3,4,8–12. This overexpression is associated with increased mTORC1 signaling, enhanced protein synthesis, and accelerated cell growth, correlating with poor prognosis in multiple cancer types3,12–19. Consequently, LAT1 has emerged as a promising target for cancer therapy, aiming to exploit the differential expression between cancer cells and normal tissues3,20,21.
Recent research efforts have focused on developing selective LAT1 inhibitors to ensure therapeutic efficacy while minimizing off-target effects3,22,23. However, achieving high selectivity for LAT1 over its closely related isoform LAT2 (SLC7A8)24 and other amino acid transporters has remained challenging. Nanvuranlat (JPH203 or KYT-0353) has shown particular promise among the various compounds developed25. Nanvuranlat has demonstrated potent antitumor activity in preclinical studies across various LAT1-overexpressing cancer cells in vitro and in vivo3,17,25–33. In clinical trials, nanvuranlat has exhibited an acceptable safety profile and shown evidence of antitumor activity in patients with advanced solid tumors34. Notably, a recent phase II clinical trial in patients with biliary tract cancer met its primary endpoint of improving progression-free survival (PFS), further highlighting nanvuranlat’s potential as a therapeutic agent35.
Understanding the pharmacological properties of LAT1 inhibitors and the structural bases of their interaction with LAT1 is crucial for optimizing their therapeutic potential and predicting their behavior in vivo. The impact of metabolic modifications on inhibitor efficacy and selectivity is also of particular interest. Recently, cryo-electron microscopy analyses have revealed the three-dimensional structure of LAT1 and its binding complexes with multiple ligands, including nanvuranlat36–41. These structural insights now enable us to understand the pharmacological properties of ligands and their metabolites from a structural perspective. Nanvuranlat is rapidly metabolized to its N-acetyl derivative in the liver42. While this metabolite has been detected in plasma and tumor tissues, its pharmacological activity and contribution to the overall therapeutic effect of nanvuranlat remain unclear42,43. In this study, we comprehensively characterize nanvuranlat and its N-acetyl derivative, the primary metabolite of nanvuranlat, focusing on their inhibitory properties against LAT1 and related transporters, their inhibition kinetics, and the structural basis of their interaction with LAT1. This study provides the first quantitative characterization of the kinetic properties and selectivity of nanvuranlat and its N-acetyl metabolite, offering insights into the impact of N-acetylation on LAT1 interactions based on recently available structural information.
Results
Selective inhibition of LAT1 over LAT2 by nanvuranlat and its N-acetyl derivative
The inhibitory effect of nanvuranlat on LAT1 and LAT2, closely related structurally and functionally, was examined. The N-acetyl derivative of nanvuranlat (Fig. 1), a primary metabolite of nanvuranlat, was evaluated to determine whether N-acetylation affects its inhibitory potency or selectivity for LAT1 and LAT2.
Fig. 1.

Chemical structures of nanvuranlat and N-acetyl-nanvuranlat.
First, we assessed the inhibitory effect of nanvuranlat and N-acetyl-nanvuranlat (0.1, 1 and 10 µM) on the uptake of l-[14C]leucine (1 µM), a typical substrate of LAT1, in HEK293-hLAT1 cells expressing human LAT1 and l-[14C]alanine (1 µM), a typical substrate of LAT2, in HEK293-hLAT2 cells expressing human LAT2. As shown in Fig. 2a, both nanvuranlat and N-acetyl-nanvuranlat inhibited LAT1 in a concentration-dependent manner. In contrast, neither compound exhibited inhibitory effects on LAT2 at the same concentrations (Fig. 2b). When comparing nanvuranlat and N-acetyl-nanvuranlat, the latter showed a weaker inhibitory effect on LAT1 than nanvuranlat (Fig. 2a).
Fig. 2.

The inhibitory effect of nanvuranlat and N-acetyl-nanvuranlat. (a) and (b), inhibition of l-[14C]leucine uptake in HEK293-hLAT1 cells (a) and l-[14C]alanine uptake in HEK293-hLAT2 cells (b). Nanvuranlat and N-acetyl-nanvuranlat at 0.1, 1 and 10 µM inhibited LAT1-mediated uptake of l-[14C]leucine (1 µM), whereas they did not inhibit LAT2-mediated uptake of l-[14C]alanine (1 µM). (c) and (d), growth inhibition of HEK293-hLAT1 and HEK293-hLAT2 cells by nanvuranlat and N-acetyl-nanvuranlat. Nanvuranlat and N-acetyl-nanvuranlat suppressed the proliferation of HEK293-hLAT1 cells in a concentration-dependent manner with GI50 values of 1.83 ± 0.31 µM for nanvuranlat (c). For N-acetyl-nanvuranlat, the GI50 value could not be determined, as the inhibition did not reach 50% at the maximum concentration tested (30 µM). Neither nanvuranlat nor N-acetyl-nanvuranlat inhibited the proliferation of HEK293-hLAT2 cells (d). Data are presented as mean ± S.D., n = 4. Nanv.: nanvuranlat; N-Ac-Nanv.: N-acetyl-nanvuranlat.
Next, we examined the effects of nanvuranlat and N-acetyl-nanvuranlat (0.003-30 µM) on the proliferation of HEK293-hLAT1 and HEK293-hLAT2 cells. As shown in Fig. 2c, both compounds inhibited the proliferation of HEK293-hLAT1 cells in a concentration-dependent manner (Fig. 2c). In contrast, neither compound exhibited an inhibitory effect on the proliferation of HEK293-hLAT2 cells within the same concentration range (Fig. 2d). Consistent with their inhibition of LAT1-mediated l-[14C]leucine uptake, both compounds exhibited concentration-dependent inhibition of HEK293-hLAT1 cell proliferation. However, the GI50 value for N-acetyl-nanvuranlat in HEK293-hLAT1 cells could not be determined, as 50% inhibition was not achieved within the tested concentration range (up to 30 µM), indicating that its inhibitory effect is much weaker than that of nanvuranlat (GI50 = 1.83 ± 0.31 µM) (Fig. 2c).
These results demonstrate that nanvuranlat and N-acetyl-nanvuranlat exhibit a higher selectivity for LAT1 over LAT2 in inhibiting amino acid uptake despite the high structural similarity between the two transporters. The N-acetylation of nanvuranlat does not significantly affect its selectivity between LAT1 and LAT2. However, N-acetylation does reduce the inhibitory activity of nanvuranlat on LAT1. Furthermore, the extent of inhibition on LAT1- and LAT2-mediated uptake by nanvuranlat and N-acetyl-nanvuranlat is consistent with the observed effects on cell proliferation in LAT1- and LAT2-expressing cells.
Competitive inhibition and sustained effects of nanvuranlat and its N-acetyl derivative on LAT1
As both nanvuranlat and N-acetyl-nanvuranlat were found to inhibit LAT1 in a concentration-dependent manner without affecting LAT2 (Fig. 2a and b), we conducted a quantitative comparison of their inhibitory effects on LAT1. To this end, we measured the concentration-dependent uptake of l-[14C]leucine in the presence and absence of each inhibitor (Fig. 3a and c). Lineweaver-Burk plot analyses confirmed that nanvuranlat and N-acetyl-nanvuranlat act as competitive inhibitors of LAT1 (Fig. 3b and d). The calculated Ki values were 38.7 ± 5.1 nM for nanvuranlat and 1.68 ± 0.23 µM for N-acetyl-nanvuranlat, indicating that N-acetylation reduces the affinity for LAT1 by approximately 43.4-fold.
Fig. 3.

Inhibition kinetics of nanvuranlat and N-acetyl-nanvuranlat on LAT1. The uptake of l-[14C]leucine (1–400 µM) was measured in the absence or presence of nanvuranlat (0.1 and 0.5 µM) and N-acetyl-nanvuranlat (1 and 5 µM) in HEK293-hLAT1 cells. Uptake data were plotted against leucine concentration and fitted to Michaelis–Menten curves (a and c). Lineweaver–Burk plot analysis was performed to evaluate the inhibition kinetics of nanvuranlat and N-acetyl-nanvuranlat (b and d), revealing competitive inhibition for both compounds. The calculated Ki values were 38.7 ± 5.1 nM for nanvuranlat and 1.68 ± 0.23 µM for N-acetyl-nanvuranlat. Data are presented as mean ± S.D., n = 4. Nanv.: nanvuranlat; N-Ac-Nanv.: N-acetyl-nanvuranlat.
Of particular interest, it was previously suggested that nanvuranlat may sustain its inhibitory effect on LAT1 even after drug exposure and subsequent washing44. We assessed the residual LAT1 inhibitory activity following treatment with nanvuranlat to verify this prolonged inhibition. We also examined N-acetyl-nanvuranlat to determine whether it maintains a prolonged inhibitory effect on LAT1. HEK293-hLAT1 cells were treated with 20 µM of either compound for 30 min, followed by washing, after which residual LAT1 inhibition was assessed. For nanvuranlat, immediately after washing, LAT1-mediated uptake of l-[14C]leucine (1 µM) was approximately 20% of the control level. Although this inhibition gradually decreased, it remained substantial, with l-[14C]leucine uptake at about 60% of the control after 60 min (Fig. 4a). After 24 h, the inhibitory effect was vastly diminished, with uptake at around 90% of the control, still significantly lower than the control (Fig. 4b). N-Acetyl-nanvuranlat also showed residual inhibition, albeit to a lesser extent than nanvuranlat, with initial post-wash uptake at around 65% of control, which nearly returned to baseline after 60 min (Fig. 4a).
Fig. 4.

Sustained inhibition of LAT1-mediated leucine transport by nanvuranlat and N-acetyl-nanvuranlat. HEK293-hLAT1 cells were treated with nanvuranlat, N-acetyl-nanvuranlat (20 µM), or 0.6% DMSO (control) for 30 min. After washing out the compounds, cells were incubated in a fresh culture medium and assessed for leucine uptake at 0, 10, 20, 30, and 60 min (a) and 24 h (b). The leucine uptake values were normalized to the control at each time point, with the control uptake value set at 100%. Experiments were performed independently three times, each with four replicates. Data are presented as mean ± S.D. Nanv.: nanvuranlat; N-Ac-Nanv.: N-acetyl-nanvuranlat. The inhibition reduction rate was similar for both compounds: 29.8% for nanvuranlat and 30.8% for N-acetyl-nanvuranlat. This rate represents the percentage reduction in inhibition over the first 10 min, relative to the inhibition value at 0 min, calculated as follows: Inhibition reduction rate = (% Leu uptake at 10 min – % Leu uptake at 0 min)/(100 – % Leu uptake at 0 min) × 100.
Nanvuranlat as a non-transportable inhibitor of LAT1: evidence from temperature-dependent uptake assays
Inhibition experiments of l-[14C]leucine uptake in HEK293-hLAT1 cells indicated that nanvuranlat competitively inhibits LAT1 (Fig. 3b). To investigate whether nanvuranlat is transported after binding to the LAT1 substrate binding site, we conducted temperature-dependent uptake assays. We compared the cell-associated radioactivity of 14C-labeled nanvuranlat at 37 °C and on ice in HEK293-hLAT1 cells. Since transporters typically exhibit temperature sensitivity, with reduced transport rates at low temperatures and near cessation at 0 °C, this method provides a means to assess transporter-mediated uptake by examining differences in cell-associated radioactivity between 37 °C and ice-cold conditions45–47.
As illustrated in Fig. 5, the cell-associated radioactivity of l-[14C]leucine, a substrate of LAT1, was significantly higher at 37 °C than on ice, consistent with transporter-mediated uptake. In contrast, the radioactivity associated with 14C-nanvuranlat showed no significant difference between on ice and 37 °C. These findings indicate that nanvuranlat is not transported by LAT1 but acts as a non-transportable inhibitor, binding to the LAT1 substrate site without undergoing transport.
Fig. 5.
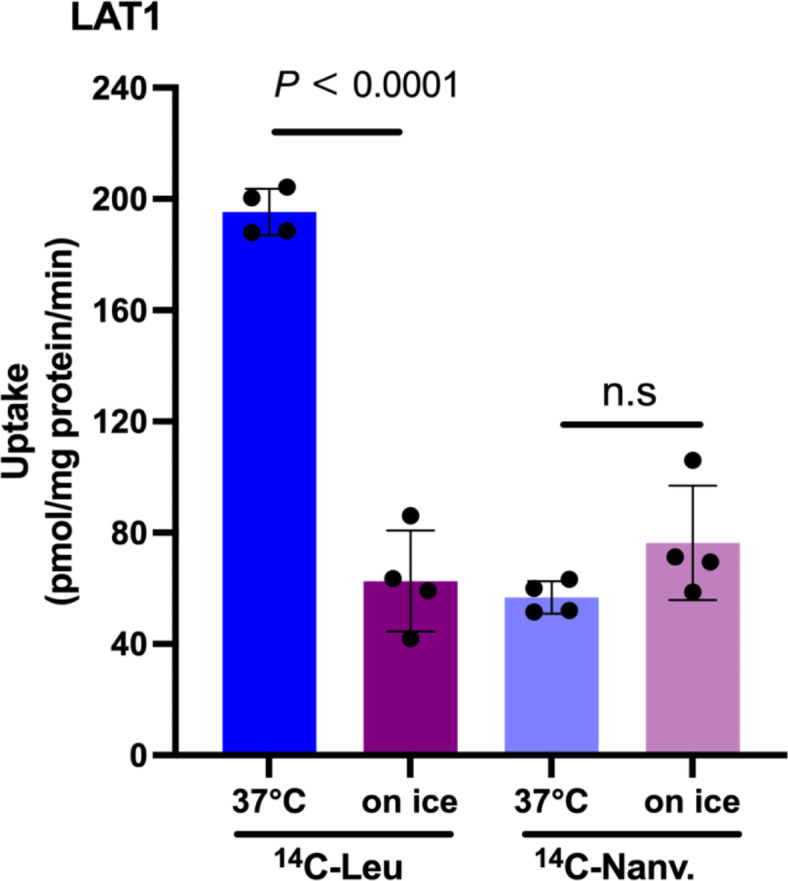
Absence of 14C-nanvuranlat uptake in HEK293-hLAT1 cells. Uptake measurements of 1 µM l-[14C]leucine (14C-Leu) and 1 µM 14C-nanvuranlat (14C-Nanv.) were conducted at 37 °C and on ice in HEK293-hLAT1 cells. The cell-associated counts of l-[14C]leucine were temperature-dependent, with significantly lower uptake observed on ice than 37 °C. In contrast, the cell-associated counts of 14C-nanvuranlat were unaffected by temperature, indicating that LAT1 does not transport 14C-nanvuranlat. Data are presented as mean ± S.D., n = 4.
Selective action of nanvuranlat and its N-acetyl derivative on LAT1 among amino acid transporters
Given the high affinity of nanvuranlat and N-acetyl-nanvuranlat for LAT1, with no observed inhibition of LAT2 at the tested concentrations, we extended our investigation to assess their effects on other amino acid transporters. Amino acid transporters generally recognize amino acid derivative ligands through interactions with amino and carboxy groups and the side chain’s steric properties and charge1. Since nanvuranlat and N-acetyl-nanvuranlat possess neutral bulky side chains, we examined their inhibitory effects on amino acid transporters specific to neutral amino acids. The transporters tested included those that handle branched-chain or aromatic amino acids, such as the systems L (LAT1-4), T (TAT1), B0 (B0AT1), B0,+ (ATB0,+), b0,+, and y+L (y+LAT1, y+LAT2) (Supplementary Table S1). Additionally, we evaluated systems ASC (ASCT1, ASCT2) and N/A (SNAT1-5), which predominantly transport small neutral amino acids, thereby covering the majority of neutral amino acid transporters (Supplementary Table S1). For each transporter, we measured the uptake of a radiolabeled typical substrate (50 µM) in the presence and absence of nanvuranlat and N-acetyl-nanvuranlat at 1 and 10 µM concentrations.
As depicted in Fig. 6a, among the transporters from system L for large neutral amino acids, nanvuranlat at both 1 and 10 µM, and N-acetyl-nanvuranlat at 10 µM, significantly inhibited the LAT1-mediated uptake of l-[14C]leucine (50 µM). Neither compound affected l-[14C]leucine uptake in control oocytes that did not express LAT1. Consistent with observations in HEK293-hLAT2 cells, neither nanvuranlat nor N-acetyl-nanvuranlat inhibited LAT2-mediated l-[14C]alanine uptake in Xenopus oocytes expressing LAT2 (Fig. 6b). Similarly, these compounds exhibited no inhibitory effects on LAT3 or LAT4 (Fig. 6c and d).
Fig. 6.

Inhibitory effect of nanvuranlat and N-acetyl-nanvuranlat on system L transporters. Uptakes of typical substrates (50 µM l-[14C]leucine for LAT1 (a), LAT3 (c), and LAT4 (d); 50 µM l-[14C]lalanine for LAT2 (b)) were measured in the presence of 1 µM and 10 µM of nanvuranlat and N-acetyl-nanvuranlat for 30 min in Xenopus oocyte expression system. Measurements were conducted in Na+-free uptake solution using control Xenopus oocytes injected with water instead of cRNA (“Water”) and the oocytes injected with cRNA for expressing each transporter. Uptake rates are expressed as mean ± S.D. (n = 8–12). Nanv.: nanvuranlat; N-Ac-Nanv.: N-acetyl-nanvuranlat. The data shown in (a), (c), and (d) were obtained using the same batch of oocytes. The l-[14C]leucine uptake data from control oocytes are shared across (a), (c), and (d), but are presented in each panel (a, c, and d) to allow comparison on the same scale as the l-[14C]leucine uptake in oocytes expressing each respective transporter.
We further assessed the inhibitory effects of nanvuranlat and N-acetyl-nanvuranlat on other amino acid transporters that mediate the transport of branched-chain or aromatic amino acids, including y+LAT1, y+LAT2, ATB0,+, B0AT1, TAT1 and b0,+. Neither compound inhibited these transporters (Fig. 7a–f). To ensure comprehensive coverage of neutral amino acid transporters, we also evaluated systems ASC (ASCT1, ASCT2) and N/A (SNAT1-5), which mainly transport small neutral amino acids (Supplementary Fig. S1 and S2). Both nanvuranlat and N-acetyl-nanvuranlat exhibited no significant inhibition of these transporters, with the sole exception being SNAT4, which displayed a non-significant trend towards inhibition by nanvuranlat at 10 µM (Supplementary Fig. S2d).
Fig. 7.

Effect of nanvuranlat and N-acetyl-nanvuranlat on neutral amino acid transporters other than system L that transport branched chain or aromatic amino acids. Uptakes of 50 µM l-[14C]leucine (Leu) by y+LAT1 (a), y+LAT2 (b), ATB0,+ (c), B0AT1 (d); the uptake of 50 µM l-[14C]tyrosine (Tyr) by TAT1 (e); and the uptake of 50 µM l-[14C]cystine by b0,+ (f) were measured in the presence of 1 µM and 10 µM of nanvuranlat and N-acetyl-nanvuranlat for 30 min in Xenopus oocyte expression system. Measurements were conducted using control Xenopus oocytes injected with water instead of cRNA (“Water”) and the oocytes injected with cRNA for expressing each transporter. Uptake rates are presented as mean ± S.D. (n = 8–12). For the functional expression of b0,+ in (f), rBAT, an auxiliary subunit of system b0,+ transporter that strongly induces system b0,+ activity in Xenopus oocytes, was expressed. Thus, the induced system b0,+ activity was of Xenopus origin, although human rBAT was expressed. Na+-free uptake solution was used for TAT1 and b0,+, whereas ND96 solution was used for the other transporters. Nanv.: nanvuranlat; N-Ac-Nanv.: N-acetyl-nanvuranlat. The data shown in panels (a) and (b) and those shown in panels (c) and (d) were obtained using the same respective batches of oocytes. The l-[14C]leucine uptake data from control oocytes are shared between panels (a) and (b) and separately between panels (c) and (d), but they are presented in each panel (a and b, and c and d) to allow for comparison on the same scale as the l-[14C]leucine uptake in oocytes expressing each respective transporter.
Discussion
The present study comprehensively characterizes nanvuranlat and its N-acetyl derivative, elucidating their pharmacological profiles concerning LAT1 inhibition and selectivity across various amino acid transporters. Our findings not only corroborate the potent and selective inhibition of LAT1 by nanvuranlat but also reveal novel insights into its mechanism of action and the impact of N-acetylation on its pharmacological properties.
A striking observation in our study is the remarkable selectivity of nanvuranlat for LAT1 over LAT2 despite the high structural similarity between these two transporters2. The selectivity was evident in both the inhibition of amino acid uptake and the suppression of cell proliferation (Fig. 2). This selectivity is consistent with previous findings by Oda et al.25, who demonstrated that KYT-0353 (nanvuranlat) potently inhibited the growth of LAT1-expressing S2-LAT1 cells, while showing minimal effects on LAT2-expressing S2-LAT2 cells. The ability of nanvuranlat to discriminate between LAT1 and LAT2 is particularly noteworthy, given that many previously reported inhibitors, such as BCH, exhibit activity against both transporters3,4,6,24. This high selectivity for LAT1 over LAT2 is crucial for potential therapeutic applications, as it may minimize off-target effects associated with LAT2 inhibition in normal tissues where LAT2 is more widely expressed3,24.
The high selectivity of nanvuranlat for LAT1 over LAT2 represents a significant potential advancement in the field of LAT1-targeted therapeutics. Previous efforts to develop LAT1-selective compounds have included both substrates and inhibitors, aiming for clinical application3,48,49. As examples of LAT1-selective substrates, fluoro-α-methyl-l-tyrosine (FAMT) and iodo-α-methyl-l-tyrosine (IMT) have been developed as PET and SPECT probes, respectively, for LAT1-specific imaging50–54. Because adding an α-methyl group to aromatic amino acids makes them LAT1-selective, these compounds exhibit high selectivity for LAT1 52–54. More recently, a bicyclic phenylalanine analog, (R)-2-amino-1,2,3,4-tetrahydro-2-naphthoic acid, has been reported to exhibit high LAT1 selectivity comparable to α-methyl-phenylalanine23. However, while these substrate-type compounds show high selectivity, their affinity for LAT1 is significantly lower than that of nanvuranlat. In addition to nanvuranlat, other LAT1-selective inhibitors based on the structure of T3 thyroid hormone, such as SKN103, have been developed55. However, these compounds also demonstrated lower affinity compared to nanvuranlat. Other recent inhibitors, JX-119, JX-078, and JX-075, are high-affinity inhibitors with IC50 values between 100 and 250 nM for inhibition of l-[14C]leucine uptake, appearing to have slightly lower LAT1 affinity than nanvuranlat based on these IC50 values40. Their selectivity for LAT1 over LAT2 has not been thoroughly evaluated. Additionally, compounds based on dithiazole and dithiazine scaffolds have been identified as potent LAT1 inhibitors, though their selectivity profiles require further investigation56. Thus, nanvuranlat’s exceptional selectivity for LAT1 over LAT2, coupled with its high affinity, positions it as a leading compound in the development of LAT1-targeted therapies.
Our kinetic studies (Fig. 3) provide the first direct evidence of the competitive nature of LAT1 inhibition by nanvuranlat and its N-acetyl derivative, offering critical mechanistic insights. The Ki value of 38.7 ± 5.1 nM for nanvuranlat indicates a high-affinity interaction with LAT1, consistent with its potent inhibitory effects in cell growth inhibition assays. This high-affinity, competitive interaction with LAT1 is likely facilitated by the compound’s structural features, which closely mimic the natural substrates of LAT1 in the binding pocket while precluding transport3,38,41. Recent structural insights into LAT1 inhibition, as reported by Lee et al.41, provide additional support for our findings on the competitive nature of nanvuranlat’s inhibition. Their cryo-electron microscopy analyses of LAT1 captured in association with various inhibitors reveal that nanvuranlat occupies the substrate-binding pocket, directly competing with amino acid substrates36,38,40,41. The structural data show that the α-carboxy and α-amino groups of nanvuranlat bind to the unwound parts of TM1 and TM6, respectively, corresponding to the interaction sites of the amino acid substrates (Supplementary Fig. S3a)38,41. Moreover, the proximal benzene ring of nanvuranlat occupies the same position as the side chain of phenylalanine, a typical LAT1 substrate41. This precise structural mimicry explains the compound’s ability to effectively compete with natural amino acid substrates. The binding mode involves fundamental interactions with residues such as Phe252 and Tyr259, which may explain the compound’s high affinity (Supplementary Fig. S3a)41.
Our study also provides crucial insights into the non-transportable nature of nanvuranlat as a LAT1 inhibitor. As demonstrated in Fig. 5, the lack of temperature-dependent uptake of 14C-nanvuranlat in HEK293-hLAT1 cells indicates that nanvuranlat acts as a non-transportable blocker rather than a competitive substrate. This characteristic is fundamental to its mechanism of action and distinguishes it from other amino acid-like inhibitors, such as BCH, that LAT1 may transport3,41. The structural data from Lee et al. offer a molecular explanation for this non-transportable nature41. Importantly, the amino-phenylbenzoxazol moiety of nanvuranlat (Fig. 1), particularly its amino group, induces a kink in TM10 of LAT1 41. This kink is proposed to be crucial in preventing the rotation of TM10, which is a critical conformational change in the series of structural transitions involved in the transport cycle, thereby contributing significantly to the inhibitory action of nanvuranlat41.
In light of these structural findings, our observations regarding the decreased affinity of N-acetyl nanvuranlat gain new significance. The N-acetylation of nanvuranlat occurs on the amino group of the amino-phenylbenzoxazol moiety (Fig. 1), the very group implicated in TM10 kinking41. In the docking simulation of N-acetyl-nanvuranlat with LAT1, the introduced N-acetyl group occupies a small pocket (triangle in Supplementary Fig. S3f) within the binding site, altering the placement of the amino group in nanvuranlat and the N-acetyl group in N-acetyl-nanvuranlat (Supplementary Fig. S3f). This shift also affects the positioning of their terminal benzene rings (double asterisks in Supplementary Fig. S3), weakening the interaction with Phe252 in N-acetyl-nanvuranlat (Supplementary Fig. S3). Since the interaction between the terminal benzene ring and Phe252 is critical for the high-affinity binding of nanvuranlat, this reduction likely contributes to the decreased affinity of N-acetyl-nanvuranlat. The N-acetyl group may also introduce steric hindrance, hindering the compound’s ability to access and occupy the binding site fully. Additionally, N-acetylation could alter the amino group’s hydrogen bonding and electrostatic interactions, weakening the overall interaction network with LAT1. The increased bulk of the N-acetyl group might further force the inhibitor into a suboptimal binding pose within the substrate pocket, reducing the efficiency of key interactions, including the critical kinking of TM10.
The sustained inhibitory effect of nanvuranlat on LAT1 function persisting after compound removal (Fig. 4) is a particularly intriguing finding. A noteworthy aspect of our results is the unexpected similarity in dissociation behavior between nanvuranlat and N-acetyl-nanvuranlat, despite their significant difference in binding affinity as reflected by their Ki values. Specifically, nanvuranlat and N-acetyl-nanvuranlat exhibited remarkably similar inhibition reduction rates of 29.8% and 30.8%, respectively (Fig. 4). These nearly identical rates, which reflect slow dissociation from LAT1, imply that N-acetylation does not significantly influence the dissociation process. This suggests that the structural elements responsible for the slow dissociation are likely distinct from those affected by N-acetylation. The similar dissociation rates indicate that both compounds, once bound, form complexes with LAT1 that are equally stable over time. This sustained inhibition is reminiscent of the concept of residence time in drug-target interactions, which could explain the apparent discrepancy between equilibrium binding (as reflected by Ki values) and dissociation kinetics57.
Structural studies have revealed that nanvuranlat adopts a U-shaped conformation when bound to LAT1, with both ends of the molecule interacting with different regions of the binding site (Supplementary Fig. S3a and b)38,41. This conformation allows the inhibitor to form multiple interactions, particularly at the distal ends of the side chains41. The regions distant from the N-acetylation site may form stable interactions within the binding pocket that are less affected by the N-acetylation. The U-shaped binding mode enables the inhibitor to fit snugly into the binding pocket, creating a tight and stable complex resistant to rapid dissociation. This structural arrangement likely contributes to the observed slow dissociation rate of the inhibitor from LAT1 38,41. Preserving dissociation kinetics despite reduced affinity has essential implications for the pharmacological properties of these compounds. N-acetyl-nanvuranlat, while requiring higher concentrations to achieve the same level of inhibition as nanvuranlat, might still provide a similar duration of effect once bound to the transporter. This characteristic could be particularly relevant in clinical contexts where the compound’s concentration may fluctuate, potentially influencing the design of dosing strategies for LAT1 inhibitors.
The exceptional selectivity of nanvuranlat for LAT1 extends beyond its discrimination between LAT1 and LAT2. Our comprehensive evaluation of multiple amino acid transport systems (Figs. 6 and 7, and Supplementary Figs. S1, S2) reveals a remarkable specificity for LAT1 among many transporters. We tested nanvuranlat and N-acetyl-nanvuranlat against systems L, T, B0, B0,+, b0,+, y+L, ASC, and N/A, encompassing both SLC7 family members and other amino acid transporters. Strikingly, both compounds showed significant inhibition only for LAT1, maintaining this high selectivity even after N-acetylation, albeit with reduced potency. This high selectivity is particularly noteworthy when compared to the classical system L inhibitor BCH, which has been shown to inhibit not only all system L transporters (LAT1, LAT2, LAT3, LAT4) but also B0AT1 (system B0) and ATB0,+ (system B0,+)3. In contrast, nanvuranlat and N-acetyl-nanvuranlat demonstrate a remarkably refined selectivity, targeting only LAT1 among the various amino acid transporters tested. This exceptional specificity underscores the potential of nanvuranlat and its N-acetyl derivative as highly selective LAT1 inhibitors, offering a significant advantage over less selective compounds like BCH in research and potential therapeutic applications.
This broad-spectrum selectivity is particularly noteworthy given the structural and functional similarities among many amino acid transporters. It suggests that nanvuranlat’s binding mode exploits unique features of the LAT1 binding pocket, features that are not shared by other transporters. The maintained LAT1 selectivity of N-acetyl-nanvuranlat, despite its reduced affinity, provides valuable insights into the structural determinants of LAT1 specificity. While the interaction between the amino group of the amino-phenylbenzoxazol moiety and TM10 appears crucial for high-affinity binding, our results indicate it may not be the primary determinant of LAT1 selectivity. This observation aligns with the structural insights provided by Lee et al., who identified specific residues in LAT1, such as Ser144 and Phe252, as potential contributors to inhibitor selectivity37,41.
The exceptional selectivity of nanvuranlat for LAT1 over such a wide range of amino acid transporters has significant implications for its potential as an anticancer agent3. This high specificity suggests a reduced likelihood of off-target effects related to amino acid transport in normal tissues, potentially leading to a more favorable safety profile. Moreover, the ability to selectively target LAT1, which is overexpressed in many cancers, could provide a therapeutic window for effective treatment while minimizing systemic toxicity. The maintained selectivity of the N-acetyl metabolite, albeit with reduced potency, further supports the potential for sustained therapeutic effects even after hepatic metabolism42,43.
The clinical implications of our findings are particularly noteworthy in light of the recent phase II clinical trial results for nanvuranlat in biliary tract cancer35. The successful meeting of the primary endpoint in this trial underscores the therapeutic potential of nanvuranlat. Our results provide several key insights that may explain the observed clinical efficacy. Firstly, we demonstrated that N-acetyl-nanvuranlat, rapidly generated in the liver retains significant LAT1 inhibitory activity despite its reduced affinity. Additionally, our findings suggest that nanvuranlat itself is likely taken up into hepatocytes via OATP transporters, potentially increasing exposure in the hepatobiliary system. Furthermore, the sustained inhibitory effect we observed for nanvuranlat and its N-acetyl derivative is particularly relevant. Upon administration, nanvuranlat undergoes rapid N-acetylation by N-acetyltransferase 2 in the liver34,35,42,43. Although this metabolic transformation reduces its LAT1 inhibitory activity, the resulting N-acetyl-nanvuranlat accumulates preferentially in the biliary system. The localized concentration of the parent compound and its metabolite in the target tissue, combined with their sustained inhibitory effects even retained in the N-acetyl metabolite, likely contributes to the observed therapeutic effect. This multifaceted mechanism—comprising active hepatic uptake and metabolic conversion of nanvuranlat, along with sustained inhibition and local accumulation of both the parent compound and its N-acetyl metabolite—may explain the observed efficacy of nanvuranlat in biliary tract cancer, even with the reduced LAT1 inhibitory potency of the metabolite.
In conclusion, our study provides a comprehensive characterization of nanvuranlat as a potent, selective, and non-transportable inhibitor of LAT1. The high selectivity for LAT1 over LAT2 and other amino acid transporters, coupled with its sustained inhibitory effect and non-transportable nature, positions nanvuranlat as a promising candidate for targeted cancer therapy. The impact of N-acetylation on its pharmacological properties highlights the importance of considering metabolic modifications in drug development. It underscores the critical role of precise structural complementarity in achieving optimal inhibitor potency and selectivity. Future studies combining detailed structural analyses with kinetic measurements could provide deeper insights into the molecular basis of these observations, paving the way for developing more effective and selective anticancer strategies targeting amino acid transport systems.
Materials and methods
Chemicals
Nanvuranlat ((2S)-2-amino-3-[4-[(5-amino-2-phenyl-1,3-benzoxazol-7-yl)methoxy]-3,5-dichlorophenyl]propanoic acid, CAS No.: 1037592–40 − 7) (2HCl salt; purity > 99%), N-acetyl-nanvuranlat and 14C-nanvuranlat (1.098 GBq/mmol)43 were provided by J-Pharma Co., Ltd (Yokohama, Japan). l-[14C]leucine (12.5 GBq/mmol) was purchased from Moravek Biochemicals (Brea, USA). l-[14C]alanine (2.07 GBq/mmol) and l-[14C]cystine (3.7 GBq/mmol) were obtained from American Radiolabeled Chemicals (St. Louis, USA). l-[14C]glutamine (10.1 GBq/mmol) was sourced from PerkinElmer (Boston, MA). l-[14C]tyrosine (17.83 GBq/mmol) was acquired from Amersham Biosciences (Buckinghamshire, U.K.). MS222 (ethyl 3-aminobenzoate methanesulfonate, tricaine) was purchased from Sigma Aldrich (St Louis, USA). Other general chemicals were purchased from Wako (Osaka, Japan).
Transport measurement in stable cell lines
HEK293 stably expressing human LAT1 (HEK293-hLAT1) and human LAT2 (HEK293-hLAT2) were constructed as described previously using HEK293 cell line (HSRRB, Osaka, Japan) by transfection with plasmids encoding human LAT1 (GenBank/EMBL/DDBJ accession no. AB018009) and human LAT2 (GenBank/EMBL/DDBJ accession no. AB037669)58 and maintained in Minimum Essential Medium (Nacalai Tesque, Kyoto, Japan) containing 10% (v/v) heat-inactivated fetal bovine serum (Thermo Fisher Scientific, Grand Island, NY), 100 units/mL penicillin, 100 µg/mL streptomycin (Nacalai Tesque, Kyoto, Japan), 1% Non-Essential Amino Acids (Wako, Osaka, Japan) and 0.9 mg/mL G418 disulfate (Nacalai Tesque, Kyoto, Japan), under 5% CO₂ at 37 °C with high humidity. Cells were seeded at a density of 2.3 × 105 cells/well for HEK293-hLAT1 and 2.4 × 105 cells/well for HEK293-hLAT2 in poly-d-lysine-coated 24-well plates and cultured for 2 days.
After the culture medium was removed, the cells were pre-incubated in 500 µL of pre-warmed Na+-free HBSS (Hank’s balanced salt solution) containing 125 mM choline chloride, 4.8 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.3 mM CaCl2, 5.6 mM glucose, and 25 mM HEPES (pH 7.4) at 37 °C for 10 min. Uptake of radiolabeled compounds was measured for 1 min in Na+-free HBSS containing 1 µM l-[14C]leucine for HEK293-hLAT1 or 1 µM l-[14C]alanine for HEK293-hLAT2 58. The inhibitory effect of nanvuranlat and N-acetyl-nanvuranlat on the uptake was evaluated by adding test compounds diluted from dimethyl sulfoxide (DMSO) stock solutions (30 mM for nanvuranlat, 10 mM for N-acetyl-nanvuranlat), concurrently with the radio-labeled substrates.
Cells were then washed three times with ice-cold HBSS and lysed with 500 µL of 0.1 N NaOH. The lysate was mixed with 500 µL of Emulsifier safe cocktail (PerkinElmer, Waltham, MA, USA), and radioactivity was measured by liquid scintillation counting (LSC-3100; Aloka, Tokyo, Japan). Protein concentration in the cell lysate was determined using the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Rockford, IL). Kinetic parameters (Ki) for nanvuranlat and N-acetyl-nanvuranlat were determined by assessing the uptake of l-[14C]leucine at various concentrations. As indicated in the figure legend, a wide range of leucine concentrations was used to ensure comprehensive kinetic coverage. Control experiments with DMSO were performed parallel to confirm that the effects were specific to the test compounds.
For 14C-nanvuranlat uptake measurement, HEK293-hLAT1 cells were incubated with 1 µM 14C-nanvuranlat in Na+-free HBSS. The temperature dependence of uptake was examined by performing measurements with 1 µM 14C-nanvuranlat or 1 µM l-[14C]leucine for 1 min either on ice or at 37 °C.
Cell proliferation assay
To evaluate the effects of nanvuranlat and N-acetyl-nanvuranlat on cell proliferation, a cell proliferation assay was conducted using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Kumamoto, Japan), according to the manufacturer’s instructions. Cells were seeded at a density of 1.5 × 103 cells/well in poly-d-lysine-coated 96-well plates. The following day, cells were treated with various concentrations of nanvuranlat or N-acetyl-nanvuranlat for 72 h. The degree of proliferation inhibition was calculated as (C – T)/(C – T0) × 100 (%), where T0 represents the cell count immediately before starting treatment (0 h), C is the cell count for the vehicle control at 72 h, and T is the cell count at the indicated concentrations of nanvuranlat or N-acetyl-nanvuranlat at 72 h. The sensitivity of cells to nanvuranlat or N-acetyl-nanvuranlat was evaluated based on the 50% growth inhibition (GI50) value, defined as the compound concentration required to achieve 50% inhibition of cell proliferation. The GI50 value was calculated by the GraphPad Prism 10.1.0 (GraphPad Software, Boston, MA, USA).
Sustained inhibition assay
To assess the sustained inhibition—defined as the inhibitory effect that persists after compound washout—HEK293-hLAT1 cells were preincubated in a culture medium containing 20 µM nanvuranlat or N-acetyl-nanvuranlat for 30 min at 37 °C. The cells were then washed three times with pre-warmed, fresh culture medium and incubated in 500 µL of culture medium for 0, 10, 20, 30, and 60 min, as well as 24 h. Following this incubation, cells were washed three times with Na⁺-free HBSS and then subjected to uptake assays by incubation with 1 µM l-[14C]leucine for 1 min. l-[14C]Leucine uptake was quantified as a percentage relative to the control (cells not treated with nanvuranlat or N-acetyl-nanvuranlat) to evaluate the extent of sustained inhibition.
cDNAs and complementary RNA (cRNA) synthesis
cDNAs for human transporters used in this study are listed in Supplementary Table S1. cRNAs were synthesized in vitro from the linearized plasmids using mMessage mMachine Kit, polyadenylated with Poly(A) Tailing Kit, and purified with MEGAclear Kit (Ambion, Austin, TX), following the manufacturer’s instructions. Details on the plasmid vectors, restriction enzyme sites for subcloning, linearization enzymes, and RNA polymerases used for cRNA synthesis are provided in Supplementary Table S1.
Expression in Xenopus laevis oocytes and transport measurements
The animal experiments were performed in accordance with the guidelines of the Research Institutes of Animal Experimentation and approved by the Animal Experiments Committee of Osaka University. This study complied with the Animal Research: In Vivo Reporting of Experiments (ARRIVE) guidelines. Xenopus laevis (Saitama Experimental Animals Supply Co.,Ltd., Saitama, Japan) were anesthetized in 0.1% MS222 for surgical oocyte harvest as described4,53. Defolliculated oocytes were injected with polyadenylated cRNA (25 ng/oocyte) for oocyte expression4,7,53. For the functional expression of LAT1, LAT2, y+LAT1 and y+LAT2, equimolar 4F2hc cRNA was co-injected53. For B0AT1, equimolar collectrin cRNA was also co-injected53. Transport measurements were performed 2–4 days after injection as previously described53. Briefly, oocytes were incubated at room temperature with 500 µL uptake solution containing 14C-labeled compounds. ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.5) was used as the uptake solution for Na+-dependent transport. For Na+-free uptake solution, NaCl was replaced by choline chloride. Radioactivity was determined by liquid scintillation counting. Functional expression of each transporter in Xenopus oocytes was confirmed by measuring the transport of its typical substrate as described in the figure legends. Each data point represents the mean ± S.D. (n = 5–13). To confirm reproducibility, three independent experiments were conducted using different batches of oocytes.
Statistical analysis
GI50 values and the kinetic parameters, including Ki values, were obtained by the GraphPad Prism 10.1.0. Statistical analyses were performed using the unpaired two-tailed Student’s t-test between two groups. For multiple groups, one-way ANOVA followed by Dunnett’s multiple comparisons test were used. Differences were considered significant when p-values were < 0.05. Data are presented as mean ± S.D.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
C.J. and Y.K. conceived the study. C.J. and X.Z. conducted experiments. C.J., X.Z., and Y.K. performed data analysis. C.J. and Y.K. wrote the manuscript with contributions from all authors. All authors have approved the final version of the manuscript.
Funding
This research was supported by Japan Society for the Promotion of Science, Grants-in-Aid for Scientific Research to Y.K. ([19H03407], [22H02809], and [22K19404]) and C.J. ([23K19451]), Japan Agency for Medical Research and Development Project for Cancer Research and Therapeutic Evolution to Y.K. ([JP19cm0106151], [JP20cm0106151], [JP21cm0106151], and [JP23ama221121], and [JP24ama221121]), and a collaborative research grant from J-Pharma Co., Ltd (December 2017–March 2023).
Data availability
All data generated or analyzed during this study can be found in the manuscript and Supplementary Information files. The raw data are available from the corresponding author upon reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Christensen, H. N. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol. Rev.70, 43–77 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Fotiadis, D., Kanai, Y. & Palacín, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Aspects Med.34, 139–158 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Kanai, Y. Amino acid transporter LAT1 (SLC7A5) as a molecular target for cancer diagnosis and therapeutics. Pharmacol. Ther.230, 107964 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Kanai, Y. et al. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem.273, 23629–23632 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Mastroberardino, L. et al. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature395, 288–291 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Wagner, C. A., Lang, F. & Bröer, S. Function and structure of heterodimeric amino acid transporters. Am. J. Physiol. Cell. Physiol.281, C1077–C1093 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Yanagida, O. et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta1514, 291–302 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Matsuo, H. et al. Expression of a system L neutral amino acid transporter at the blood-brain barrier. Neuroreport11, 3507–3511 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Tărlungeanu, D. C. et al. Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell167, 1481–1494 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohgaki, R. et al. Essential roles of L-type amino acid transporter 1 in syncytiotrophoblast development by presenting fusogenic 4F2hc. Mol. Cell. Biol.37, e00427–e00416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaira, K. et al. l-type amino acid transporter 1 and CD98 expression in primary and metastatic sites of human neoplasms. Cancer Sci.99, 2380–2386 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaira, K. et al. Clinical significance of L-type amino acid transporter 1 expression as a prognostic marker and potential of new targeting therapy in biliary tract cancer. BMC Cancer. 13, 482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang, Q. & Holst, J. L-type amino acid transport and cancer: Targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res.5, 1281–1294 (2015). [PMC free article] [PubMed] [Google Scholar]
- 14.Lu, X. The role of large neutral amino acid transporter (LAT1) in cancer. Curr. Cancer Drug Targets. 19, 863–876 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Okanishi, H., Ohgaki, R., Xu, M., Endou, H. & Kanai, Y. Phosphoproteomics revealed cellular signals immediately responding to disruption of cancer amino acid homeostasis induced by inhibition of L-type amino acid transporter 1. Cancer Metab.10, 18 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikubo, K. et al. Pharmacologic inhibition of LAT1 predominantly suppresses transport of large neutral amino acids and downregulates global translation in cancer cells. J. Cell. Mol. Med.26, 5246–5256 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou, X. et al. Inhibition of amino acid transporter LAT1 in cancer cells suppresses G0/G1-S transition by downregulating cyclin D1 via p38 MAPK activation. J. Pharmacol. Sci.154, 182–191 (2024). [DOI] [PubMed] [Google Scholar]
- 18.Kaira, K. et al. Prognostic significance of L-type amino acid transporter 1 expression in resectable stage I-III nonsmall cell lung cancer. Br. J. Cancer98, 742–748 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaira, K. et al. Prognostic significance of L-type amino-acid transporter 1 expression in surgically resected pancreatic cancer. Br. J. Cancer107, 632–638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scalise, M., Galluccio, M., Console, L., Pochini, L. & Indiveri, C. The human SLC7A5 (LAT1): The intriguing histidine/large neutral amino acid transporter and its relevance to human health. Front. Chem.6, 243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kandasamy, P., Gyimesi, G., Kanai, Y. & Hediger, M. A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci.43, 752–789 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Jin, C. et al. Interaction of halogenated tyrosine/phenylalanine derivatives with organic anion transporter 1 in the renal handling of tumor imaging probes. J. Pharmacol. Exp. Ther.375, 451–462 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Chen, S. et al. Structure-activity characteristics of phenylalanine analogs selectively transported by L-type amino acid transporter 1 (LAT1). Sci. Rep.14, 4651 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segawa, H. et al. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J. Biol. Chem.274, 19745–19751 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Oda, K. et al. L-Type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci.101, 173–179 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cormerais, Y. et al. Genetic disruption of the multifunctional CD98/LAT1 complex demonstrates the key role of essential amino acid transport in the control of mTORC1 and Tumor Growth. Cancer Res.76, 4481–4492 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Okanishi, H., Ohgaki, R., Okuda, S., Endou, H. & Kanai, Y. Proteomics and phosphoproteomics reveal key regulators associated with cytostatic effect of amino acid transporter LAT1 inhibitor. Cancer Sci.112, 871–883 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quan, L. et al. Amino acid transporter LAT1 in tumor-associated vascular endothelium promotes angiogenesis by regulating cell proliferation and VEGF-A-dependent mTORC1 activation. J. Exp. Clin. Cancer Res.39, 266 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi, Z. et al. Inhibition of cancer-type amino acid transporter LAT1 suppresses B16-F10 melanoma metastasis in mouse models. Sci. Rep.13, 13943 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishikubo, K. et al. Combination effects of amino acid transporter LAT1 inhibitor nanvuranlat and cytotoxic anticancer drug gemcitabine on pancreatic and biliary tract cancer cells. Cancer Cell. Int.23, 116 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma, Y. et al. Upregulation of ATF4 mediates the cellular adaptation to pharmacologic inhibition of amino acid transporter LAT1 in pancreatic ductal adenocarcinoma cells. J. Pharmacol. Sci.155, 14–20 (2024). [DOI] [PubMed] [Google Scholar]
- 32.Saito, S. et al. The LAT1 inhibitor JPH203 suppresses the growth of castration-resistant prostate cancer through a CD24-mediated mechanism. Cancer Sci.115, 2461–2472 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosilio, C. et al. L-type amino-acid transporter 1 (LAT1): A therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia29, 1253–1266 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Okano, N. et al. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Invest. New Drugs38, 1495–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Furuse, J. et al. A phase II placebo-controlled study of the effect and safety of nanvuranlat in patients with advanced biliary tract cancers previously treated by systemic chemotherapy. Clin. Cancer Res.30, 3990–3995 (2024). [DOI] [PubMed] [Google Scholar]
- 36.Yan, R., Zhao, X., Lei, J. & Zhou, Q. Structure of the human LAT1-4F2hc heteromeric amino acid transporter complex. Nature568, 127–130 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Lee, Y. et al. Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat. Struct. Mol. Biol.26, 510–517 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Hu, Z. & Yan, R. Structural basis for the inhibition mechanism of LAT1-4F2hc complex by JPH203. Cell. Discov. 10, 73 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu, D. et al. The complete assembly of human LAT1-4F2hc complex provides insights into its regulation, function and localisation. Nat. Commun.15, 3711 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan, R. et al. Mechanism of substrate transport and inhibition of the human LAT1-4F2hc amino acid transporter. Cell. Discov.7, 16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee, Y. et al. Structural basis of anticancer drug recognition and amino acid transport by LAT1. Nat. Commun.. 10.1101/2023.12.03.567112v1.
- 42.Wempe, M. et al. Metabolism and pharmacokinetic studies of JPH203, an L-amino acid transporter 1 (LAT1) selective compound. Drug Metab. Pharmacokinet.27, 155–161 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Toyoshima, J., Kusuhara, H., Wempe, M. F., Endou, H. & Sugiyama, Y. Investigation of the role of transporters on the hepatic elimination of an LAT1 selective inhibitor JPH203. J. Pharm. Sci.102, 3228–3238 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Okunushi, K. et al. JPH203, a newly developed anti-cancer drug, shows a preincubation inhibitory effect on L-type amino acid transporter 1 function. J. Pharmacol. Sci.144, 16–22 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Ohtsu, N. et al. Functional coupling of organic anion transporter OAT10 (SLC22A13) and monocarboxylate transporter MCT1 (SLC16A1) influencing the transport function of OAT10. J. Pharmacol. Sci.150, 41–48 (2022). [DOI] [PubMed] [Google Scholar]
- 46.Ohgaki, R. et al. Interaction of the sodium/glucose cotransporter (SGLT) 2 inhibitor canagliflozin with SGLT1 and SGLT2. J. Pharmacol. Exp. Ther.358, 94–102 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Shiraya, K. et al. A novel transporter of SLC22 family specifically transports prostaglandins and co-localizes with 15-hydroxyprostaglandin dehydrogenase in renal proximal tubules. J. Biol. Chem.285, 22141–22151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh, N. & Ecker, G. F. Insights into the structure, function, and ligand discovery of the large neutral amino acid transporter 1, LAT1. Int. J. Mol. Sci.19, 32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uchino, H. et al. Transport of amino acid-related compounds mediated by L-type amino acid transporter 1 (LAT1): Insights into the mechanisms of substrate recognition. Mol. Pharmacol.61, 729–737 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Kaira, K. et al. Fluorine-18-α-methyltyrosine positron emission tomography for diagnosis and staging of lung cancer: A clinicopathologic study. Clin. Cancer Res.13, 6369–6378 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Nobusawa, A. et al. Diagnostic usefulness of 18F-FAMT PET and L-type amino acid transporter 1 (LAT1) expression in oral squamous cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging. 40, 1692–1700 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Wiriyasermkul, P. et al. Transport of 3-fluoro-L-α-methyl-tyrosine by tumor-upregulated L-type amino acid transporter 1: A cause of the tumor uptake in PET. J. Nucl. Med.53, 1253–1261 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Wei, L. et al. Specific transport of 3-fluoro-L-α-methyl-tyrosine by LAT1 explains its specificity to malignant tumors in imaging. Cancer Sci.107, 347–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shikano, N. et al. Isoform selectivity of 3-125I-iodo-alpha-methyl-L-tyrosine membrane transport in human L-type amino acid transporters. J. Nucl. Med.44, 244–246 (2003). [PubMed] [Google Scholar]
- 55.Kongpracha, P. et al. Structure-activity relationship of a novel series of inhibitors for cancer type transporter L-type amino acid transporter 1 (LAT1). J. Pharmacol. Sci.133, 96–102 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Napolitano, L. et al. Potent inhibitors of human LAT1 (SLC7A5) transporter based on dithiazole and dithiazine compounds for development of anticancer drugs. Biochem. Pharmacol.143, 39–52 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Copeland, R., Pompliano, D. & Meek, T. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov.5, 730–739 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Khunweeraphong, N. et al. Establishment of stable cell lines with high expression of heterodimers of human 4F2hc and human amino acid transporter LAT1 or LAT2 and delineation of their differential interaction with α-alkyl moieties. J. Pharmacol. Sci.119, 368–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study can be found in the manuscript and Supplementary Information files. The raw data are available from the corresponding author upon reasonable request.