

Abstract
Insertion sequences are compact and pervasive transposable elements found in bacteria, which encode only the genes necessary for their mobilization and maintenance1. IS200- and IS605-family transposons undergo ‘peel-and-paste’ transposition catalysed by a TnpA transposase2, but they also encode diverse, TnpB- and IscB-family proteins that are evolutionarily related to the CRISPR-associated effectors Cas12 and Cas9, respectively3,4. Recent studies have demonstrated that TnpB and IscB function as RNA-guided DNA endonucleases5,6, but the broader biological role of this activity has remained enigmatic. Here we show that TnpB and IscB are essential to prevent permanent transposon loss as a consequence of the TnpA transposition mechanism. We selected a family of related insertion sequences from Geobacillus stearothermophilus that encode several TnpB and IscB orthologues, and showed that a single TnpA transposase was broadly active for transposon mobilization. The donor joints formed upon religation of transposon-flanking sequences were efficiently targeted for cleavage by RNA-guided TnpB and IscB nucleases, and co-expression of TnpB and TnpA led to substantially greater transposon retention relative to conditions in which TnpA was expressed alone. Notably, TnpA and TnpB also stimulated recombination frequencies, surpassing rates observed with TnpB alone. Collectively, this study reveals that RNA-guided DNA cleavage arose as a primal biochemical activity to bias the selfish inheritance and spread of transposable elements, which was later co-opted during the evolution of CRISPR–Cas adaptive immunity for antiviral defence.
Some of the largest and most rapid forms of genetic diversification result from transposable elements7,8 (TEs). TE propagation imposes fitness costs on host cells, leading to the emergence of pathways that manage this genetic conflict, but TEs and host cells also engage in interactions that can result in co-operation or even co-option of TE sequences and genes9. Indeed, many critical cellular processes evolved directly from mobile genetic elements—including mRNA intronic splicing10, telomere maintenance11, immunoglobulin diversification12 and spacer acquisition13—highlighting the molecular opportunities afforded by exaptation and domestication of transposon-encoded enzymes14,15. Recent research efforts similarly showcase the technological utility of transposon genes for DNA cleavage/joining reactions and genome-engineering applications16.
Bacterial and archaeal adaptive immune systems encoded by CRISPR–Cas loci represent a compelling example where recurring gene exchange between host and transposon has had a pervasive evolutionary role15. CRISPR arrays are themselves the products of sequential DNA integration reactions catalysed by Cas1, a homologue of transposases found within casposon TEs, suggesting that the early origins of adaptive immunity required the co-option of cas1 genes by proto-CRISPR–Cas systems17. In the more recent evolutionary past, mature CRISPR–Cas systems have been repurposed by diverse types of mobile genetic elements, including plasmids18, phages19 and transposons20–22. Finally, the hallmark RNA-guided DNA cleaving enzymes that define type II and type V CRISPR–Cas systems—Cas9 and Cas12—are evolutionarily related to IscB and TnpB enzymes encoded by IS200 and IS605 (IS200/IS605)-family TEs3,4. Thus, although CRISPR–Cas systems canonically offer their hosts immunity from foreign mobile genetic elements, their very genesis is intimately linked with mobile genetic element gene domestication events that provided critical biochemical capabilities15.
Bacterial TEs that encode only the gene(s) necessary for transposition are referred to as insertion sequences (ISs), and IS elements within the IS200/IS605 superfamily are mobilized by an HUH endonuclease-superfamily transposase known as TnpA23. Additionally, they often bear an accessory nuclease gene known as tnpB or iscB2. An early study demonstrated that TnpB is dispensable for TnpA-mediated transposition, but did not convincingly reveal a biological function for TnpB24. More recently, it was reported that TnpB and IscB nucleases both function as RNA-guided DNA nucleases that use non-coding RNAs encoded by the IS element itself5,6. These findings demonstrated that the ability for single-effector, CRISPR-associated proteins to target double-stranded DNA (dsDNA) using an RNA guide first arose in bacterial transposons. However, how this molecular property plays a part within the transposition pathway of these elements has remained enigmatic (Fig. 1a).
Fig. 1 |. Pervasive distribution of IS200/IS605-like elements in G. stearothermophilus.

a, Schematic of a representative IS200/IS605 element. tnpA encodes a Y1-family tyrosine transposase that is responsible for DNA excision and integration; tnpB/iscB encodes RNA-guided nucleases whose biological roles are unknown. b, Bottom, schematic of a non-autonomous IS element encoding TnpB and its associated overlapping ωRNA. Top, a model of ωRNA structural covariation in the indicated region. The green rectangle (bottom) indicates the transposon boundaries, and the guide portion of the ωRNA (top) is shown in blue. c, Genome-wide distribution of IS200/IS605-family elements in G. stearothermophilus strain DSM 458. Five distinct families are shown (ISGst2–6), based on sequence similarity of transposon ends and the encoded nuclease. d, Read coverage from small RNA-seq data of G. stearothermophilus strain ATCC 795333, demonstrating expression of putative ωRNAs from each of the indicated G. stearothermophilus IS families. TnpB-associated ωRNAs are encoded within or downstream of the ORF, whereas IscB-associated ωRNAs are encoded upstream of the ORF.
Here we show that retention of IS200/IS605 transposons at the donor site after DNA excision—and thus long-term transposon survival—relies on DNA cleavage by TnpB or IscB (TnpB/IscB) nucleases. By exploiting transposon-encoded guide RNAs to specifically recognize excision products and generate genomic DNA double-strand breaks (DSBs), TnpB/IscB trigger host-mediated recombination that reinstalls the transposon, providing an alternative pathway to achieve proliferative transposition that we term ‘peel-and-paste, cut-and-copy’. Beyond uncovering an elegant mechanism of transposon maintenance, this work advances our understanding of the biological function of a large family of RNA-guided nucleases that are encoded within diverse transposable elements found in all domains of life25–27.
Diversity of TnpB and IscB proteins
We set out to explore the evolutionary diversity of TnpB and IscB proteins—which, similar to Cas12 and Cas9, contain RuvC or RuvC and HNH nuclease domains, respectively—and identify conserved genes and sequence elements within their genetic neighbourhood, as an entry point to investigating their function. We first mined the NCBI NR database for homologues and built phylogenetic trees that highlight the diversity of both protein families (Extended Data Fig. 1a,d). When we extracted flanking genomic regions, we identified only a sporadic association with Y1 tyrosine transposases, with around 25% of tnpB genes containing an identifiable tnpA nearby, indicative of autonomous transposons. Notably, iscB genes were much less abundant than tnpB and rarely associated with tnpA (approximately 1.5%). This suggests that the majority of tnpB/iscB genes are encoded within transposons lacking tnpA, suggesting a non-autonomous state that requires transposases encoded elsewhere to mobilize them in trans (Extended Data Fig. 1a,d). TnpB, but not iscB, genes were also found associated with an unrelated serine resolvase (also denoted tnpA), a hallmark of IS607-family transposons28, albeit at a much lower frequency (around 8%) (Extended Data Fig. 1d).
In our initial analyses, we also observed a conspicuous, highly conserved intergenic region upstream of iscB that was bounded by the transposon RE, which bore marked similarity to a non-coding RNA termed HEARO29. Similarly, non-coding RNA molecules termed sotRNAs were detected downstream of tnpB genes in Halobacteria30,31, and both IscB and TnpB use these transposon-encoded RNAs—referred to hereafter as ωRNAs—as guides to direct cleavage of complementary dsDNA substrates, in a mechanism analogous to Cas9 and Cas125,6. We generated covariation models for TnpB- and IscB-specific ωRNAs, highlighting conserved secondary structural motifs unique to each element (Fig. 1b and Extended Data Fig. 1b). These motifs act as scaffolds encoded within the transposon and extend beyond its boundaries to generate guide RNA sequences located just outside the transposon right end (RE)5,6, such that ωRNAs are reprogrammed after each transposition event. We used these models to demonstrate the tight genetic linkage between tnpB/iscB genes and flanking ωRNA loci (Extended Data Fig. 1a,d).
To investigate whether ωRNA production might be sensitive to local genetic context, we analysed the orientation of genes upstream of tnpB/iscB and observed a strong bias for genes encoded in the same orientation as iscB but not tnpB (Extended Data Fig. 1c). IscB-specific ωRNAs rely on a specific structure consisting of a constant scaffold sequence derived from the transposon RE, along with a guide region encoded outside the transposon boundary and positioned at the ωRNA 5′ end. This implies that ωRNA biogenesis requires transcription initiation outside the IS element, proceeding towards the IscB open reading frame (ORF) (Extended Data Fig. 1b). Our analyses suggest that transposon insertions in transcriptionally active regions facilitate the production of functional ωRNAs, and that these insertions are either preferentially generated during transposition and/or preferentially retained during evolution. By contrast, TnpB-specific ωRNAs are processed from the TnpB mRNA transcribed within the transposon itself31,32 and, thus, TnpB-containing IS elements do not exhibit appreciable orientation bias with respect to neighbouring genes (Extended Data Fig. 1c).
To select candidates for experimental study, we sifted through our phylogenetic trees, prioritizing homologues that were found in the same species within related and high-copy TEs, indicative of recent transposition events. We converged on G. stearothermophilus, a thermophilic soil bacterium that has yielded useful thermostable proteins33 and whose genome revealed a substantial expansion of five, IS605-family elements encoding both TnpB and IscB, denoted ISGst2–6, collectively comprising around 1% of the genome (Fig. 1c). Analysis of RNA-sequencing (RNA-seq) data from two distinct G. stearothermophilus strains revealed consistent ωRNA expression from multiple elements33 (Fig. 1d and Extended Data Fig. 1e), and we found that the transposon left end (LE) and RE boundaries of these IS elements were highly similar in DNA sequence (Extended Data Fig. 2a–d), suggesting a common mechanism of mobilization. Using this information, we identified a candidate tnpA gene responsible for transposing these elements, as well as minimal non-autonomous IS elements that lacked protein-coding genes altogether and resembled palindrome-associated transposable elements1 (PATEs) (Extended Data Fig. 2e).
In addition to sharing similar sequences within the LE and RE, ISGst2–6 elements exhibited conserved, clade-specific transposon-adjacent motifs (TAMs) and transposon-encoded motifs (TEMs) (Extended Data Fig. 2a–d). Prior studies of TnpA transposases from Helicobacter pylori IS608 (HpyTnpA) and Deinococcus radiodurans ISDra2 (DraTnpA) revealed that these motifs constitute the target and cleavage sites recognized during insertion and excision reactions, respectively. Yet, rather than being recognized exclusively through protein–DNA recognition, these motifs form non-canonical base-pairing interactions with a DNA ‘guide’ sequence located in the sub-terminal ends of the IS element34–36 (Fig. 2a). Focusing on multiple sequence alignments between ISGst2–6 elements, we observed covarying mutations between both the TAM and TEM sequences and their associated DNA guide sequences (Fig. 2a and Extended Data Fig. 2a,b), further suggesting that these elements would be active for transposition.
Fig. 2 |. TnpA catalyses DNA excision for multiple families of IS elements.

a, Schematic of ISGst3, highlighting the sub-terminal palindromic ends (top). TAM and TEM motifs are highlighted in yellow and orange, respectively, DNA guide sequences are shown in orange, and their putative base-pairing interactions are indicated; red dashed lines indicate transposon boundaries and thus the sites of ssDNA cleavage and religation. The donor joint formed upon transposon loss is shown at the bottom and comprises the TAM abutting RE-flanking sequence (nucleotides denoted with N). b, Schematic of the heterologous transposon excision assay in Escherichia coli. Plasmids encode TnpA and a mini-transposon (mini-Tn) substrate, whose loss is monitored by PCR using the indicated primers. c, TnpA is active in recognizing and excising all five families of G. stearothermophilus IS elements. Cell lysates were tested after overnight expression of TnpA with the indicated G. stearothermophilus IS mini-Tn substrates, and PCR products were resolved by agarose gel electrophoresis. Marker denotes a positive excision control; U, unexcised; E, excised; M, a Y125A TnpA mutant. d, Excision products from c exhibit the expected ‘donor joint’ architecture, as demonstrated by Sanger sequencing. Dashed lines denote the religation site following excision; the TAM is highlighted in yellow. e, Transposon excision requires intact LE and RE sequences, as shown via testing of the mutagenized mini-Tn substrates indicated on the right. Experiments were performed as in c using ISGst3; substrates 1, 3, 4 and 5 encode the transposon on the lagging-strand template strand. Transposon ends and TAMs are indicated with green triangles and yellow boxes, respectively; M denotes Y125A TnpA mutant. f, Transposon excision is dependent on cognate pairing between compatible TAM and guide sequences. Excision experiments were performed as in c using ISGst3 with indicated mutations in the TAM or TEM (blue) or DNA guide (orange). Substrate 4 has mutations to cognate sequences from IS608. Mut., mutation.
TnpA catalyses transposon excision and integration
TnpA is part of the HUH endonuclease superfamily and recognizes DNA hairpin structures in a sequence- and structure-specific way to mediate single-stranded DNA (ssDNA) cleavage and ligation23. Biochemical and genetic studies of HpyTnpA and DraTnpA revealed a peel-out-paste-in transposition mechanism, in which IS elements first excise as a circular ssDNA intermediate with abutted LE and RE sequences, coincident with precise rejoining of the flanking DNA to produce a scarless donor joint that regenerates the original genomic sequence2. Transposition to a new target site occurs downstream of a TAM recognized through base-pairing with the LE DNA guide, via insertion of the circular ssDNA in a manner that requires subsequent second-strand synthesis but does not create a target site duplication. Excision and integration reactions both rely heavily on ssDNA and the formation of conserved, intramolecular LE and RE stem-loop structures (Fig. 2a), implicating DNA replication as a major opportunity for transposon activation2,37,38.
We designed a DNA excision assay to test the activity of GstTnpA on a mini-Tn substrate derived from its native autonomous IS element, ISGst2. We cloned E. coli expression vectors that encoded GstTnpA upstream of the mini-Tn, which comprised an antibiotic resistance gene flanked by full-length LE and RE sequences and 50 bp of genomic G. stearothermophilus sequences upstream and downstream of the predicted transposon boundaries. Primers were designed to bind outside the mini-Tn, such that PCR from cellular lysates would amplify either the starting substrate or a shorter reaction product resulting from transposon excision and DNA religation (Fig. 2b). We also generated a parallel panel of substrates containing LE and RE sequences derived from ISGst3–6, which natively encode IscB, TnpB or ωRNA only, to determine the breadth of GstTnpA substrate recognition. GstTnpA was active on all five families of IS elements, with excision dependent on the predicted catalytic tyrosine residue (Fig. 2c), but did not cross-react with a substrate derived from the H. pylori IS608 element (Extended Data Fig. 3a,b). Sanger sequencing of excision products revealed that in each case, TnpA precisely re-joined sequences flanking the mini-Tn to generate a scarless donor joint (Fig. 2d). Using a quantitative PCR (qPCR)-based strategy to prime directly off the donor joint sequence, we calculated excision frequencies of around 0.70% directly from overnight cultures (Extended Data Fig. 3c–e).
We next investigated sequence determinants of transposon excision in greater detail, focusing on the ISGst3 element that natively encodes TnpB and its associated ωRNA. Excision proceeded regardless of whether the mini-Tn was encoded on the leading-strand or lagging-strand template, but was ablated when we scrambled either the LE or RE sequence, confirming the critical importance of these regions for TnpA recognition. Excision was also strongly dependent on the presence of a cognate TAM adjacent to the LE as well as a compatible DNA guide sequence located within the LE, as mutation of either region led to a loss of product formation (Fig. 2e). Interestingly, simultaneous mutation of both the TAM and LE guide sequence to the corresponding motifs found in IS608 restored excision activity with GstTnpA (Fig. 2f), confirming the importance of these complementary base-pairing interactions to mediate DNA excision. Similar base-pairing interactions occur between a DNA guide sequence within the RE and a matching TEM found within the RE boundary, with only minor differences between the TAM and TEM (TAM/TEM) motif at positions 3 and 5 (Extended Data Fig. 2a–d). Whereas the excision reaction did not tolerate mutation of the TAM sequence to the TEM sequence, we were surprised to find that mutations to the TEM were still tolerated, despite ablating predicted base-pairing interactions with the RE guide sequence (Fig. 2f). However, closer inspection revealed that these excision events resulted from erroneous selection of an alternative mini-Tn boundary downstream of the native RE, at a sequence matching the wild-type TEM (TTCAC) (Extended Data Fig. 3f,g). These results indicate that IS200/IS605-family elements tolerate flexible spacing between the TAM/TEM motif and corresponding guide sequences, allowing for occasional capture of additional sequences outside of the native LE and RE boundaries.
We also investigated the ability of GstTnpA to catalyse insertions at new target sites. Using a traditional mating-out assay with the ISGst3 mini-Tn (Extended Data Fig. 4a), in which transposition events into a conjugative plasmid are isolated via drug selection, we measured transposition efficiencies of 2.5 × 10−7, which were several orders of magnitude lower than the observed rates of excision (Extended Data Figs. 3e and 4b,d). Similar transposition frequencies were also observed for autonomous elements that encoded both tnpA and tnpB within the same element. These results suggest that, under the tested experimental conditions, TnpA expression would eventually lead to permanent transposon loss from the population, absent any active mechanisms for maintaining transposons at their donor sites during or after excision (see below). Long-read sequencing of drug-resistant transconjugants confirmed the presence of new mini-Tn insertions, which were invariably located downstream of endogenous TAM sites on the F-plasmid, confirming the essentiality of this motif (Extended Data Fig. 4c). Collectively, these experiments demonstrate that GstTnpA is active in mobilizing a large network of diverse, IS605-like elements found in the G. stearothermophilus genome, but that its intrinsic enzymatic properties render transposons vulnerable to being permanently lost from the population without an active mechanism for donor site preservation. Thus, we next focused on the molecular properties of TnpB and IscB, given their frequent presence as accessory factors encoded within the same transposons.
TnpB and IscB function as RNA-guided nucleases
We hypothesized that TnpB and IscB nucleases would function with transposon-encoded ωRNAs to target the donor joint produced upon scarless transposon excision (Fig. 3a), forcing cells to survive otherwise lethal DSBs by restoring the transposon through recombination6,39. With knowledge that GstTnpA was active in mobilizing diverse IS elements, we turned our attention to reconstituting nuclease activity for the associated GstTnpB and GstIscB proteins. We used a plasmid interference assay in which successful targeting results in plasmid cleavage and loss, leading to cell lethality under antibiotic selection (Fig. 3b). We designed expression plasmids (denoted pEffector) encoding both TnpB/IscB and the corresponding ωRNA guides derived from their native G. stearothermophilus IS elements, alongside target plasmids (pTarget) containing donor joints that were bioinformatically identified and experimentally verified in TnpA excision assays (Figs. 2d and 3b). After screening various promoter combinations driving expression of the nuclease and ωRNA (Extended Data Fig. 5a), we found that GstIscB and three distinct GstTnpB homologues were highly active for RNA-guided DNA cleavage of their native donor joints (Fig. 3c,d). TnpB1 initially displayed weak activity against its native donor site (Fig. 3c,d), but further screening of lacZ-specific guide sequences uncovered robust cleavage activity at elevated temperatures (Extended Data Fig. 5f,g), a result that probably reflects the wide temperature range (30–75 °C) at which G. stearothermophilus natively grows. Notably, HpyTnpB encoded by the well-studied IS608 element was inactive when tested under standard conditions, whereas we recapitulated the recently described activity6 for DraTnpB (Extended Data Fig. 5b).
Fig. 3 |. TnpB and IscB target donor joint molecules produced by TnpA.

a, Schematic representation of four IS families (coloured rectangles), alongside homologous sites from related G. stearothermophilus strains that lack the transposon insertion. TAMs are highlighted in the donor joint sequences shown below each element. b, Schematic of E. coli-based plasmid interference assay. Protein–RNA complexes are encoded by pEffector, and targeted cleavage of pTarget results in a loss of kanamycin resistance and cell lethality on selective LB-agar plates. c, G. stearothermophilus TnpB and IscB homologues are highly active for RNA-guided DNA cleavage, as assessed by plasmid interference assays. Transformants with a targeting (T) or non-targeting (NT) ωRNA–pTarget combination were serially diluted, plated on selective media and cultured at 37 °C for 24 h. d, Quantification of the data in c, normalized to the non-targeting plasmid control for each G. stearothermophilus IS element. CFU, colony-forming units; ND, not detected. Bars indicate mean ± s.d. (n = 3). e, DNA cleavage by TnpB2 is highly sensitive to TAM mutations, as assessed by plasmid interference assays. Data were quantified and plotted as in d for the indicated TAM mutations; TTTAT denotes the wild-type TAM. f, DNA cleavage by IscB is highly sensitive to TAM mutations, as assessed by plasmid interference assays. Data were quantified and plotted as in d for the indicated TAM mutations; TTCAT denotes the wild-type TAM. g, Schematic of E. coli-based genome targeting assay, in which RNA-guided DNA cleavage of lacZ by TnpB/IscB results in cell death. Spec, spectinomycin; specR, spectinomycin resistance gene. h, TnpB2 and IscB are active for targeted genomic DNA cleavage, as assessed by genome targeting assay. Transformants with a targeting or non-targeting ωRNA were serially diluted, plated on selective media and cultured at 37 °C for 24 h. dTnpB2, D196A mutant; dIscB, D58A/H209A/H210A mutant.
An elegant feature of IS605-family elements is that the same LE-abutting TAM sequence that TnpA requires for transposon excision and integration is also required for DNA targeting and cleavage by TnpB/IscB, akin to the role of PAM sequences for CRISPR–Cas9 and CRISPR–Cas125,6,40. We systematically mutagenized the TAM and found that DNA cleavage was ablated with even single base pair changes, which would also render the site of ωRNA biogenesis at the transposon RE—where the motif differs from the cognate TAM in only two positions—completely unrecognizable (Fig. 3e,f and Extended Data Fig. 5c,d). TnpB and IscB were both also functional for genomic targeting and cleavage, and point mutations in the predicted HNH and/or RuvC nuclease domains completely ablated activity (Fig. 3g,h). Of note, a panel of three TnpB-specific ωRNAs targeting lacZ showed varying levels of activity (Extended Data Fig. 5e), suggesting additional unknown requirements that impact the efficiency of DNA targeting and cleavage.
As our initial screening of TnpB/IscB homologues involved the use of engineered expression cassettes, we further explored the targeting capabilities of a fully native transposon encoding TnpB2. We cloned ISGst3 directly from the G. stearothermophilus genome and found that TnpB2–ωRNA complexes expressed from this context were highly active in recognizing and cleaving plasmid and genomic targets in E. coli, including the empty donor joint substrate (Extended Data Fig. 5h–k). To investigate whether this native transposon remained active for transposition, we inserted tnpA upstream of tnpB to replicate the typical gene arrangement found in autonomous IS elements and then measured mobilization using a conventional mating-out assay. TnpA catalysed transposition in cis at similar levels to experiments performed in the absence of TnpB (Extended Data Fig. 4b,d), demonstrating that IS elements retain similar transposition functionality in cis and in trans, and do not exhibit appreciable differences in targeting new sites for integration in the presence or absence of a TnpB nuclease.
To investigate binding specificity, we next performed chromatin immunoprecipitation with sequencing (ChIP–seq) experiments to map all chromosomal binding sites of nuclease-dead IscB and TnpB programmed with lacZ-specific ωRNAs (Fig. 4a). The resulting data revealed strong enrichment at the on-target site and numerous off-targets (Extended Data Fig. 6a–d), and the majority of peaks shared highly conserved consensus motifs of 5′-TTCAT-3′ (IscB from ISGst6) and 5′-TTTAT-3′ (TnpB2 from ISGst3) (Fig. 4b,c), which precisely matched the TAM motifs neighbouring the native ISGst6 and ISGst3 elements, respectively (Extended Data Fig. 2a). Similar consensus motifs emerged when we tested cleavage activity in cells using pTarget libraries containing degenerate TAM sequences (Fig. 4d,e). Neither TnpB nor IscB exhibited a strong requirement for extensive complementarity within the seed sequence for the off-target sites analysed (Extended Data Fig. 6a,b), and this absence was notable in comparison to matched experiments with Cas9 and Cas12a, which were strongly dependent on 3–7 nt of PAM-adjacent sequence matching the guide RNA41 (Extended Data Fig. 6c,d). These results suggest the possibility that Cas9 and Cas12 may have evolved a greater degree of reliance on RNA–DNA complementarity for stable DNA binding, whereas IscB and TnpB may be strictly dependent on a more extensive TAM motif42,43.
Fig. 4 |. Unbiased identification of TnpB/IscB TAM specificity by ChIP–seq and library assays.

a, Schematic of ChIP–seq workflow to monitor genome-wide binding specificity of TnpB/IscB. E. coli cells were transformed with plasmids encoding catalytically inactive dTnpB2 or dIscB and a genome-targeting or non-targeting ωRNA. After induction, cells were collected, protein–DNA cross-links were immunoprecipitated, and next-generation sequencing (NGS) libraries were prepared and sequenced. b, Left, genome-wide representation of ChIP–seq data for dIscB with target site (blue triangle) shown, for targeting and non-targeting samples alongside the input control. Coverage is shown as reads per kilobase per million mapped reads (RPKM), normalized to the highest peak in the targeting sample. Right, off-target binding events were analysed by MEME-ChIP, which revealed a strongly conserved consensus motif consistent with the wild-type TAM (TTCAT) but weak seed sequence bias; part of the ωRNA guide sequence is shown below. Consensus motifs are oriented 5′ of the IS element LE. n is the number of peaks contributing to the motif; E is the E-value significance. c, Representative ChIP–seq data for dTnpB2, plotted as in b. d, Schematic of TAM library cleavage assay, in which plasmids expressing nuclease-active TnpB/IscB and an associated ωRNA (pEffector) were designed to cleave a target sequence flanked by randomized 6-mer (pTarget). Plasmid cleavage results in plasmid elimination, loss of cell viability and depletion of the particular TAM upon library sequencing. kanR, kanamycin resistance gene. e, WebLogo representation of the ten most depleted sequences upon deep sequencing of plasmid samples from the TAM library cleavage assay for TnpB2 and IscB. Consensus motifs are oriented 5′ of the IS element LE.
Together with data highlighting the importance of the TAM in TnpA-mediated excision and integration, these findings implicate the TAM as a critical hub of DNA interrogation during multiple stages of the transposon life cycle for IS200/IS605-family elements. Despite the chemically diverse reactions being catalysed during DNA transposition and DNA cleavage, TnpB/IscB nucleases and TnpA transposases were both constrained by selective pressures to faithfully recognize overlapping sequence motifs, presumably for selfish interests to promote transposon proliferation. We therefore focused our subsequent efforts on investigating how TnpA and TnpB coordinate their biochemical activities to modulate transposon maintenance and spread.
RNA-guided DSBs drive transposon retention
We hypothesized that co-expression of IscB or TnpB nucleases with compatible ωRNAs would rapidly intercept the donor joint products generated upon transposon excision by TnpA, thereby creating a targeted DSB at the site of excision. In bacteria, DSBs are typically lethal unless they are repaired via homologous recombination, often involving a sister chromosome39, and the strand-specific excision of IS200/IS605-family transposons as ssDNA would ensure that the sister chromosome produced during DNA replication can always serve as a homologous donor template. Thus, the mechanism we envisioned would be akin to the role of homing endonucleases in promoting lateral mobility through DSB-triggered recombination44, but rather than specifying new target sites for transposon insertion, TnpB/IscB would promote reinstallation of transposon copies at pre-existing donor sites.
To test this, we first generated an E. coli strain harbouring a lacZ-interrupting mini-Tn that was inserted downstream of a TnpB-compatible TAM, such that scarless excision by TnpA would result in a phenotypic switch from non-functional lacZ− (white colony phenotype) to functional lacZ+ (blue colony phenotype; Fig. 5a). We transformed strains with expression plasmids encoding TnpA (or an inactive mutant) and TnpB (or an inactive mutant), programmed with either a non-targeting ωRNA or a lacZ-targeting ωRNA designed to cleave the donor joint generated upon TnpA-mediated mini-Tn excision. After enriching for excision events by growing on MacConkey agar, we plated cells on medium containing X-gal and performed blue-white colony screening. We immediately observed the emergence of a large fraction of blue colonies in the presence of wild-type TnpA, but not a catalytically inactive mutant (Fig. 5b and Extended Data Fig. 7a), and colony PCR analysis confirmed that these colonies had indeed permanently lost the transposon at the donor lacZ locus (Fig. 5b,c). When we plated a similar population of cells onto X-gal plates that also contained kanamycin, thus selecting for the presence of the mini-Tn, blue colonies were 1,000× less abundant (Fig. 5b), confirming our earlier findings that the frequency of transposon excision at the donor site vastly exceeds the frequency of transposon integration at a new target site.
Fig. 5 |. RNA-guided nucleases promote transposon survival via recombination at donor sites.

a, Workflow to measure transposon fate in E. coli with TnpA and TnpB. A mini-Tn was inserted at a compatible TAM in lacZ, and cells were transformed with plasmids expressing wild-type or mutant TnpA and/or TnpB. White or blue colonies indicate transposon retention or excision, respectively. Kanamycin-resistant (KanR) blue colonies result from transposition to new sites. Carb, carbenicillin; Kan, kanamycin; Spec, spectinomycin. b, Bar graph showing the frequency of transposon excision for each condition, quantified by blue/white colony screening. Orange bars represent colonies that underwent transposition events. M, TnpA Y125A mutant; d, TnpB D196A mutant. Data are mean ± s.d. (n = 3). c, PCR and gel analysis of lacZ genotypes from b. Lanes ME and MU denote excised and unexcised marker controls; U, unexcised mini-Tn; E, excised mini-Tn product. d, Workflow to measure transposon recombination in E. coli with TnpA and TnpB. Native ISGst3 transposons with TnpB or both TnpA and TnpB were inserted at a compatible TAM in plasmid-encoded lacZ. Plasmids were used to transform E. coli cells harbouring a wild-type lacZ locus. RNA-guided DNA cleavage of genomic lacZ triggers recombination with the ectopic ISGst3-lacZ, leading to white colonies. Tet, tetracycline. e, TnpA and TnpB co-operate for efficient self-mobilization into a vacant donor site via recombination. Bar graph shows the plasmid transformation efficiency for each condition; white bars report colonies with a lacZ− phenotype. Active TnpA enhances survival and recombination through an unknown mechanism. Data are mean ± s.d. (n = 3). f, PCR and gel analysis of genomic lacZ genotypes, confirming parental loci or recombination products containing integrated ISGst3. g, Model for peel-and-paste, cut-and-copy transposition mechanism catalysed by TnpA and TnpB in IS200/IS605-family transposons; a similar mechanism is expected for IS elements encoding IscB. TnpA mediates scarless transposon excision, often during replication, leading to loss at one of two sister chromosomes. Target sites recognized by TnpB–ωRNA (or IscB–ωRNA) complexes are interrupted by the transposon but regenerated at the excised donor joint, leading to RNA-guided DNA cleavage. The resulting DSBs trigger recombination with a homologous chromosome, faithfully preserving the transposon at the donor site. Transposition events at new target sites produce new ωRNA guides, facilitating further transposon spread. Transposons are shown in dark green; TAMs are yellow; guide and target sequences are in blue and orange.
Co-expression of TnpB and a lacZ-specific ωRNA completely eliminated the emergence of blue colonies under otherwise identical conditions, and colony PCR confirmed that transposons were uniformly maintained at their original genomic location (Fig. 5b,c and Extended Data Fig. 7a–d). This effect was dependent on both a targeting ωRNA and an intact TnpB nuclease domain, indicating that targeted binding alone is insufficient for transposon retention at the donor site, but that targeted cleavage and local DSB generation are necessary.
Because these experiments assumed that transposon excision was occurring normally with and without TnpB, but could not directly observe consecutive loss and gain, we designed another set of experiments to more directly test the hypothesis that RNA-guided DNA cleavage of the donor joint by TnpB would trigger copying of the transposon between homologous sites via recombination. Rather than monitoring retention of the transposon at the donor site, we sought an alternative approach that would mimic the cellular state after replication-dependent excision of the ssDNA transposon from one parental strand, leading to the presence of homologous sister chromosomes with only one chromosomal copy containing the IS element. We therefore developed an assay to monitor recombination events occurring between a plasmid-encoded IS element inserted into full-length lacZ, and its corresponding lacZ donor joint site encoded in the genome (Fig. 5d). We dispensed with synthetic expression constructs for these experiments and instead used the ISGst3 element cloned directly from G. stearothermophilus gDNA, enabling native relative expression of TnpB and ωRNA, with or without TnpA, in a more biologically relevant context.
Upon E. coli transformation, TnpB and ωRNA expression should cause targeted DSBs within the genomic lacZ locus, leading to one of two potential outcomes: cell death from unresolved DNA damage, or survival via homologous recombination with the lacZ locus on the ectopic plasmid, effectively copying the ISGst3 element and disrupting the target site. We scored these outcomes by quantifying the number of surviving colonies that were lacZ+ (uncleaved or unmutated, blue colony phenotype) or lacZ− (recombination products, white colony phenotype) (Fig. 5d).
Consistent with our previous findings, we observed a reduction of approximately 500× in cell survival after transformation with the wild-type ISGst3 element, highlighting the selective pressure imposed by ωRNA-guided TnpB, and this effect was ablated with inactivating nuclease mutations (Fig. 5e and Extended Data Fig. 7e). Notably, an autonomous element that also encoded TnpA led to a 50× increase in colony counts, and 98% of the surviving colonies were lacZ−, indicating a disruption of the target site; this effect was dependent on the catalytic activity of TnpA (Fig. 5e and Extended Data Fig. 7e). To verify that genomic lacZ disruption resulted from insertion of the plasmid-encoded ISGst3 element, we performed colony PCR and long-read Nanopore sequencing of multiple isolates, which revealed the occurrence of scarless recombination events (Fig. 5f).
Collectively, these results reveal the potent roles of transposon-encoded nucleases in both preserving and mobilizing themselves into homologous (vacant) target sites, through the combined action of RNA-guided DNA cleavage and ensuing recombination (Fig. 5g). Although our experiments focused on one specific TnpB homologue and IS element, the same mechanism would apply to IscB nucleases as well as other minimal transposons (for instance, PATEs) that encode ωRNAs but are acted upon by a nuclease in trans. RNA-guided nuclease activity is thus essential to avoid the precarious transposon loss that is otherwise unavoidable during TnpA-mediated excision. Our experiments also revealed an unexpected role of TnpA in facilitating recombination-based installation of the transposon back into the original donor site, though more experiments will be necessary to decipher the specific mechanism. Together, these observations highlight how mutually beneficial co-evolution of TnpA transposases and RNA-guided TnpB/IscB nucleases aided in the survival and dissemination of these transposable elements. (Fig. 5g).
Discussion
Prokaryotic insertion sequences are among the simplest transposable elements, containing only the gene or genes required for transposition. Paradoxically, however, large families of abundant IS elements carry genes that do not encode active transposases, but rather encode diverse accessory genes that provide myriad selective advantages for survival of the transposable element, including resolution systems, antibiotic resistance genes and pathogenicity functions1. In the case of IS200/IS605-family elements, the presence of a pervasive accessory gene accompanying tnpA has long been observed and defined with variable nomenclatures (tnpB, orfB, tlpB and so on), but aside from experiments demonstrating that these gene products are not required for transposition24,28,45, the molecular function of TnpB-family proteins has remained elusive. Two recent studies provided critical new insights into the biochemical activities of TnpB and the related accessory protein IscB, demonstrating that both enzymes use transposon-encoded guide RNAs (ωRNAs) to catalyse RNA-guided DNA cleavage5,6. Yet, despite revealing insights into the evolutionary origins of CRISPR–Cas9 and CRISPR–Cas12 effector nucleases, these studies did not explore the role of TnpB/IscB in the context of transposition.
Our results reveal that RNA-guided DNA endonucleases encoded within IS200/IS605-family transposons direct site-specific DSBs and homologous recombination, ensuring the retention and proliferative spread of these elements. This mechanism is essential to counteract transposon loss by the TnpA transposase. IS200/IS605-family transposons rely on ssDNA secondary structures for efficient excision23, which occur during host replication37,46 and DSB repair38. As a result, donor templates containing the transposon often remain available on the sister chromosome for recombination, and the scarless excision and integration products ensure accurate homology. This pathway is crucial for preserving the transposon at the original donor site and may also facilitate its spread to homologous sequences lacking insertion events, including sister chromosomes, horizontally transferred elements or homologous chromosomes in eukaryotes. This pathway probably increases the transposon copy number over many generations, as new donor substrates become available to be mobilized in trans. Indeed, the genome of G. stearothermophilus contains nearly 50 elements that lack tnpA but that we predict could be mobilized in trans, based on sequence conservation of the transposon ends. The very presence of PATEs, which often encode ωRNAs that would themselves need to be acted on in trans by TnpB, supports the modularity of these molecular components1,5. It is noteworthy, however, that IS transposon expansion also increases the diversity of potential genome-targeting ωRNAs, imposing an additional mutational burden on the cell due to increased chances of off-target cleavage activity.
Class II (DNA) transposons have evolved diverse mechanisms of mobilization, which differ critically in the fate of DNA at the donor site, the form of the mobile DNA itself, and the chemical mechanism by which the mobile DNA is integrated at the new target site. Conventional cut-and-paste transposons are excised as dsDNA molecules that can be reintegrated elsewhere in the genome, leaving behind DSBs at the donor site that must be repaired for cell survival47. Certain cut-and-paste transposons, such as Tn5 and Tn10, have evolved mechanisms to mobilize preferentially during DNA replication48,49. In copy-and-paste transposons such as Tn3, the donor and target DNA sequences become linked by a single-strand transfer, creating a forked structure that is acted upon by DNA polymerase to generate a double-stranded cointegrate; dedicated resolution systems can then resolve this cointegrate, resulting in a new copy of the transposon without loss of the original transposon50. Last but not least, peel-out-paste-in transposons such as IS200/IS605 excise as ssDNA molecules from only one strand of the donor molecule, concomitant with rejoining of the flanking sequences to create a scarless donor joint that lacks the transposon2. Despite these distinct lifestyles, evolutionary survival of the selfish element ultimately requires that the rate of transposon loss—whether by permanent excision, inactivation or silencing—is compensated by transposon gain.
Our experiments demonstrate the TnpA catalyses transposon excision (that is, loss) at rates that exceed transposon integration (that is, gain) by orders of magnitude, which would be seemingly incompatible with the long-term survival of these elements. Yet the powerful combination of TnpA and TnpB/IscB-family nucleases, which evolved to use localized guide RNAs to force recombination at the sites of excision and integration, offers a unique solution to this problem by directing peel-and-paste, cut-and-copy transposition: TnpA-mediated excision/integration coupled with TnpB/IscB-mediated DNA cleavage (Fig. 5g). Our finding that TnpA stimulates recombination beyond the levels achieved by TnpB alone suggests a further synergistic effect (Fig. 5e), potentially related to its ability to interact with Holliday junction-like structures and the beta clamp37. The association of TnpB-family nucleases with diverse transposase families26, alongside the potential for transposon homing or mobilization to homologous target sites, presents a compelling avenue for future research that could provide valuable tools for enhancing site-specific recombination.
There are interesting parallels between the transposon retention mechanism by TnpB/IscB-family nucleases, and the mobilization mechanism of group I introns that encode site-specific homing endonucleases44. Homing endonucleases target conserved sites for DSB formation, thereby promoting a form of transposition that relies entirely on recombination to copy the element from the donor to the target site. IS200/IS605-family elements have instead harnessed programmable nucleases as ‘adaptive’ site-specific enzymes that target the empty donor site programmed after each transposition event, to selfishly bias the inheritance and retention of the transposable element for its continued spread. Of note, IS200/IS605-family elements themselves are sometimes encompassed within group I introns45, highlighting the diversity of these elements and their associated gene products. Collectively, these convergent nuclease–recombination-based strategies can achieve super-Mendelian inheritance by increasing the frequency of alleles that carry the mobile element.
The combination of DNA-guided transposition activity (TnpA) and RNA-guided nuclease activity (TnpB/IscB) has facilitated the pervasive spread of IS200/IS605-family elements throughout bacteria and archaea. However, TnpB homologues have also been identified within diverse transposable elements that mobilize DNA through a variety of different mechanisms. Furthermore, these homologues have been identified in all domains of life, including higher eukaryotes, suggesting that the very biochemical property of RNA-guided DNA targeting and cleavage is not restricted to CRISPR-associated enzymes (Cas9 and Cas12) and IS element-associated enzymes (IscB and TnpB), but includes nucleases that are broadly present in Bacteria, Archaea and Eukarya26,27. In this regard, CRISPR–Cas systems represent just one powerful example whereby RNA-guided DNA targeting nucleases within the TnpB superfamily have been exapted and repurposed for antiviral defence. The diverse and widespread phylogenetic distribution of these enzymes strongly suggests that other cellular pathways will be identified where RNA-guided targeting enzymes were similarly co-opted for new molecular functions.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
IscB and TnpB detection and database curation
Homologues of IscB proteins were comprehensively detected using the amino acid sequence of a Ktedonobacter racemifer homologue (NCBI accession: WP_007919374.1) as the seed query in a JackHMMER part of the HMMER suite (v3.3.2). To minimize false homologues, a conservative inclusion and reporting threshold of 10−30 was used in the iterative search against the NCBI NR database (retrieved on 11 June 2021), resulting in 5,715 hits after convergence. These putative homologues were then annotated to profiles of known protein domains from the Pfam database (retrieved on 29 June 2021) using hmmscan with an E-value threshold of 10−5. Proteins that did not contain the RRXRR, RuvC, RuvC_III or the RuvX domain were discarded. Although the HNH domain was annotated, proteins without the HNH were not removed. The variation in the presence of the HNH domain was preserved to better represent the natural diversity of IscBs. From the remaining set, proteins that were less 250 amino acids in size were removed to eliminate partial or fragmented sequences, resulting in a database of 4,674 non-redundant IscB homologues. Contigs of all putative iscB loci were retrieved from NCBI for downstream analysis using the Bio.Entrez package.
TnpB homologues were comprehensively detected similarly to IscB, using both the H. pylori (HpyTnpB) amino acid sequence (NCBI accession: WP_078217163.1) and the G. stearothermophilus (GstTnpB2) amino acid sequence (NCBI accession: WP_047817673.1) as seed queries for two independent, iterative jackhammer searches against the NR database, with an inclusion and reporting threshold of 10−30. The union of the two searches were taken, and proteins that were less than 250 amino acids in size were removed to trim partial or fragmented sequences, resulting in a database of 95,731 non-redundant TnpB homologues. Contigs of all putative tnpB loci were retrieved from NCBI for downstream analysis using the Bio.Entrez package.
Phylogenetic analyses
IscB protein sequences were clustered with at least 95% length coverage and 95% alignment coverage using CD-HIT51 (v4.8.1). The clustered representatives were taken and aligned using MAFFT52 (v7.508) with the E-INS-I method for four rounds. Post-alignment cleaning consisted of using trimAl53 (v1.4.rev15) to remove columns containing more than 90% of gaps and manual inspection. The phylogenetic tree was created using IQ-Tree 254 (v2.1.4) with the WAG model of substitution. Branch support was evaluated with 1,000 replicates of SH-aLRT, aBayes, and ultrafast bootstrap support from the IQ-Tree package. The tree with the highest maximum-likelihood was used as the reconstruction of the IscB phylogeny.
Putative TnpB sequences were clustered by 50% length coverage and 50% alignment coverage using CD-HIT51. Similar to IscB, the clustered representatives were taken and aligned using MAFFT52 with the E-INS-I method for four rounds. Post-alignment cleaning consisted of using trimAl53 to remove columns containing more than 90% of gaps and manual inspection. The phylogenetic tree was created using IQ-Tree 254 with the WAG model of substitution. Branch support was evaluated with 1,000 replicates of SH-aLRT, aBayes, and ultrafast bootstrap support from the IQTREE package. The tree with the highest maximum-likelihood was used as the reconstruction of the TnpB phylogeny.
ωRNA covariation analyses
Initial searches of the Rfam database indicated a potential non-coding RNA belonging to the HNH endonuclease-associated RNA and ORF (HEARO) RNA (RF02033)29,55. A covariance model of HEARO RNA (retrieved 24 June 2021) was initially used to discover all HEAROs within our curated IscB-associated contig database using cmsearch from the Infernal package (v1.1.4)56. A liberal minimum bit score of 15 was used in an attempt to capture distant or degraded HEAROs, and the identification of a HEARO as a putative ωRNA was supported by its proximity, orientation and relative location to the nearest identified IscB ORF. The remaining hits were considered ωRNAs if they were upstream of an IscB ORF and within 500 bp or overlapping with the nearest IscB ORF. After inspecting the RF02033 model, it appeared to lack additional structural elements located downstream. To address this, the ωRNA boundaries were refined and used to generate a more accurate, comprehensive covariance model. Hits to the RF02033 model described above were retrieved, expanded 200 bp downstream, and clustered by 80% length coverage and 80% alignment coverage using CD-HIT51. CMfinder57 (v0.4.1.9) was then used with recommended parameters to discover new motifs de novo. Additional structures were discovered and present in over 80% of the expanded sequences. This covariance model was used to expand the 3′ coordinates of previously identified ωRNAs to encompass the second stem-loop using cmsearch on expanded ωRNAs. These refined ωRNA boundaries and sequences were then used to create a new ωRNA model. The refined ωRNAs were clustered by 99% length coverage and 99% alignment coverage using CD-HIT to remove duplicates. A structure-based multiple alignment was then performed using mLocARNA58 (v1.9.1) with the following parameters:
--max-diff-am 25 --max-diff 60 --min-prob 0.01 --indel −50 --indel-open −750 --plfold-span 100 --alifold-consensus-dp
The resulting alignment with structural information was used to generate a new ωRNA covariance model with the Infernal suite, refined with Expectation-Maximization from CMfinder, and verified with R-scape at an E-value threshold of 10−5. The resulting ωRNA covariance model was used with cmsearch to discover new ωRNAs within our curated IscB-associated contig database. The resulting sequences were aligned to generate a new CM model that was used to again search our IscB-associated contig database. This process was repeated three times for our final, IscB-associated ωRNA model.
While covariance models of TnpB-associated ωRNAs were available through Rfam (RF03065 and RF02998), these models appeared to only include a very small subset of TnpB-associated ωRNA and contained very few hits. Based on small RNA-seq analyses that suggested a non-coding RNA often overlapped with the TnpB ORF and extended into the RE boundary of the IS element, we extracted sequences 150 bp downstream of the last nucleotide of the TnpB ORF to define the RE and transposon boundaries. The ~150-bp sequences were clustered by 99% length coverage and 99% alignment coverage using CD-HIT54 to remove duplicates. The remaining sequences were then clustered again by 95% length coverage and 95% alignment coverage using CD-HIT54. This was done to identify clusters of sequences that were closely related but not identical, as expected of IS elements that have recently mobilized to new locations. For the 300 largest clusters, which all had a minimum of 10 sequences, MUSCLE59 (v3.8.1551) with default parameters was used to align each cluster of sequences. Then, each cluster alignment was manually inspected for the boundary between high conservation and low conservation, or where there was a stark drop-off in mean pairwise identity over all sequences. This point was annotated for each cluster as the putative 3′ end of the IS elements. If there was no conservation boundary, sequences in these clusters were expanded by another 150 bp, in order to capture the transposon boundaries, and realigned. The consensus sequence of each alignment (defined by a 50% identity threshold up until the putative 3′ end) was extracted, and rare insertions that introduced gaps in the consensus were manually removed. With the 3′ boundary of the IS element, and thus the 3′ boundary of the TnpB ωRNA properly defined, covariance models of the TnpB ωRNA could be built.
From a randomly selected member of each of the 300 clusters, a 250-bp window of sequence 5′ of the 3′ end of the ωRNA was extracted. A structure-based multiple sequence alignment was then performed using mLocARNA and used to generate a TnpB-specific ωRNA covariance model with Infernal, refined with CMfinder, and verified with R-scape60 at an E-value threshold of 10−5. This was iterated twice to generate the final generic model of TnpB-associated ωRNA. In addition, more localized ωRNA covariance models were created for each of the four TnpB homologues used in this study (GstTnpB1–4). Each protein was used as a seed query in a phmmer (v3.3.2) search against the NR database, with an inclusion and reporting threshold of 10−30 to identify close relatives of each protein. The steps described above were used to define transposon boundaries and generate ωRNA models using sequences identified in our phmmer search.
TnpA detection and autonomous element identification
For both IscB- and TnpB-associated contigs, TnpA was detected using the Pfam Y1_Tnp (PF01797) for a hmmsearch from the HMMR suite (v3.3.2), with an E-value threshold of 10−4. This search was performed independently on both the curated coding sequences (CDSs) of each contig from NCBI and on the ORFs predicted by Prodigal61 with default settings. The union of these searches was used as the final set of detected TnpA proteins. IS elements that encoded IscB homologues within 1,000 bp of a detected TnpA, or that encoded TnpB homologues within 10,000 bp of a detected TnpA, were defined as autonomous. Analyses which uncovered association with serine resolvases (PF00239) were performed with the same parameters mentioned above.
Orientation bias analysis
The closest NCBI-annotated or predicted CDS upstream of each transposon-encoded gene (tnpB, iscB or the IS630 transposase) was retrieved and analysed relative to the gene itself. Initially, the metadata for every NCBI-annotated CDS within contigs containing these genes (tnpB, iscB or the IS630 transposase) were retrieved, including coordinates and strandedness. Using this information, the closest upstream CDS was identified for each gene based on distance. Then, the annotated orientation of the closest upstream CDS was compared to the annotated orientation of the respective transposon-encoded gene (tnpB, iscB or the IS630 transposase), to determine whether they were matching. This analysis was performed for gene/CDS pairs at all distances between 0 and 1,000 bp upstream (5′) of the transposon-encoded gene ORF, where 0 bp was defined as overlapping, using a custom Python script.
Transposon boundary and TAM/TEM motif determination for G. stearothermophilus IS elements
IS200/IS605-family elements found in G. stearothermophilus strain DSM 458 (NCBI accession: NZ_CP016552.1) that encoded iscB or tnpB were identified by a protein homology-based search, as described above. Initial identification of transposon boundaries was performed by multiple sequence alignment of each unique tnpB or iscB gene using DNA sequences flanking the TnpB/IscB ORF, and alignments were performed using the MUSCLE (5.1) PPP algorithm in Geneious (2023.0.1). To build covariance models of the transposon ends, cmfinder was used to detect structural motifs for each end of ISGst2, ISGst3 and ISGst6 (LE and RE separately) and produce an alignment based on secondary structure. This model was then used for further searches (CMSearch), to identify structurally similar positions within the genome of G. stearothermophilus strain DSM 458. All transposon ends were initially paired with the most similar query end and then manually curated, to ensure that each LE and RE within a given pair were correctly positioned relative to each other. This analysis identified several PATE-like elements lacking any protein-coding genes, and a total of 47 IS elements were identified with similar LE and RE sequences. Fifty base pairs upstream and downstream were extracted and aligned using the MUSCLE (5.1) PPP algorithm in Geneious and trimmed using trimAl (v1.4.rev15), to capture transposon boundaries and identify TAM/TEM motifs based on previous literature describing the location of these essential motifs34. Transposon DNA guide regions were predicted based on structural similarities to the transposon ends of H. pylori IS608 and covarying mutations at those predicted locations. TAM motifs, which function as target sites for the transposon insertion event, were confirmed by blastn analysis of DNA sequences flanking predicted transposon boundaries to the nucleotide or whole-genome database. Phylogenetic trees of transposon ends were built using FastTree62 (2.1.11) with default parameters.
RNA-seq
G. stearothermophilus strain DSM 458 was obtained from DSMZ and cultured at 55 °C in liquid nutrient broth (3 g l−1 beef extract and 5 g l−1 peptone) and grown to saturation. Cells were pelleted and RNA extracted using a hot phenol extraction. Illumina sequencing libraries were generated using the NEBNext Small RNA Library Prep kit, with the following modifications: total RNA was diluted in FastAP buffer (Thermo Fisher Scientific) supplemented with SUPERase•In RNase Inhibitor (Thermo Fisher Scientific) and fragmented by incubating at 92 °C for 1.5 min. The fragmented RNA was then DNase treated and dephosphorylated with TURBO DNase (Thermo Fisher Scientific) and FastAP (Thermo Fisher Scientific). Next, 5′ end phosphorylation and 3′ end repair were carried out using T4 PNK (NEB) in 1× T4 DNA ligase buffer (NEB). The RNA was column-purified using RNA Clean and Concentrator-5 (Zymo), and the concentration was determined using the DeNovix RNA Assay. Illumina adapter ligation and cDNA synthesis were performed using the NEBNext Small RNA Library Prep kit, with substitution of a custom i7 adapter containing a 10-mer unique molecular identifier (UMI). Dual-index barcodes were added by PCR amplification (10 cycles), and the cDNA libraries were purified using the Monarch PCR and DNA Cleanup Kit (NEB). High-throughput sequencing was performed on an Illumina MiniSeq in paired-end mode, with 75 cycles per end with automated demultiplexing and adapter trimming.
RNA-seq analysis of G. stearothermophilus DSM 458
RNA-seq data were processed using cutadapt v4.263 to trim low-quality ends from reads and exclude reads shorter than 20 bp. UMIs were extracted from read 2 using UMI-tools v1.1.464 before mapping reads to the reference genome (NZ_CP016552.1) using bwa-mem2 v2.2.165 with default parameters. Mapped reads were sorted and indexed using SAMtools v1.1766 prior to UMI deduplication with UMI-tools. Coverage tracks were generated with bamCoverage v3.5.167 using a bin size of 1 and extending reads to fragment size. Coverage over selected genomic regions was visualized in IGV.
Small RNA-seq analysis of G. stearothermophilus ATCC 7953
Small RNA-seq reads were retrieved from NCBI SRA database under accession SRX3260293. Reads were downloaded using the SRA toolkit (2.11.0) and mapped to genomic regions encoding G. stearothermophilus IscB and TnpB homologues used in this study, using G. stearothermophilus strain ATCC 7953 (GCA_000705495.1) from which small RNA-seq data derive33. Reads were mapped using Geneious RNA assembler at medium sensitivity and visualized using IGV68.
Plasmid construction
All strains and plasmids used in this study are described in Supplementary Tables 1 and 2, respectively, and a subset is available from Addgene. In brief, genes encoding TnpA, TnpB and IscB homologues from G. stearothermophilus, H. pylori and D. radiodurans (Supplementary Table 3) were synthesized by GenScript, along with mini-Tn elements containing a chloramphenicol resistance gene. To generate mini-Tn plasmids, we cloned a gene fragment encoding the transposase (TnpA) downstream of a lac and T7 promoter, along with a mini-Tn cassette. The mini-Tn cassette comprised 50-bp genomic DNA sequences flanking the transposon ends derived from native elements, as well as a chloramphenicol resistance gene. To generate pEffector plasmids, gene fragments (Genscript) of ωRNA encoded downstream of T7 promoter, along with tnpB or iscB also encoded downstream of T7 promoter, were cloned into pCDF-Duet1 vectors at PfoI and Bsu36I sites. Oligonucleotides containing J23-series promoters were cloned into SalI and KpnI sites, replacing the T7 promoter for ωRNA expression, or into PfoI–XhoI sites, replacing the T7 promoter for tnpB expression. pTarget plasmids were generated using a minimal pCOLADuet-1, generated by around-the-horn PCR, to create a minimal pCOLADuet-1 containing only the ColA origin of replication and kanamycin resistance gene. This vector was then used to generate pTargets encoding 45-bp target sites by around-the-horn PCR. Derivatives of these plasmids were cloned using a combination of methods, including Gibson assembly, restriction digestion-ligation, ligation of hybridized oligonucleotides and around-the-horn PCR. Plasmids were cloned, propagated in NEB Turbo cells (NEB), purified using Miniprep Kits (Qiagen) and verified by Sanger sequencing (GENEWIZ).
Recombineering
Lambda Red (λ-Red) recombination was used to generate genomically integrated mini-Tn cassettes. In brief, E. coli strain MG1655 (sSL0810) was transformed with pSIM6 (pSL2684) carrying a temperature-sensitive vector encoding λ-Red recombination genes, to generate strain sSL2681, and cells were made electrocompetent using standard methods. Fragments for recombineering were generated using standard PCR amplification with primers to append 50-bp overhangs homologous to the sites of integration. PCR fragments were gel-extracted and used to electroporate sSL2681, and cells were recovered for 24 h in LB medium. Cells were spun down and plated onto LB-agar containing kanamycin (50 μg ml−1) to select for mini-Tn cassette integration. Single colonies were isolated and confirmed to contain a genomically integrated mini-Tn within the lacZ locus by colony PCR and Sanger sequencing.
Transposon excision assays
For each excision experiment involving a plasmid-based IS element, a single plasmid encoding TnpA and a chloramphenicol resistance gene-containing mini-Tn IS element was used to transform E. coli strain MG1655. Cultures were grown overnight at 37 °C on LB-agar under antibiotic selection (100 μg ml−1 carbenicillin, 25 μg ml−1 chloramphenicol). Next, three colonies were picked from each agar plate and used to inoculate 5 ml LB supplemented with 0.05 mM IPTG and antibiotic for only the backbone marker (100 μg ml−1 carbenicillin). The liquid cultures were incubated at 37 °C for 24 h. Cell lysates were generated, as described previously18. In brief, the optical density at 600 nm was measured for liquid cultures. Approximately 3.2 × 108 cells (equivalent to 200 μl of cultures with an optical density at 600 nm (OD600) = 2.0) were transferred to a 96-well plate. Cells were pelleted by centrifugation at 4,000g for 5 min and resuspended in 80 μl of H2O. Next, cells were lysed by incubating at 95 °C for 10 min in a thermal cycler. The cell debris was pelleted by centrifugation at 4,000g for 5 min, and 10 μl of lysate supernatant was removed and serially diluted with 90 μl of H2O to generate 10- and 100-fold lysate dilutions for PCR and qPCR analyses.
IS element excision from the plasmid backbone was detected by PCR using OneTaq 2X Master Mix with Standard Buffer (NEB) and 0.2 μM primers, designed to anneal upstream and downstream of the IS element. PCR reactions contained 0.5 μl of each primer at 10 μM, 12.5 μl of OneTaq 2X Master Mix with Standard Buffer, 2 μl of 100-fold diluted cell lysate serving as template, and 9.5 μl of H20. The total volume per PCR was 25 μl. PCRs were performed in a BioRad T100 thermal cycler using the following thermal cycling parameters: DNA denaturation (94 °C for 30 s), 35 cycles of amplification (denaturation: 94 °C for 20 s, annealing: 52 °C for 20 s, extension: 68 °C for 30 s), followed by a final extension (68 °C for 5 min). Products were resolved by 1.5% agarose gel electrophoresis and visualized by staining with SYBR Safe (Thermo Fisher Scientific). IS element excision events were confirmed by Sanger sequencing of gel-extracted, column-purified (Qiagen) PCR amplicons (GENEWIZ/Azenta Life Sciences).
For excision events involving genomically integrated IS elements, lysates were prepared as described above but collected from LB-agar containing carbenicillin (100 μg ml−1), spectinomycin (100 μg ml−1) and X-gal (200 μg ml−1) in transposition assays combining TnpA and TnpB, as described below. PCRs were performed in a BioRad T100 thermal cycler using the following thermal cycling parameters: DNA denaturation (94 °C for 30 s), 26 cycles of amplification (denaturation: 94 °C for 20 s, annealing: 52 °C for 20 s, extension: 68 °C for 75 s), followed by a final extension (68 °C for 5 min). Products were resolved by 1.5% agarose gel electrophoresis and visualized by staining with SYBR Safe (Thermo Fisher Scientific). IS element excision events were confirmed by Sanger sequencing of gel-extracted, column-purified (Qiagen) PCR amplicons (GENEWIZ/Azenta Life Sciences).
qPCR measurements of IS element excision
IS element excision frequency from a plasmid backbone was detected by qPCR using SsoAdvanced Universal SYBR Green Supermix. qPCR analyses (Extended Data Fig. 3c–e) were performed using a donor joint-specific primer along with a flanking primer designed to amplify only the excision product; genome-specific primers for relative quantification were designed to amplify the E. coli reference gene, rssA. 10 μl qPCR reactions containing 5 μl of SsoAdvanced Universal SYBR Green Supermix, 2 μl of 2.5 μM primer pair, 1 μl H2O, and 2 μl of tenfold-diluted lysate were prepared as described for transposon excision assays. Reactions were prepared in 384-well clear/white PCR plates (BioRad), and measurements were performed on a CFX384 RealTime PCR Detection System (BioRad) using the following thermal cycling parameters to selectively amplify excision products: polymerase activation and DNA denaturation (98 °C for 2.5 min), 40 cycles of amplification (98 °C for 10 s, 62 °C for 20 s) and terminal melt-curve analysis (65–95 °C in 0.5 °C per 5 s increments).
To confirm the sensitivity of qPCR-based measurements from plasmid-encoded mini-Tn substrates, we prepared lysates from cells harbouring a plasmid containing a mock excised mini-Tn substrate (pSL4826) and a plasmid containing the mini-Tn but lacking an active TnpA transposase required for excision (pSL4735). We simulated variable IS element excision frequencies across five orders of magnitude (ranging from 0.002% to 100%) by mixing cell lysates from the control strain and the IS-encoding strain in various ratios. These measurements demonstrated accurate detection of excision products in genomic IS element excision assays in vivo to a frequency of 0.001 (Extended Data Fig. 3d).
Similarly, IS element excision frequencies of genomically integrated mini-Tn substrates were quantified by qPCR using SsoAdvanced Universal SYBR Green Supermix (BioRad) (Extended Data Fig. 7b–d). Cells were collected from LB medium containing carbenicillin (100 μg ml−1), spectinomycin (100 μg ml−1) and X-gal (200 μg ml−1), as described above. qPCR analyses were performed using transposon-flanking and genome-specific primers. Transposon-flanking primers were designed to amplify an approximately 209-bp fragment upon excision; an unexcised product would yield a 1,661-bp unexcised fragment. A separate pair of genome-specific primers was designed to amplify an E. coli reference gene (rssA) for normalization purposes (Supplementary Table 4). Ten μl qPCR reactions containing 5 μl of SsoAdvanced Universal SYBR Green Supermix, 2 μl of 2.5 μM primer pair, 1 μl H2O and 2 μl of tenfold-diluted lysate were prepared, as described for transposon excision assays. Reactions were prepared in 384-well clear/white PCR plates (BioRad), and measurements were performed on a CFX384 RealTime PCR Detection System (BioRad) using the following thermal cycling parameters to selectively amplify excision products: polymerase activation and DNA denaturation (98 °C for 2.5 min), 40 cycles of amplification (98 °C for 10 s, 60 °C for 20 s) and terminal melt-curve analysis (65–95 °C in 0.5 °C per 5 s increments).
To confirm the sensitivity of qPCR-based measurements from genomically integrated mini-Tn, we prepared lysates from a control MG1655 strain, and a strain containing a genomically encoded IS element that disrupts the lacZ gene. Similar to the plasmid-based assay (Extended Data Fig. 3d), we simulated variable IS element excision frequencies across five orders of magnitude (ranging from 0.002% to 100%) by mixing cell lysates from the control strain and the IS-encoding strain in various ratios, and showed that we could accurately detect excision products in genomic IS element excision assays in vivo to a frequency of 0.001 (Extended Data Fig. 7d).
Mating-out assays
sSL1592 (a gift from J. E. Peters) harbours a mini-F plasmid derivative with an integrated spectinomycin cassette. This strain was transformed with either pDonor1, a plasmid carrying a mini-Tn harbouring a kanamycin marker and either GstTnpA (pSL4245) or catalytically inactive GstTnpA (pSL4974); or pDonor2, a plasmid carrying ISGst3 with various combinations of active GstTnpA and GstTnpB2 (pSL5492–pSL5495). Cells were selected on LB medium containing spectinomycin (100 μg ml−1), carbenicillin (100 μg ml−1) and kanamycin (50 μg ml−1) to generate a donor strain. Three independent colonies were inoculated in liquid LB medium containing spectinomycin (100 μg ml−1), carbenicillin (100 μg ml−1), kanamycin (50 μg ml−1) and 0.05 mM IPTG to induce expression of TnpA for 12 h at 37 °C. In parallel, the recipient strain harbouring genomically encoded resistance for rifampicin and nalidixic acid were grown in liquid LB medium containing rifampicin (100 μg ml−1) and nalidixic acid (30 μg ml−1) for 12 h at 37 °C. Cells were diluted 100-fold into fresh liquid LB media with respective antibiotics and grown for 2 h to about 0.5 OD. Cells were then washed with H2O and mixed at a concentration of 5 × 107 for both donor and recipient cells, and plated onto solid LB-agar medium with no antibiotic selection. Cells were grown for 20 h at 37 °C, scraped off plates, and resuspended in H2O. Cells were then serially diluted and plated onto LB medium containing rifampicin (100 μg ml−1), nalidixic acid (30 μg ml−1), spectinomycin (100 μg ml−1) and kanamycin (50 μg ml−1) to monitor transposition. In addition, cells were also plated to rifampicin (100 μg ml−1), nalidixic acid (30 μg ml−1) and spectinomycin (100 μg ml−1), to determine the entire transconjugant population. The frequency of transposition was calculated by taking the number of colonies that exhibited NalR + RifR + SpecR + KanR phenotype (that is, transposition-positive), divided by the number of transconjugants that exhibited a NalR + RifR + SpecR phenotype. Transconjugants showing resistance to nalidixic acid, rifampicin, spectinomycin and kanamycin were isolated, and DNA was then isolated using the Zymo Research ZR BAC DNA miniprep kit and sequenced using Nanopore long-read sequencing (Plasmidsaurus). Reads were analysed in Geneious Prime (2023.0.1) using a custom blast database to identify reads containing mini-Tn and flanking mini-F plasmid sequences. Insertion events were aligned to the mini-F-plasmid reference to identify sites of integration.
Plasmid interference assays
Plasmid interference assays were performed in E. coli BL21 (DE3) (Fig. 3c,f and Extended Data Fig. 5a,b,d,g) or E coli strain K-12 substrain MG1655 (sSL0810) for all other experiments. For Fig. 3c (TnpB homologues), BL21 (DE3) cells were transformed with pTarget plasmids, and single colony isolates were selected to prepare chemically competent cells. Four hundred nanograms of pEffector plasmids were then delivered via transformation. After 3 h, cells were spun down at 4,000g for 5 min and resuspended in 20 μl of H2O. Cells were then serially diluted (10×) and transferred to LB medium containing spectinomycin (100 μg ml−1), kanamycin (50 μg ml−1) and 0.05 mM IPTG, and grown for 24 h at 37 °C. For all remaining spot assays using MG1655 strains, chemically competent cells were first prepared with pEffector plasmid and then transformed with 400 ng of pTarget plasmids. After 3 h, cells were spun down at 4,000g for 5 min and resuspended in 20 μl of H2O. Cells were then serially diluted (10×) and transferred to LB medium containing spectinomycin (100 μg ml−1), kanamycin (50 μg ml−1) and 0.05 mM IPTG, and grown for 14 h at 37 °C. Plates were imaged in an Amersham Imager 600.
Quantification of plasmid interference assays was performed by determining the number of colony-forming units (CFU) following transformation. Cells were first transformed with pEffector plasmids and prepped as chemically competent cells for a second round of transformation with 200 ng of pTarget. Cells were then spun down at 4000 g for 5 min and resuspended in 100 μl H2O. Cells were then serially diluted and plated onto LB-agar medium containing spectinomycin (100 μg ml−1) and kanamycin (50 μg ml−1). 0.05 mM IPTG was added to the medium when a T7 promoter was used. CFUs were counted following 24 h of growth at 37 °C. Frequencies were normalized relative to a non-targeting guide RNA control condition.
Genome targeting and cell-killing assays
Cell-killing assays via genomic targeting with TnpB or IscB (Fig. 3h and Extended Data Fig. 5e,k) were performed by transforming E. coli strain K-12 substrain MG1655 (sSL0810) with spectinomycin or tetracycline-resistant plasmids that constitutively expressed TnpB/IscB and either genome-targeting or non-targeting guide RNAs; experiments presented in Extended Data Fig. 5g used E. coli strain BL21 (DE3). Cells were transformed with 400 ng plasmid, recovered for 2 h in LB, spun down at 4,000g for 5 min, and then resuspended in 20 μl of H2O. Cells were then serially diluted (10×), plated onto LB-agar medium containing either spectinomycin (100 μg ml−1) or tetracycline (10 μg ml−1), and grown for 24 h at 37 °C.
ChIP–seq experiments and library preparation
ChIP–seq experiments were generally performed as described previously69. The following active site mutations were introduced to inactivate the endonuclease domains of the respective 3×FLAG-tagged proteins: GstIscB (D87A, H238A, H239A); GstTnpB (D196A); SpyCas9 (D10A, H840A); AsCas12a (D908A). E. coli BL21(DE3) cells were transformed with a single plasmid encoding the catalytically inactive effector and either a lacZ-targeting ωRNA or non-targeting ωRNA. After incubation for 16 h at 37 °C on LB-agar plates with antibiotic (200 μg ml−1 spectinomycin), cells were scraped and resuspended in 1 ml of LB. The OD600 was measured, and approximately 4.0 × 108 cells (equivalent to 1 ml with an OD600 of 0.25) were spread onto two LB-agar plates containing antibiotic (200 μg ml−1 spectinomycin) and supplemented with 0.05 mM IPTG. Plates were incubated at 37 °C for 24 h. All cell material from both plates was then scraped and transferred to a 50-ml conical tube.
Cross-linking was performed by mixing 1 ml of formaldehyde (37% solution; Thermo Fisher Scientific) with 40 ml of LB medium (about 1% final concentration) followed by immediate resuspension of the scraped cells by vortexing and 20 min of gentle shaking at room temperature. Cross-linking was stopped by the addition of 4.6 ml of 2.5 M glycine (about 0.25 M final concentration), followed by 10 min incubation with gentle shaking. Cells were pelleted at 4 °C by centrifuging at 4,000g for 8 min. The following steps were performed on ice using buffers that had been sterile-filtered: the supernatant was discarded, and the pellets were fully resuspended in 40 ml TBS buffer (20 mM Tris-HCl pH 7.5, 0.15 M NaCl). After centrifuging at 4,000g for 8 min at 4 °C, the supernatant was removed, and the pellet was resuspended in 40 ml TBS buffer again. Next, the OD600 was measured for a 1:1 mixture of the cell suspension and fresh TBS buffer, and a standardized volume equivalent to 40 ml of OD600 = 0.6 was aliquoted into fresh 50-ml conical tubes. A final 8 min centrifugation step at 4,000g and 4 °C was performed, cells were pelleted and the supernatant was discarded. Residual liquid was removed, and cell pellets were flash-frozen using liquid nitrogen and stored at −80 °C or kept on ice for the subsequent steps.
Bovine serum albumin (GoldBio) was dissolved in 1× PBS buffer (Gibco) and sterile-filtered to generate a 5 mg ml−1 BSA solution. For each sample, 25 μl of Dynabeads Protein G (Thermo Fisher Scientific) slurry (hereafter, beads or magnetic beads) were prepared for immunoprecipitation. Up to 250 μl of the initial bead slurry were prepared in a single tube, and washes were performed at room temperature, as follows: the slurry was transferred to a 1.5-ml tube and placed onto a magnetic rack. The supernatant was removed, 1 ml BSA solution was added, and the beads were fully resuspended by vortexing, followed by rotating for 30 s. This was repeated for three more washes. Finally, the beads were resuspended in 25 μl (× no. of samples) of BSA solution, followed by addition of 4 μl (× no. of samples) of monoclonal anti-FLAG M2 antibodies produced in mouse (Sigma-Aldrich). The suspension was moved to 4 °C and rotated for >3 h to conjugate antibodies to magnetic beads. While conjugation was proceeding, cross-linked cell pellets were thawed on ice, resuspended in FA lysis buffer 150 (50 mM HEPES-KOH pH 7.5, 0.1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, 1% (v/v) Triton X-100, 150 mM NaCl) with protease inhibitor cocktail (Sigma-Aldrich) and transferred to a 1 ml milliTUBE AFA Fiber (Covaris). The samples were sonicated on a M220 Focused-ultrasonicator (Covaris) with the following SonoLab 7.2 settings: minimum temperature, 4 °C; set point, 6 °C; maximum temperature, 8 °C; peak power, 75.0; duty factor, 10; cycles/bursts, 200; 17.5 min sonication time. After sonication, samples were cleared of cell debris by centrifugation at 20,000g and 4 °C for 20 min. The pellet was discarded, and the supernatant (about 1 ml) was transferred into a fresh tube and kept on ice for immunoprecipitation. For non-immunoprecipitated input control samples, 10 μl (about 1%) of the sheared cleared lysate were transferred into a separate 1.5 ml tube, flash-frozen in liquid nitrogen and stored at −80 °C.
After >3 h, the conjugation mixture of magnetic beads and antibodies was washed four times with BSA solution, as described above, but at 4 °C. Next, the beads were resuspended in 30 μl (× no. of samples) FA lysis buffer 150 with protease inhibitor, and 31 μl of resuspended antibody-conjugated beads were mixed with each sample of sheared cell lysate. The samples rotated overnight for 12–16 h at 4 °C for immunoprecipitation of FLAG-tagged proteins. The next day, tubes containing beads were placed on a magnetic rack, and the supernatant was discarded. Then, six bead washes were performed at room temperature, as follows, using 1 ml of each buffer followed by sample rotation for 1.5 min: (1) two washes with FA lysis buffer 150 (without protease inhibitor); (2) one wash with FA lysis buffer 500 (50 mM HEPES-KOH pH 7.5, 0.1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, 1% (v/v) Triton X-100, 500 mM NaCl); (3) one wash with ChIP wash buffer (10 mM Tris-HCl pH 8.0, 250 mM LiCl, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, 1% (v/v) Triton X-100, 500 mM NaCl); and (4) two washes with TE buffer 10/1 (10 mM Tris-HCl pH 8.0, 1 mM EDTA). The beads were then placed onto a magnetic rack, the supernatant was removed and the beads were resuspended in 200 μl of fresh ChIP elution buffer (1% (w/v) SDS, 0.1 M NaHCO3). To release protein–DNA complexes from beads, the suspensions were incubated at 65 °C for 1.25 h with gentle vortexing every 15 min to resuspend settled beads. During this incubation, the non-immunoprecipitated input samples were thawed, and 190 μl of ChIP Elution Buffer was added, followed by the addition of 10 μl of 5 M NaCl. After the 1.25 h incubation of the immunoprecipitated samples was complete, the tubes were placed back onto a magnetic rack, and the supernatant containing eluted protein–DNA complexes was transferred to a new tube. Then, 9.75 μl of 5 M NaCl was added to ~195 μl of eluate, and the samples (both immunoprecipitated and non-immunoprecipitated controls) were incubated at 65 °C overnight to reverse-cross-link proteins and DNA. The next day, samples were mixed with 1 μl of 10 mg ml−1 RNase A (Thermo Fisher Scientific) and incubated for 1 h at 37 °C, followed by addition of 2.8 μl of 20 mg ml−1 proteinase K (Thermo Fisher Scientific) and 1 h incubation at 55 °C. After adding 1 ml of buffer PB (QIAGEN recipe), the samples were purified using QIAquick spin columns (QIAGEN) and eluted in 40 μl TE buffer 10/0.1 (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA).
ChIP–seq Illumina libraries were generated for immunoprecipitated and input samples using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB). Sample concentrations were determined using the DeNovix dsDNA Ultra High Sensitivity Kit. Starting DNA amounts were standardized such that an approximately equal mass of all input and immunoprecipitated DNA was used for library preparation. After adapter ligation, PCR amplification (12 cycles) was performed to add Illumina barcodes, and approximately 450 bp DNA fragments were selected using two-sided AMPure XP bead (Beckman Coulter) size selection, as follows: the volume of barcoded immunoprecipitated and input DNA was brought up to 50 μl with TE Buffer 10/0.1; in the first size-selection step, 0.55× AMPure beads (27.5 μl) were added to the DNA, the sample was placed onto a magnetic rack, and the supernatant was discarded and the AMPure beads were retained; in the second size-selection step, 0.35× AMPure beads (17.5 μl) were added to the DNA, the sample was placed onto a magnetic rack, and the AMPure beads were discarded and the supernatant was retained. The concentration of DNA was determined for pooling using the DeNovix dsDNA High Sensitivity Kit.
Illumina libraries were sequenced in paired-end mode on the Illumina MiniSeq and NextSeq platforms, with automated demultiplexing and adapter trimming (Illumina). For each ChIP–seq sample, >1,000,000 raw reads (including genomic and plasmid-mapping reads) were obtained.
ChIP–seq data analyses
ChIP–seq data analyses were generally performed as described previously69. In brief, ChIP–seq paired-end reads were trimmed and mapped to an E. coli BL21(DE3) reference genome (GenBank: CP001509.3). Genomic lacZ and lacI regions partially identical to plasmid-encoded genes were masked in all alignments (genomic coordinates: 335,600–337,101 and 748,601–750,390). In the ChIP–seq analyses of Cas9 and Cas12a, the rrnB t1 terminator genomic sequence was masked (genomic coordinates: 4,121,275–4,121,400). Mapped reads were sorted and indexed, and multi-mapping reads were excluded. Aligned reads were normalized by RPKM and visualized in IGV68. For genome-wide views, maximum read coverage values were plotted in 1-kb bins. Peak calling was performed using MACS3(3.0.0a7)70 with respect to non-immunoprecipitated control samples of TnpB and Cas9. The peak summit coordinates in the MACS3 output summits.bed file were extended to encompass a 200-bp window using BEDTools (v2.30.0)71. The corresponding 200-bp sequence for each peak was extracted from the E. coli reference genome using the command bedtools getfasta. Sequence motifs were determined using MEME-ChIP (5.4.1)72. Individual off-target sequences (Extended Data Fig. 6) represent sequences from the top enriched peaks determined by MACS3 that contain the MEME-ChIP motif.
TAM library cloning
TAM libraries containing a 6-bp randomized sequence between the native target sequences for GstIscB (ISGst6) and GstTnpB2 (ISGst3) were cloned. In brief, two partially overlapping oligos (oSL9404 and oSL9405) were annealed by heating to 95 °C for 2 min and then cooled to room temperature. One of these oligos (oSL9404) contained a 6-nt degenerate sequence flanked by target sites for GstTnpB2 and GstIscB. Annealed DNA was treated with DNA Polymerase I, Large (Klenow) Fragment (NEB) in 40-μl reactions and incubated at 37 °C for 30 min, then gel-purified (QIAGEN Gel Extraction Kit). Double-stranded insert DNA and vector backbone (pSL4031) were digested with BamHI and HindIII (37 °C, 1 h). The digested insert was cleaned-up (Qiagen MinElute PCR Purification Kit), and the digested backbone was gel-purified (Qiagen QIAquick Gel Extraction Kit). The backbone and insert were ligated with T4 DNA Ligase (NEB). Ligation reactions were used to transform electrocompetent NEB 10-beta cells according to the manufacturer’s protocol. After recovery (37 °C for 1 h), cells were plated on large bioassay plates containing LB-agar and kanamycin (50 μg ml−1). Approximately 5 million CFUs were scraped from each plate, representing 1,000× coverage of each library member, and plasmid DNA was isolated using the Qiagen CompactPrep Midi Kit.
TAM library assays and NGS library prep
DNA solutions containing 500 ng of the TAM plasmid library (pSL4841) and 500 ng of plasmids encoding either GstTnpB2 (pSL4369) or GstIscB (pSL4514) were used to co-transform electrocompetent E. coli BL21(DE3) cells according to the manufacturer’s protocol (Sigma-Aldrich). Cells were serially diluted on large bioassay plates containing LB-agar with spectinomycin (100 μg ml−1) and kanamycin (50 μg ml−1). Approximately 600,000 CFUs were scraped from plates, representing 100× coverage of each library member, and plasmid DNA was isolated using the Qiagen CompactPrep Midi Kit. Illumina amplicon libraries for NGS were prepared through a 2-step PCR amplification. In brief, ~50 ng of plasmid DNA recovered from TAM assays was used in each PCR-1 amplification reaction with primers flanking the degenerate TAM library sequence and containing universal Illumina adapters as 5′ overhangs. Amplification was carried out using high-Fidelity Q5 DNA Polymerase (NEB) for 16 thermal cycles. Samples from PCR-1 amplification were diluted 20-fold and amplified for PCR-2 in 10 thermal cycles with primers containing indexed p5/p7 sequences. Reactions were verified by analytical gel electrophoresis. Sequencing was performed with a paired-end run using a MiniSeq High Output Kit with 150-cycles (Illumina).
Analyses of NGS TAM library data
Analyses of TAM library experiments were performed using a custom Python script. Demultiplexed reads were filtered to remove reads that did not contain a perfect match to the 58-bp sequence upstream of the degenerate sequence for any i5-reads. For reads that passed this filtering step, the 6-nt degenerate sequence was extracted and counted. The relative abundance of each degenerate sequence in a sample was determined by dividing the degenerate sequence count by the total number of sequence counts for that sample. Then, the fold-change between the output and input libraries was calculated by dividing the relative abundance of each degenerate sequence in the output library by its relative abundance in the input library, followed by log2 transformation. Sequence logos were constructed by taking the 10-most depleted sequences and generating the logo using WebLogo73 (v2.8).
Transposition assays combining TnpA and TnpB
E. coli strain K-12 substrain MG1655 (sSL0810) was engineered to carry a genomically integrated mini-Tn containing a kanamycin resistance cassette inserted into lacZ by recombineering, as described above to generate sSL2771. This strain was transformed with either pCDFDuet-1 (pSL0007) or various GstTnpB vectors (pSL4369, pSL4664, pSL4518 or pSL4740, see Supplementary Table 2 for descriptions), followed by selection on LB-agar containing spectinomycin (100 μg ml−1) and kanamycin (50 μg ml−1). Single colony isolates of cells harbouring each plasmid were used to prepare chemically competent cells, which were then transformed with a TnpA expression vector (pSL4529) or a catalytically inactive mutant TnpA expression vector (pSL4534), followed by selection on LB-agar containing carbenicillin (100 μg ml−1), spectinomycin (100 μg ml−1) and kanamycin (50 μg ml−1). Three single colony isolates of each transformant were grown in liquid LB containing carbenicillin (100 μg ml−1), spectinomycin (100 μg ml−1) and kanamycin (50 μg ml−1) for 14 h at 37 °C. The optical density (OD) of each culture was measured, and approximately 107 cells were plated onto MacConkey-agar medium containing carbenicillin (100 μg ml−1), spectinomycin (100 μg ml−1) and 0.05 mM IPTG for TnpA induction. Importantly, the medium did not contain kanamycin, to allow for excision of the mini-Tn. Cells were grown at 37 °C for 4 days on MacConkey medium to enrich for mini-Tn excision events. Cells were then collected, serially diluted, and plated onto LB-agar containing carbenicillin (100 μg ml−1), spectinomycin (100 μg ml−1) and X-gal (200 μg ml−1), or carbenicillin (100 μg ml−1), spectinomycin (100 μg ml−1), kanamycin (50 μg ml−1) and X-gal (200 μg ml−1), and grown for 18 h at 37 °C. The total number of colonies was counted, along with the number of blue colonies to determine the frequency of excision and reintegration events. In addition, genomic lysates were collected from cells as described above for PCR analyses.
Recombination assays combining TnpA and TnpB
E. coli strain K-12 substrain MG1655 (sSL0810) cells were transformed by heat shock with 400 ng of plasmid encoding G. stearothermophilus IS elements with TnpA and/or TnpB–ωRNA, recovered for 1 h at 37 °C in liquid LB, and plated on LB-agar plates containing 10 μg ml−1 tetracycline. Colonies were counted and converted to CFUs per μg of DNA. Tetracycline plates were then replica plated to LB-agar plates containing both tetracycline (10 μg ml−1) and X-gal (200 μg ml−1) for blue/white colony screening, and white colonies were counted to determine the frequency of recombination events at the genomic lacZ locus. Genotypes were confirmed by selecting three independent colonies for each condition, extracting genomic lysate as described above, and performing diagnostic PCR using primers that annealed outside of the homology arms located on the ISGst3-lacZ plasmid, such that only the genomic locus was amplified. PCR results were visualized by gel electrophoresis and confirmed by long-read Nanopore sequencing (Plasmidsaurus).
Statistics and reproducibility
qPCR and analytical PCRs resolved by agarose gel electrophoresis gave similar results in three independent biological replicates. Sanger sequencing of excision products was performed once for each isolate. Next-generation sequencing of PCR amplicons was performed once. Plasmid interference assays were performed in three independent replicates. Transposition assays combining TnpA and TnpB were performed with three independent biological replicates.
Extended Data
Extended Data Fig. 1 |. Bioinformatic analyses of IscB and TnpB homologs.

a, Phylogenetic tree of IscB and IsrB protein homologs; IscB contains HNH and RuvC nuclease domains, whereas IsrB lacks the HNH nuclease. Genetic neighborhood analyses demonstrate that most homologs are encoded proximal to a predicted ωRNA (inner ring), whereas the vast majority do not reside near a predicted tyrosine-family TnpA transposase gene (outer ring). The GstIscB homolog encoded by ISGst6 used in this study is indicated. Bootstrap values are indicated for major nodes. b, Schematic of a non-autonomous IS element encoding IscB and its associated ωRNA; a structural covariation model is shown in the inset (top). The red rectangle and dotted black line indicate the transposon boundaries, and the guide portion of the ωRNA is shown in blue. LE and RE, transposon left end and right end. c, Orientation bias of the nearest upstream ORFs to the indicated protein-coding gene (iscB, tnpB, or IS630 transposase), demonstrating that IS elements encoding IscB are preferentially integrated (or retained) in an orientation matching that of the upstream gene. The y-axis indicates the frequency of ORFs containing the same orientation, at a distance from the gene start codon defined by the x-axis. 242 bp represents the average length of IscB-associated ωRNAs upstream of IscB ORF. The spike at ~0-bp for TnpB corresponds to IS elements that encode adjacent/overlapping tnpA and tnpB genes. IS630 transposase genes are included as a representative gene from unrelated transposable elements. d, Phylogenetic tree of TnpB homologs, with bootstrap values shown for major nodes. Genetic neighborhood analyses demonstrate that most homologs are encoded proximal to a predicted ωRNA (inner ring), whereas the vast majority do not reside near a predicted tyrosine- or serine-family TnpA transposase genes (outer rings). Interestingly, TnpB homologs are associated with two distinct transposase families in prokaryotes: tyrosine transposases (denoted TnpA (Y)) within IS200/605-family elements, and serine transposases (denote TnpA (S)) within IS607-family elements. GstTnpB homologs used in this study are highlighted, along with the predicted structures of their associated ωRNAs based on covariance modeling. The ωRNA encoded by ISGst2 did not show strong covariation in structure and was therefore omitted. e, Read coverage for RNA-seq data from G. stearothermophilus strain DSM 458, demonstrating expression of putative ωRNAs from each of the indicated ISGst families. ISGst5 is a PATE-like element that lacks any protein-coding ORFs. Other TnpB-associated ωRNAs are encoded within/downstream of the ORF, whereas IscB-associated ωRNAs are encoded upstream of the ORF.
Extended Data Fig. 2 |. Classification of IS605-family elements encoded by G. stearothermophilus strain DSM 458.
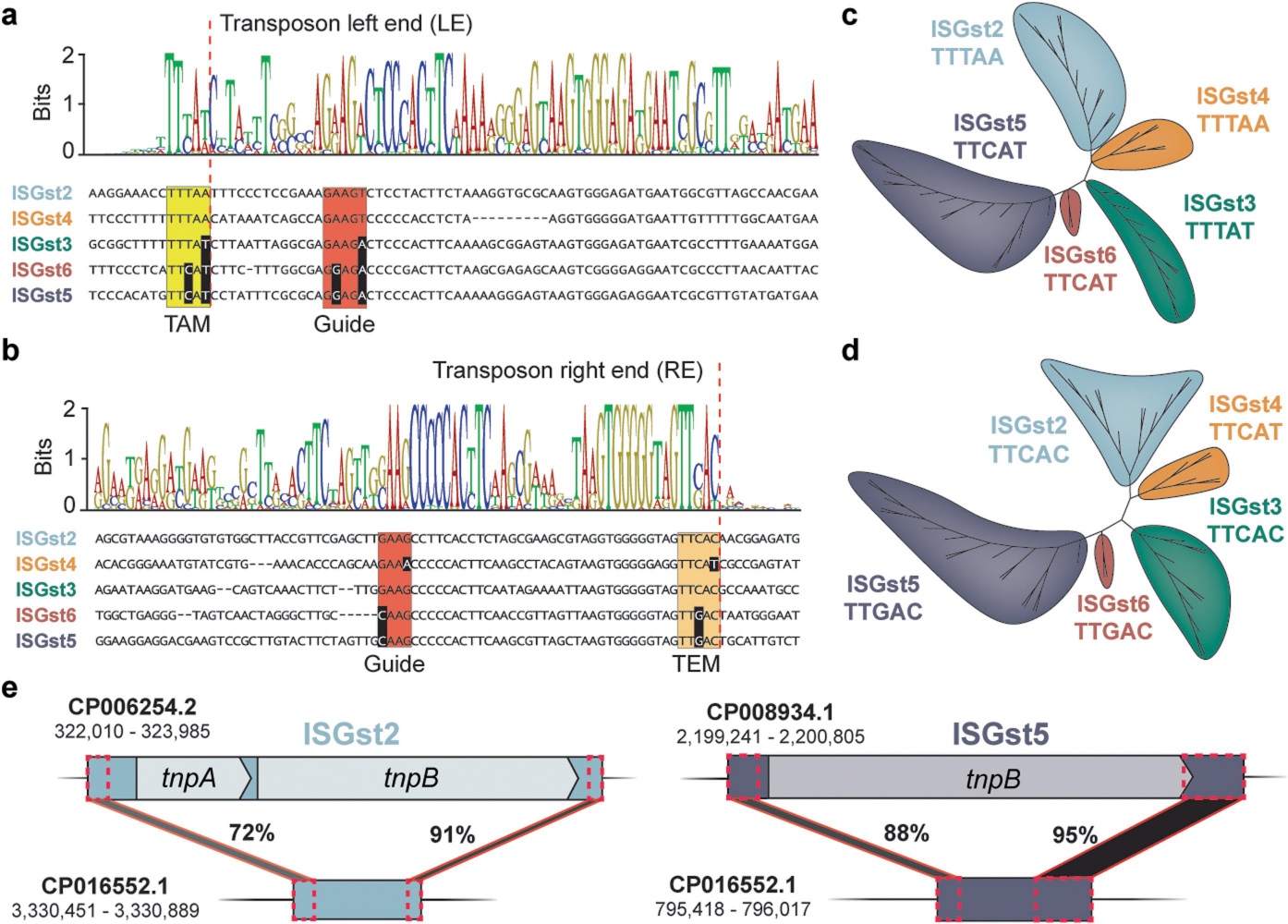
a, DNA multiple sequence alignment of transposon left ends (LE) for IS200/IS605-family elements from G. stearothermophilus. The weblogo (top) is built from 47 unique elements (Supplementary Figs. 2 & 3), and one representative sequence from each family is shown below, with the TAM highlighted in yellow and DNA guide sequences highlighted in red. Nucleotides highlighted in black exhibit covarying mutations, relative to ISGst2. TAM, transposon-adjacent motif; the dotted red line indicates the transposon boundary. b, DNA multiple sequence alignment of transposon right ends (RE) for IS200/IS605-family elements from G. stearothermophilus, shown as in a. TEM, transposon-encoded motif is shown in orange. c, Phylogenetic tree of ISGst elements based on the transposon left end. Each colored clade encodes an associated TnpB/IscB protein homolog and is flanked by the indicated TAM sequence. d, Phylogenetic tree of ISGst elements based on the transposon right end, shown as in b but with TEM sequence in lieu of TAM. e, Schematic of PATEs (palindrome-associated transposable elements) related to ISGst2 and ISGst5, which contain similar transposon ends but no protein-coding genes. The percent sequence identity between shaded regions (black) is shown, as are the genomic accession IDs and coordinates.
Extended Data Fig. 3 |. Specificity and efficiency of transposon DNA excision by TnpA.

a, Schematic of heterologous E. coli transposon excision assay. Plasmids encode TnpA and a mini-transposon (Mini-Tn) substrate, whose loss is monitored by PCR using the indicated primers. The expected sizes of PCR products generated from donor joints that are produced upon religation of flanking sequences are shown, for both ISGst2 and H. pylori IS608. b, TnpA homologs do not cross-react with distinct IS elements, as assessed by analytical PCR. Cell lysates were tested after overnight expression of TnpA in combination with a mini-Tn substrate from either G. stearothermophilus (G) or H. pylori (H), and PCR products were resolved by agarose gel electrophoresis. M refers to catalytically inactive mutants. Note that HpyTnpA is substantially more active for DNA excision than GstTnpA under the tested conditions. U, unexcised; E, excised. c, Schematic of qPCR assay to quantify excision frequencies, in which one of the two primers anneals directly to the donor joint formed upon mini-Tn excision and religation. d, Comparison of simulated excision frequencies, generated by mixing clonally excised and unexcised lysate in known ratios, versus experimentally determined integration efficiencies measured by qPCR. e, qPCR-based quantification of TnpA-mediated excision of an ISGst2 mini-Tn substrate in E. coli. Excised refers to a cloned excision product; TnpAM denotes a TnpA mutant (Y125A); ND, not detected above a 0.0001% threshold. Bars indicate mean ± standard deviation (n = 3). f, Schematic of mini-Tn for ISGst3 element, highlighting the subterminal palindromic transposon ends located on the top strand (top). Transposon-adjacent and transposon-encoded motifs (TAM and TEM) are shown in yellow and orange, respectively; DNA guides are shown in orange, and their putative base-pairing interactions are indicated; dotted lines indicate transposon boundaries and thus the sites of ssDNA cleavage and religation. Sanger sequencing of excision events confirms the identity of the expected donor joint product formed upon transposon loss (bottom). Sanger sequencing results are duplicated from Fig. 2d. g, Schematic and Sanger sequencing data as in f, but for a modified ISGst3 substrate containing TEM mutations. Experimentally detected products erroneously excise at an alternative, aberrant TEM-like sequence located outside of the native transposon boundary (orange), presumably because of the need to maintain cognate base-pairing between the DNA guide (orange and TEM (blue).
Extended Data Fig. 4 |. Mating-out assay to monitor transposition of IS Gst3.

a, Schematic of mating-out assay, in which transposition events into the F-plasmid are monitored via drug selection. E. coli donor cells carrying an F-plasmid were transformed with a plasmid encoding either TnpA and ISGst3-derived mini-Tn (pDonor1) or an autonomous ISGst3 element with tnpA and tnpB (pDonor2). After induction of TnpA, conjugation was used to transfer the F-plasmid into the recipient strain, and transposition events were quantified by selecting for recipient cells (RifR) containing spectinomycin (F+) and kanamycin (mini-Tn+) resistance. b, Transposition frequency of ISGst3 mini-Tn deriving from pDonor1 into the F-plasmid was measured with and without tnpA. Bars indicate mean ± s.d. (n = 6). c, Drug-selected cells from mating-out assays from pDonor1 contain TAM-proximal IS insertions, as evidenced by long-read Nanopore sequencing. A genetic map of the F-plasmid is shown, along with the location of distinct ISGst3-derived mini-Tn integration events. The insets show a zoom-in view of each integration site at the nucleotide level, with the TAM highlighted in yellow and the integration site denoted by an arrow. d, Transposition frequency of an autonomous ISGst3 deriving from pDonor2 into the F-plasmid was measured with active and catalytically inactive forms of TnpA and TnpB. M, TnpA Y125A mutant; d, TnpB D196A mutant; ND, not detected. Bars indicate mean ± s.d. (n = 3).
Extended Data Fig. 5 |. Optimization and testing of DNA cleavage parameters with TnpB/IscB nucleases.

a, Promoter screen to optimize conditions for E. coli-based interference assays using plasmid-encoded ωRNA and TnpB2. P1 indicates the promoter for ωRNA expression, whereas P2 indicates the promoter for TnpB expression. Transformants with a targeting (T) or non-targeting (NT) ωRNA-pTarget combination were serially diluted, plated on selective media, and cultured at 37 °C for 24 h. b, Results from plasmid interference assays with HpyTnpB (IS608) and DraTnpB (ISDra2) using ωRNAs that target native donor joint products, which revealed an absence of activity for HpyTnpB. Experiments were performed as in a. c, DNA cleavage by TnpB2 is highly sensitive to TAM mutations, as assessed by plasmid interference assays. Data are shown as in a, with the indicated TAM sequences; TTTAT denotes the WT TAM, and NT denotes a non-targeting control. d, DNA cleavage by IscB is highly sensitive to TAM mutations, as assessed by plasmid interference assays. Data are shown as in a, with the indicated TAM sequences; TTCAT denotes the WT TAM, and NT denotes a non-targeting control. e, TnpB2 is only active for targeted genomic DNA cleavage using select ωRNAs, as assessed by genome targeting assays. Transformants with a non-targeting (NT) or one of three lacZ-targeting guides (T1–T3) were serially diluted, plated on selective media, and cultured at 37 °C for 24 h. f, Schematic of E. coli-based genome targeting assay, in which RNA-guided DNA cleavage of lacZ by TnpB results in cell death. Guides were designed to target three sites within the lacZ coding sequence. g, TnpB1 is active for targeted genomic DNA cleavage at elevated temperatures, as assessed by genome targeting assays. Transformants with a non-targeting (NT) or one of three lacZ-targeting guides (T1–T3) were serially diluted, plated on selective media, and cultured at 37 °C or 45 °C for 24 h, as shown. h, Schematic of E. coli-based plasmid interference assay using a native ISGst3 element. The transposon was cloned from G. stearothermophilus gDNA onto the pEffector plasmid, with 40 bp of flanking sequence to preserve the natural ωRNA. Targeted cleavage of pTarget results in a loss of kanamycin resistance and cell lethality on selective LB-agar plates. i, TnpB2 encoded by a native ISGst3 transposon is active for targeted cleavage of its donor joint. Transformants with a targeting (T) or non-targeting (NT) ωRNA-pTarget combination were serially diluted, plated on selective media, and cultured at 37 °C for 24 h. dTnpB2 contains an inactivating D196A mutation, whereas ΔTnpB2 contains a stop codon at codon 32. j, Schematic of E. coli-based genome targeting assay as in e, but with pEffector containing a native ISGst3 transposon cloned into lacZ. RNA-guided DNA cleavage of genomic lacZ results in cell death. k, TnpB2 encoded by a native ISGst3 transposon is active for targeted genomic cleavage of a donor joint mimic. Transformants were serially diluted, plated on selective media, and cultured at 37 °C for 24 h. dTnpB2 contains an inactivating D196A mutation, whereas ΔTnpB2 contains a stop codon at codon 32.
Extended Data Fig. 6 |. Off-target ChIP-seq DNA binding analyses.

a, ChIP-seq experiments reveal recruitment of dIscB to the target site (blue triangle) with a targeting ωRNA. Genome-wide representation of ChIP-seq data for dIscB reshown from Fig. 4b, with addition of a second replicate. Representative off-target sites for dIscB identified by MACS3 are highlighted (OT1–4) and analyzed in the right panel. The middle panel highlights analysis of off-target binding events using MEME-ChIP, as shown in Fig. 4b. Motifs shared by off-target peaks reveal conserved TAM sequences and little conservation of the adjacent seed sequence. The sequence of the 5′ end of the corresponding ωRNA is shown below each motif. n indicates the number of peaks contributing to the motif and their percentage of total peaks called by MACS3; E, E-value significance of the motif generated from the MEME-ChIP analysis. DNA sequences corresponding to the on-target and off-target sites are shown on the right, with TAM (yellow) and mismatches (orange) highlighted. b, ChIP-seq experiments reveal recruitment of dTnpB2 to the target site (blue triangle) with a targeting ωRNA. Data shown as in a. Similar to dIscB, dTnpB2 shows limited seed sequence requirements. c, ChIP-seq experiments reveal recruitment of dCas9 to the target site (blue triangle) with a targeting ωRNA. Data shown as in a. Analysis of off-target sites reveal a short (3–4 nt) seed sequence adjacent to the PAM motif. d, ChIP-seq experiments reveal recruitment of dCas12a to the target site (blue triangle) with a targeting ωRNA. Data shown as in a. Analysis of off-target sites reveals a short (5–7 nt) seed sequence adjacent to the PAM motif.
Extended Data Fig. 7 |. qPCR analysis of IS element loss upon TnpA and TnpB co-expression.

a, TnpB promotes transposon retention at the donor site, as assessed by blue-white colony screening for the experiment in Fig. 5a–c; images of representative LB-agar plates are shown. TnpAM, TnpA Y125A mutant; dTnpB, TnpB D196A mutant. b, Schematic of qPCR-based strategy for quantifying excision. Primers are designed to flank the donor joint generated upon excision and religation. Selective PCR conditions with a shortened extension time allows for reduced amplification of the starting locus containing the mini-Tn. rssA was used as a reference gene for ΔCq calculations. c, Comparison of simulated excision frequencies, generated by mixing clonally excised and unexcised lysates in known ratios, versus experimentally determined integration efficiencies measured by qPCR. d, qPCR-based quantification of transposon excision for experiments presented in Fig. 5a–c. TnpA was present in all conditions marked with orange bars, and Empty refers to an experiment lacking TnpB; Excised and Unexcised Marker labels shown in blue correspond to clonal control strains with either wild-type lacZ or mini-Tn-interrupted lacZ, respectively. The detection limit is based on qPCR experiments tested on simulated excision frequency samples shown in b. Bars indicate mean ± s.d. (n = 3–9). e, Images of representative LB-agar plates accompanying the experiments presented in Fig. 5d–f, highlighting the roles of TnpA and TnpB in transposon maintenance and spread via recombination. TnpAM, TnpA Y125A mutant; dTnpB, TnpB D196A mutant.
Supplementary Material
Acknowledgements
The authors thank D. R. Gelsinger and A. Bernheim for helpful bioinformatics discussions, L. E. Berchowitz for helpful RNA biology discussions, J. C. Cheong for technical support, L. F. Landweber for qPCR instrument access, and the JP Sulzberger Columbia Genome Center for NGS support. C.M. was supported by NIH Postdoctoral Fellowship F32 GM143924-01A1. M.W.G.W. was supported by a National Science Foundation Graduate Research Fellowship. This research was supported by NSF Faculty Early Career Development Program (CAREER) Award 2239685, and by a generous start-up package from the Columbia University Irving Medical Center Dean’s Office and the Vagelos Precision Medicine Fund (to S.H.S.).
Footnotes
Competing interests Columbia University has filed US Patent Application Number 63/379,082 related to this work, for which C.M. and S.H.S. are inventors. S.H.S. is a co-founder and scientific advisor to Dahlia Biosciences, a scientific advisor to CrisprBits and Prime Medicine, and an equity holder in Dahlia Biosciences and CrisprBits.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-023-06597-1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
Custom scripts used for bioinformatics, TAM library analyses and ChIP–seq data analyses are available at GitHub (https://github.com/sternberglab/Meers_et_al_2023).
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-023-06597-1.
Data availability
Next generation sequencing data are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive: SRX19058888-SRX19058905, SRR23476356-SRR23476358, and SRR24994123 (BioProject Accession: PRJNA925099 and PRJNA986543) and the Gene Expression Omnibus (GSE223127). The published genome used for ChIP–seq analyses was obtained from NCBI (GenBank: CP001509.3). The published genome used for bioinformatics analyses of the G. stearothermophilus genome was obtained from NCBI (GenBank: NZ_CP016552.1). Datasets generated and analysed in the current study are available from the corresponding author upon reasonable request.
References
- 1.Siguier P, Gourbeyre E, Varani A, Ton-Hoang B & Chandler M Everyman’s guide to bacterial insertion sequences. Microbiol. Spectr. 3, MDNA3–0030-2014 (2015). [DOI] [PubMed] [Google Scholar]
- 2.He S et al. The IS200/IS605 family and “peel and paste” single-strand transposition mechanism. Microbiol. Spectr. 3, MDNA3–0039-2014 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Kapitonov VV, Makarova KS & Koonin EV ISC, a novel group of bacterial and archaeal DNA transposons that encode Cas9 homologs. J. Bacteriol. 198, 797–807 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chylinski K, Makarova KS, Charpentier E & Koonin EV Classification and evolution of type II CRISPR–Cas systems. Nucleic Acids Res. 42, 6091–6105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altae-Tran H et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 374, 57–65 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karvelis T et al. Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature 599, 692–696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haudiquet M, de Sousa JM, Touchon M & Rocha EPC Selfish, promiscuous and sometimes useful: how mobile genetic elements drive horizontal gene transfer in microbial populations. Phil. Trans. R. Soc. B 377, 20210234 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feschotte C & Pritham EJ DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 41, 331–368 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benler S & Koonin EV Recruitment of mobile genetic elements for diverse cellular functions in prokaryotes. Front. Mol. Biosci. 9, 821197 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zimmerly S & Semper C Evolution of group II introns. Mob. DNA 6, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura TM & Cech TR Reversing time: origin of telomerase. Cell 92, 587–590 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Zhang Y, Liu CC & Schatz DG Structural insights into the evolution of the RAG recombinase. Nat. Rev. Immunol. 22, 353–370 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Koonin EV & Makarova KS Origins and evolution of CRISPR–Cas systems. Phil. Trans. R. Soc. B 374, 20180087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cosby RL, Chang NC & Feschotte C Host–transposon interactions: conflict, cooperation, and cooption. Genes Dev. 33, 1098–1116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koonin EV, Makarova KS, Wolf YI & Krupovic M Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat. Rev. Genet. 21, 119–131 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Krupovic M, Makarova KS, Forterre P, Prangishvili D & Koonin EV Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR–Cas immunity. BMC Biol. 12, 36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Özcan A et al. Type IV CRISPR RNA processing and effector complex formation in Aromatoleum aromaticum. Nat. Microbiol. 4, 89–96 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Seed KD, Lazinski DW, Calderwood SB & Camilli A A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature 494, 489–491 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters JE, Makarova KS, Shmakov S & Koonin EV Recruitment of CRISPR–Cas systems by Tn7-like transposons. Proc. Natl Acad. Sci. USA 114, e7358–e7366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sternberg SH, Redding S, Jinek M, Greene EC & Doudna JA DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strecker J et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandler M et al. Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat. Rev. Microbiol. 11, 525–538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasternak C et al. ISDra2 transposition in Deinococcus radiodurans is downregulated by TnpB. Mol. Microbiol. 88, 443–455 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Filée J, Siguier P & Chandler M I am what I eat and I eat what I am: acquisition of bacterial genes by giant viruses. Trends Genet. 23, 10–15 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Bao W & Jurka J Homologues of bacterial TnpB_IS605 are widespread in diverse eukaryotic transposable elements. Mob DNA 4, 12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saito M et al. Fanzor is a eukaryotic programmable RNA-guided endonuclease. Nature 620, 660–668 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kersulyte D, Mukhopadhyay AK, Shirai M, Nakazawa T & Berg DE Functional organization and insertion specificity of IS607, a chimeric element of Helicobacter pylori. J. Bacteriol. 182, 5300–5308 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinberg Z, Perreault J, Meyer MM & Breaker RR Exceptional structured noncoding RNAs revealed by bacterial metagenome analysis. Nature 462, 656–659 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomes-Filho JV et al. Sense overlapping transcripts in IS1341-type transposase genes are functional non-coding RNAs in archaea. RNA Biol. 12, 490–500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ibrahim A, Vêncio RZN, Lorenzetti APR & Koide T Halobacterium salinarum and Haloferax volcanii comparative transcriptomics reveals conserved transcriptional processing sites. Genes 12, 1018 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nety SP et al. The transposon-encoded protein TnpB processes its own mRNA into ωRNA for guided nuclease activity. CRISPR J. 6, 232–242 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrington LB et al. A thermostable Cas9 with increased lifetime in human plasma. Nat. Commun. 8, 1424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barabas O et al. Mechanism of IS200/IS605 family DNA transposases: activation and transposon-directed target site selection. Cell 132, 208–220 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hickman AB et al. DNA recognition and the precleavage state during single-stranded DNA transposition in D. radiodurans. EMBO J. 29, 3840–3852 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morero NR et al. Targeting IS608 transposon integration to highly specific sequences by structure-based transposon engineering. Nucleic Acids Res. 46, 4152–4163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lavatine L et al. Single strand transposition at the host replication fork. Nucleic Acids Res. 44, 7866–7883 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pasternak C et al. Irradiation-induced Deinococcus radiodurans genome fragmentation triggers transposition of a single resident insertion sequence. PLoS Genet. 6, e1000799 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cox MM Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu. Rev. Genet. 35, 53–82 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Leenay RT & Beisel CL Deciphering, communicating, and engineering the CRISPR PAM. J. Mol. Biol. 429, 177–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu X et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swarts DC, van der Oost J & Jinek M Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR–Cas12a. Mol. Cell 66, 221–233.e224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang F, Zhou K, Ma L, Gressel S & Doudna JA A Cas9–guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Belfort M & Bonocora RP Homing endonucleases: from genetic anomalies to programmable genomic clippers. Methods Mol. Biol. 1123, 1–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tourasse NJ, Stabell FB & Kolstø AB Survey of chimeric IStron elements in bacterial genomes: multiple molecular symbioses between group I intron ribozymes and DNA transposons. Nucleic Acids Res. 42, 12333–12351 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ton-Hoang B et al. Single-stranded DNA transposition is coupled to host replication. Cell 142, 398–408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hickman AB & Dyda F DNA transposition at work. Chem. Rev. 116, 12758–12784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts D, Hoopes BC, McClure WR & Kleckner N IS10 transposition is regulated by DNA adenine methylation. Cell 43, 117–130 (1985). [DOI] [PubMed] [Google Scholar]
- 49.Yin JC, Krebs MP & Reznikoff WS Effect of dam methylation on Tn5 transposition. J. Mol. Biol. 199, 35–45 (1988). [DOI] [PubMed] [Google Scholar]
- 50.Nicolas E et al. The Tn3-family of replicative transposons. Microbiol. Spectr. 3, 10.1128/microbiolspec.mdna3-0060-2014 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Li W & Godzik A Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Katoh K & Standley DM MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Capella-Gutiérrez S, Silla-Martínez JM & Gabaldón T trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minh BQ et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalvari I et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–d200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nawrocki EP & Eddy SR Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yao Z, Weinberg Z & Ruzzo WL CMfinder-a covariance model based RNA motif finding algorithm. Bioinformatics 22, 445–452 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Will S, Joshi T, Hofacker IL, Stadler PF & Backofen R LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA 18, 900–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rivas E RNA structure prediction using positive and negative evolutionary information. PLoS Comput. Biol. 16, e1008387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hyatt D et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Price MN, Dehal PS & Arkin AP FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kechin A, Boyarskikh U, Kel A & Filipenko M cutPrimers: a new tool for accurate cutting of primers from reads of targeted next generation sequencing. J Comput. Biol. 24, 1138–1143 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vasimuddin M, Misra S, Li H & Aluru S Efficient architecture-aware acceleration of BWA-MEM for multicore systems. In 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS) 314–324 (IEEE, 2019). [Google Scholar]
- 66.Danecek P et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson JT, Thorvaldsdottir H, Turner D & Mesirov JP igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 39, btac830 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoffmann FT et al. Selective TnsC recruitment enhances the fidelity of RNA-guided transposition. Nature 609, 384–393 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y et al. Model-based analysis of ChIP–seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bailey TL et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Crooks GE, Hon G, Chandonia JM & Brenner SE WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next generation sequencing data are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive: SRX19058888-SRX19058905, SRR23476356-SRR23476358, and SRR24994123 (BioProject Accession: PRJNA925099 and PRJNA986543) and the Gene Expression Omnibus (GSE223127). The published genome used for ChIP–seq analyses was obtained from NCBI (GenBank: CP001509.3). The published genome used for bioinformatics analyses of the G. stearothermophilus genome was obtained from NCBI (GenBank: NZ_CP016552.1). Datasets generated and analysed in the current study are available from the corresponding author upon reasonable request.