

Abstract
A conserved transcription-translation negative feedback loop forms the molecular basis of the circadian oscillator in animals. Molecular interactions within this loop have been relatively well characterized in vitro and in cell culture; however, in vivo approaches are required to assess the functional significance of these interactions. Here, regulation of circadian gene expression was studied in vivo by using transgenic reporter mouse lines in which 6.75 kb of the mouse Period1 (mPer1) promoter drives luciferase (luc) expression. Six mPer1-luc transgenic lines were created, and all lines express a daily rhythm of luc mRNA in the suprachiasmatic nuclei (SCN). Each mPer1-luc line also sustains a long-term circadian rhythm of luminescence in SCN slice culture. A 6-h light pulse administered during the early subjective night rapidly induces luc mRNA expression in the SCN; however, high luc mRNA levels are sustained, whereas endogenous mPer1 mRNA levels return to baseline, suggesting that posttranscriptional events mediate the down-regulation of mPer1 after exposure to light. This approach demonstrates that the 6.75-kb mPer1 promoter fragment is sufficient to confer both circadian and photic regulation in vivo and reveals a potential posttranscriptional regulatory mechanism within the mammalian circadian oscillator.
Nearly all organisms express circadian (≈24-h) rhythms in behavior, physiology, and cellular activity. In mice, Drosophila, Neurospora, and cyanobacteria, extensive studies indicate that the basic molecular circadian mechanism consists of a transcription-translation feedback loop (1, 2). Recent reviews describe the mammalian model in detail (3, 4); briefly, the transcription factors CLOCK and BMAL1 (also known as MOP3) activate transcription of the mouse Period (mPer1 and mPer2) and Cryptochrome (mCry1 and mCry2) genes. The PER and CRY proteins accumulate and translocate into the nucleus where they inhibit the activity of CLOCK and BMAL1. The turnover of the inhibitory PER and CRY proteins then leads to a new cycle of activation by CLOCK and BMAL1.
Several genes in this transcriptional pathway exhibit circadian rhythms of expression, but they differ in characteristics of rhythmic expression such as circadian phase and response to light (3). For example, peak mRNA expression of mPer1, mPer2, and mCry1 occurs at different times in the suprachiasmatic nucleus (SCN), the site of the circadian pacemaker in mammals (5–7). However, the protein products of these three genes accumulate in SCN neurons around the same phase (8–10). In addition, a light pulse administered during the early subjective night leads to the rapid induction of mPer1, slower induction of mPer2, and no induction of mCry1 (11–14). Clearly, regulated circadian gene expression remains an important component of the circadian mechanism.
Transcriptional regulation of circadian promoter activity has been addressed initially in cell culture. In cell transfection/luciferase reporter assays, the CLOCK and BMAL1 proteins dimerize and bind three E-box promoter elements (5′-CACGTG-3′) within a 2.0-kb mPer1 promoter fragment to activate transcription (15). Additional studies indicate that the mPER and mCRY proteins inhibit transcriptional activity by acting on CLOCK/BMAL1 via E-box elements (10, 15–18).
Transfection-based reporter assays, although useful for the characterization of protein–DNA interactions, have not revealed the dynamics or persistence of circadian gene regulation. Such analyses of circadian gene expression require in vivo methods. Recently, mPer1-luc transgenic (P1L) rats and mice were created, and luminescence from organ cultures of SCN and peripheral tissues was measured in real time (19–21). These studies illustrate the utility of a circadian reporter system to better understand circadian physiology and molecular activity. However, in culture the photic input to the SCN is disrupted, and the in vivo context of the tissue is altered. To address such issues, we analyzed P1L mice to determine whether a 6.75-kb mPer1 promoter fragment could confer not only rhythmic expression but also light responsiveness in vivo as well as drive a circadian rhythm of reporter gene expression in mouse SCN cultures.
Materials and Methods
DNA Constructs.
mPer1-containing bacterial artificial chromosome clones were isolated, digested with BamHI, and probed by Southern hybridization using an mPer1 5′ probe (5′-TTTTCCTTATGACATCAGGGTGATACTTACCTTC-3′) to identify a 5.2-kb fragment. This BamHI fragment was purified, blunt-ended, and cloned into the SmaI site of the pGL3 Basic luciferase reporter vector (Promega) to create mPer15.2-luc. The complete insert of mPer15.2-luc was sequenced on both strands. A previously sequenced 1.96-kb SpeI–HindIII fragment was generated from an mPer1-containing λ clone (15); this fragment overlaps with the 3′ end of the BamHI fragment. A 1.33-kb HindIII–NcoI PCR product that contains the mPer1 5′-untranslated region (UTR) sequence up to the mPER1 initiator ATG was amplified by PCR and sequenced (forward primer, 5′-TTTTGTAGTACTGGCTTCCTGG-3′; reverse primer, 5′-CATGCCATGGCTGGGCCATACAGTGGAG-3′). An NcoI site (underlined) was introduced into the mPer1 fragment via the reverse primer. The mPER1 initiator ATG was engineered to coincide with the luciferase initiator ATG at the NcoI site in pGL3 Basic. mPer15.2-luc was digested with SpeI and NcoI and then ligated with the SpeI–HindII fragment and the HindIII–NcoI PCR fragment. The ligation product, mPer16.75-luc (referred to as mPer1-luc) contains 6,756 bp of the mPer1 promoter and 5′-UTR sequence and was verified by sequencing of all junctions. pcDNA3.1-mClock was as described (15). mPer1, mPer2, mCry1, and mCry2 expression plasmids (in pcDNA3.1 V5-His; Invitrogen) were provided by Dr. Steven Reppert. The mBmal1 cDNA clone was isolated from a CLONTECH mouse skeletal muscle Marathon-Ready cDNA library using human Bmal1-specific primers (forward, 5′-TGCCCACTAGGAGATGCTCTG-3′; reverse, 5′-ACTCCACCAGTAATGCACTTTG-3′). The 5′ and 3′ ends of the mBmal1 coding region were obtained by using rapid amplification of cDNA ends on the same CLONTECH Marathon-Ready library. Full-length coding mBmal1 was cloned into the EcoRI and XbaI sites of pCDNA3.1 V5-His.
Cell Culture.
NIH 3T3 fibroblasts were grown in DMEM (Mediatech, Herndon, VA) supplemented with 10% bovine calf serum (Life Technologies, Rockville, MD), penicillin/streptomycin (Sigma), Fungizone (Life Technologies), and 4 mM L-glutamine. Cells were plated the day before transfection at 2 × 10−5 cells per well in 6-well plates. Cells were transfected in OPTI-MEM (Life Technologies) with 2 ng of mPer1-luc reporter, 100 ng of pCMV-β-galactosidase for normalization, and 3.2 × 10−14 mol of each transcription factor (corresponds to 0.2 μg of pcDNA3.1-mClock, 0.164 μg of pcDNA3.1-mBmal1, 0.192 μg of pcDNA3.1-mPer1, 0.212 μg of pcDNA3.1-mPer2, 0.156 μg of pcDNA3.1-mCry1, and 0.156 μg of pcDNA3.1-mCry2) using Lipofectamine PLUS (Life Technologies) according to the manufacturer's protocol. The total mass of transfected DNA was normalized to 1.4 μg by using the pcDNA3.1 plasmid. Forty-eight hours after transfection, cells were washed with PBS and lysed in 400 μl of reporter lysis buffer (Promega). Luminescence was measured from 20 μl of lysate in the luciferase assay system (Promega). Transfection efficiency was determined by using 20 μl of lysate in the β-galactosidase reporter system (Promega).
Transgenic Mice.
Transgenic mice were generated as described (22) by using 1 ng/μl SalI–XbaI-linearized, gel-purified mPer1-luc. Transgenic mice were identified by using PCR to detect a 560-bp fragment from luciferase (luc) (TGluc forward primer, 5′-CGCCAAAAACATAAAGAAAGGC-3′; TGluc reverse primer, 5′-TGTCCCTATCGAAGGACTCTGG-3′) and confirmed by using Southern hybridization on genomic DNA with the same luc PCR probe. The transgene copy number was determined by using Southern hybridization with an mPer1 promoter region probe (bases 5,176–5,879 of mPer1-luc); the signal was quantified by using a Storm PhosphorImager (Molecular Dynamics and Amersham Pharmacia) with the IMAGEQUANT software package (Amersham Pharmacia).
Activity in Transgenic Mice.
The wheel-running activity of singly housed animals was measured as described (23). Mice were entrained to a 12-h light/12-h dark cycle for at least 7 days and then transferred to constant darkness for at least 7 days before the given experiment. Activity data were recorded and analyzed as described (14). The presence of the transgene did not affect circadian behavior: the average period was 23.7 ± 0.4 h (average ± SEM) for P1L025 (n = 6) and 23.6 ± 0.3 h for nontransgenic CD1 control mice (n = 6; data not shown). Each animal's endogenous circadian period was measured to determine the time of sacrifice for experiments performed in constant darkness. In the light-pulse experiment, light intensity was ≈300 lux (40-watt cool-white fluorescent light).
In Situ Hybridization.
Animals were killed by cervical dislocation; the brains were removed immediately, frozen on dry ice, and stored at −80°C. For the Zeitgeber time (ZT) 18 collection and constant darkness experiments, dissections were performed under infrared light by using a Find-R-Scope viewer (FJW, Palatine, IL). Sectioning, fixation, hybridization, and washing were performed as described (17). Templates for the antisense and sense luc probes were PCR-generated by using the TGluc primers with modifications. For the antisense probe, the reverse primer contained a T7 promoter (5′-TAATACGACTCACTATAGGGAGATGTCCCTATCGAAGGACTCTGG-3′), whereas for the sense probe, the forward primer contained a T3 promoter (5′-AATTACCCTCACTAAAGGGAGACGCCAAAAACATAAAGAAAGGC-3′). The endogenous mPer1 probe, the generation of 33P-labeled probes, and quantification of exposed film (2-week exposure) were performed as described (17).
Luminescence Detection in SCN Slice Culture.
SCN were cultured as described (21). Bioluminescence was measured with photomultiplier tube (PMT) detector assemblies (HC135-11 MOD, Hamamatsu Photonics, Hamamatsu City, Japan) modified from HC135-01. PMTs (R3550) were selected with dark counts below 20 counts per second at room temperature and a prescale factor of 2. The PMT position and data acquisition were as described (21).
Numerical Fitting of mRNA Abundance.
The accumulation of an mRNA molecule as a function of synthesis rate may be described as
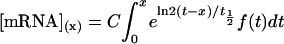 |
where [mRNA] is the concentration of mRNA, C is a constant related to transcription rate, t is the time of transcription after light exposure, x is the time of mRNA accumulation after light exposure, ƒ(t) is the function of time of transcription rates after light exposure, and t1/2 is the half-life of mRNA. This equation may be simplified and solved as
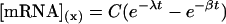 |
where λ equals ln2/t1/2, β is a constant related to accumulation rate, and ƒ(t) equals e−βt.
We used a nonlinear least-squares best fit from two independent statistical packages, SAS (SAS Institute, Cary, NC) and ORIGIN (OriginLab, Northampton, MA), to determine the parameters for λ and β for the mPer1 and luc postlight-pulse data sets. Each package generated similar values; those reported in Results were from ORIGIN.
Results
In an effort to identify mPer1 regulatory sequences upstream of the 2.0-kb mPer1 promoter region (15), a fragment from the mPer1 promoter and 5′ UTR was subcloned into the pGL3 Basic luc reporter plasmid for use in cell culture reporter and transgenic mouse studies. The product, mPer1-luc, contains 6.75 kb of the mPer1 promoter and 5′ sequence, including mPer1 exon 1, intron 1, and the untranslated portion of exon 2 up to the mPER1 initiator ATG codon (Fig. 1A). A total of five E-box elements (5′-CACGTG-3′) lie within the mPer1 promoter region; in addition, the MATINSPECTOR promoter analysis program (24) predicts binding sites for transcription factors previously implicated in the photic entrainment pathway such as the cAMP response element-binding protein and the Elk1-serum response factor (25–27). To confirm that mPer1-luc can be acted on appropriately by circadian proteins, we used transfection reporter gene assays in NIH 3T3 fibroblasts (Fig. 1B). In the presence of equimolar amounts of mCLOCK and mBMAL1, mPer1-luc expression increases ≈5.6-fold. As with previous studies, cotransfection of mCLOCK and mBMAL1 with mPER1 or mPER2 results in a modest (≈20–30%) reduction in luc expression, whereas cotransfection with mCRY1 or mCRY2 efficiently inhibits mCLOCK/mBMAL1 activity on the mPer1 promoter (10, 15–18).
Figure 1.

mPer1-luc construct and activity in transfection-based reporter assay. (A) The 6.75-kb mPer1 promoter contains five E-box sites, five putative cAMP response element-binding protein (CREB) sites, eight putative Elk1 sites, and three putative serum response factor (SRF) sites as well as exon 1 and the 5′ UTR of exon 2. (B) Transfection-based luc assays in NIH 3T3 fibroblasts using the mPer1-luc reporter. +, indicates the presence of an expression plasmid; *, indicates a significant difference from the condition of mPer1-luc reporter alone; †, indicates a significant difference from the condition of mPer1-luc reporter in the presence of only mCLOCK and mBMAL [generalized linear model ANOVA, F(5,35) = 147.91; P < 1.0 × 10−6; Scheffé's post hoc comparison, P ≤ 0.05]. Results represent two independent experiments with n = 3 of each condition.
We used this mPer1-luc reporter construct to create transgenic mice for in vivo circadian reporter studies. Six of ten transgenic founder animals successfully transmitted the transgene; from these six founders we generated individual lines named P1L005, P1L025, P1L095, P1L097, P1L128, and P1L141. The presence of the transgene was confirmed by Southern blot hybridization of genomic DNA, and transgene copy number ranged from one to ten copies (Fig. 2). Liver, heart, and tail biopsies of all six P1L lines demonstrated LUC protein activity in luciferase assays (data not shown).
Figure 2.

Diurnal variation of luc mRNA expression in the SCN of P1L mice. P1L mice were killed at ZTs 6 and 18, and coronal brain sections were hybridized in situ with a luc riboprobe. In all lines, higher expression occurs in the SCN (arrows) at ZT 6, whereas low-to-undetectable expression occurs at ZT 18. Diurnal variation was not observed in other brain regions. TG copies, indicates the transgene copy number for each line.
To characterize mPer1-luc circadian regulation in vivo, we examined luc mRNA expression in the SCN of P1L animals by using in situ hybridization. In the SCN, endogenous mPer1 mRNA expression displays a diurnal rhythm, with peak expression during the day at ZT 6 and trough expression during the night at ZT 18 (6, 7). (ZT indicates the time of day in a 12-h light/12-h dark cycle, where ZTs 0 and 12 mark the beginning of the light and dark phases, respectively. Circadian time (CT) marks subjective time in constant environmental conditions.) Transgenic animals were killed at ZTs 6 and 18, and brain sections were hybridized with a 33P-labeled luc riboprobe. All transgenic lines express luc within the SCN and display a clear diurnal pattern with high luc expression at ZT 6 and low luc expression at ZT 18 (Fig. 2). The anatomical pattern of luc expression differs among the lines; P1L025 and P1L141 exhibit highly SCN-specific expression, whereas P1L095 displays strong expression in the cortex as well as in the SCN. The hybridization signal intensities indicate a range of expression levels among transgenic lines that does not correlate well with the transgene copy number. The anatomical pattern and expression level differences may be caused by transgene position effects, but importantly, rhythmic SCN-specific expression exists in all P1L lines.
We examined SCN expression of luc mRNA in constant darkness to confirm that the 6.75-kb mPer1 fragment is sufficient to drive a circadian rhythm of expression. We chose line P1L025 because of its large colony size and relatively high SCN specificity of expression. Animals were maintained in constant darkness for at least 7 days and then were killed at 4-h CT intervals according to each animal's circadian activity rhythm. Alternate brain sections were hybridized with luc or endogenous mPer1 riboprobes. We detected a circadian rhythm of luc expression in the SCN of P1L mice that correlated with the endogenous mPer1 expression rhythm (Fig. 3). The expression of both luc and mPer1 reaches peak levels at CT 6 and displays similar amplitudes of expression (2.5- and 2.0-fold, respectively), although the expression of luc is lower than that of mPer1.
Figure 3.

Circadian rhythm of luc expression in P1L mice. Mice were maintained in constant darkness for at least 7 days before being killed, and the endogenous circadian rhythm of each animal was measured. Three to five animals were collected at each CT point beginning at 18. (A) Expression of luc. (B) Expression of endogenous mPer1.
To assess the ability of the mPer1 promoter fragment to drive a stable rhythm of luc expression over several circadian cycles, we measured luminescence in SCN explants maintained in slice culture. Each line maintained a circadian rhythm of luminescence, with peak luminescence around CT 6, for at least 5 days (Fig. 4). Bioluminescence levels roughly correlate with the level of luc mRNA expression detected by in situ hybridization in each line (Fig. 2). Bioluminescence appears to damp after the first cycle; however, the initially high signal may be caused by the release of LUC protein from damaged cells after slice culture preparation.
Figure 4.

Persistent circadian rhythms of luminescence in SCN culture. Animals were entrained to a 12-h light/12-h dark cycle. Mice were killed 1 h before lights-off, and SCN explants were cultured for 5 days in the presence of luciferin.
Finally, we used the mPer1-luc reporter in vivo to determine whether the promoter fragment contains light-responsive element(s). A brief light pulse (15–30 min) during the early or late subjective night is known to induce mPer1 expression in the SCN rapidly (12, 13, 28). To extend the characterization of mPer1 light induction, we performed a 6-h time course in which we transferred mice to a light box at CT 17 and then collected brains at 0.5, 1, 1.5, 2, 3, and 6 h of light exposure. Alternate sections were hybridized with luc and mPer1 riboprobes. In P1L mice, the light pulse at CT 17 rapidly induces expression of both luc and mPer1 with peak induction of both genes at 90 min of light exposure (Fig. 5). Endogenous mPer1 expression decreases by 2 h of light exposure and returns to baseline levels by 6 h in the light. In contrast, luc expression remains high for at least 3 h in the light, only moderately decreases by 6 h of light exposure, and remains significantly different from baseline levels at the 6-h time point. We numerically fit the mPer1 and luc data sets to determine the half-life of each mRNA. This method reported the half-life of mPer1 as 0.93 h and that of luc as 1.49 h; however, these values fell within large error margins because of the small number of collection times. Therefore, the significance of the half-life difference cannot be determined currently.
Figure 5.

Light-responsiveness of mPer1-luc expression in transgenic mice. Mice were maintained in constant darkness for at least 7 days before the experiment, and the endogenous circadian rhythm of each animal was measured. At CT 17, mice were transferred to a light box. Time-course graphs indicate the fold induction from baseline expression at CT 17. The line through each data set is the nonlinear least-squares fit to the mean for each time point assuming a biexponential function (see Results). (A) Time course of luc mRNA levels. *, denotes significant differences in signal intensity between time = 0 (dark control) and the indicated time [generalized linear model ANOVA, F(6,22) = 5.32; P = 0.004039]. Biexponential function parameters: λ = 0.46439 and β = 0.46547. (B) Time course of endogenous mPer1 mRNA levels. *, denotes significant differences in signal intensity between time = 0 (dark control) and the indicated time [generalized linear model ANOVA, F(6,22) = 9.32; P = 0.000232]. Biexponential function parameters: λ = 0.74934 and β = 0.93453.
Discussion
The luc reporter has been used as a transgene to monitor circadian rhythms in several organisms including cyanobacteria, Arabidopsis, and Drosophila (29–33). Recently this approach was applied in P1L rats by using cultured tissue explants to assess the relationship between the SCN and the periphery in response to environmental changes (21). However, luminescence in tissue culture cannot be used to monitor all changes in promoter activity that occur in vivo such as the response to light input during the subjective night.
To address these issues, we created P1L mice by using a 6.75-kb mPer1 promoter fragment and measured luc expression in vivo. All P1L mouse lines express a diurnal rhythm of luc mRNA in the SCN, which demonstrates the efficacy of this promoter in driving rhythmic expression (Fig. 2). This mPer1 promoter is sufficient to regulate circadian and light-responsive expression in the SCN (Figs. 3 and 5) as well as drive a sustained circadian rhythm of LUC protein activity in SCN culture (Fig. 4), which is similar to other transgenic studies using the mPer1 promoter to drive luc (20, 21, 34). In the SCN of P1L mice, the in vivo phase and amplitude of the luc mRNA rhythm are nearly indistinguishable from those of mPer1 (Fig. 3).
However, a 6-h light pulse during the subjective night revealed distinct patterns of mPer1 and luc mRNA abundance after prolonged exposure to light. Previous studies used a short light pulse to elicit a behavioral phase shift and then monitored mPer1 mRNA levels over several hours (12, 13, 28). The short light-pulse approach is useful to correlate threshold responses of gene activity and behavioral phase-shifting, but it does not address longer-term aspects of gene regulation. To determine the mPer1 regulatory effects of a saturating light pulse (one that elicits maximal behavioral phase shifts), we exposed mice to light for 6 h beginning at CT 17. Light rapidly induced mPer1 and luc mRNA, but after 6 h the mPer1 mRNA level decreased to baseline, whereas the luc mRNA level remained high. Three models could account for these mRNA abundance differences. The first model predicts that light initially activates both the endogenous mPer1 promoter and the transgenic mPer1-luc promoter, but that long-term light exposure leads to inhibition of mPer1 transcription, whereas luc transcription remains activated. This model assumes equal rates of mPer1 and luc mRNA decay, and it would indicate that the mPer1-luc transgene lacks light-responsive inhibitory elements. The second model predicts that light activates both the endogenous and transgenic promoters continuously for 6 h, but that mPer1 mRNA is less stable than luc mRNA. In this case, mRNA abundance could continue to rise throughout the time course or reach a steady state, depending on the half-life of the mRNA molecule. However, it is unlikely for mRNA abundance to reach a peak and then fall to basal levels in the continuous presence of the stimulus unless that same stimulus also triggers a specific degradation pathway later in the time course. Finally, the third model predicts that light initially activates both the endogenous and transgenic promoters, long-term light-induced activation is blocked later at both promoters, and mPer1 mRNA is less stable than luc mRNA. In this case, the duration of uninhibited activation and the half-life of the mRNA molecule together determine the rate of decreasing mRNA abundance.
Degradation of mRNA is an important regulatory mechanism, particularly for genes such as c-fos that are expressed transiently in response to extracellular stimuli (35). The c-fos mRNA is extremely labile, and its regulated expression is among the best understood (36, 37). Sequences within the c-fos mRNA direct its own deadenylation and rapid degradation; one such sequence lies within the AU-rich element in the 3′ UTR (38–42). Comparison of 3′ UTRs in other labile mRNAs suggested the sequence AUUUA as the destabilizing signal (43, 44). However, a subsequent study demonstrated that the nonamer UUAUUUAUU (the AU-rich destabilizing motif) is the minimal 3′-UTR signal that is capable of directing efficient mRNA degradation (45). High AU content within the 3′ UTR seems to enhance AU-rich destabilizing motif-directed destabilization, because a heterologous mRNA containing the entire c-fos 3′ AU-rich element is less stable than the same mRNA containing only the AU-rich destabilizing motif (45).
Stability of mRNA likely plays a regulatory role within the mammalian circadian oscillator as well. Similar to c-fos, mPer1 can be induced by an extracellular stimulus (light); therefore, rapid degradation of mPer1 mRNA may be involved in the regulation of message levels. Indeed, in Drosophila, the degradation rate of per mRNA appears rhythmic and contributes to the overall rhythm of per expression (46). The 3′ UTR of mPer1 is not particularly AU-rich, but it does contain the consensus UUAUUUAUU sequence 335 bases downstream of the mPER1 stop codon (GenBank accession number AF022992); this destabilizing motif is conserved in human Per1 as well, 342 bases downstream of the human PER1 stop codon (GenBank accession number AB030817). The luc 3′ UTR, in contrast, contains no sequence that resembles the AU-rich destabilizing motif signal (GenBank accession number U47295). In fact, sequence analyses show that no other known circadian gene contains the UUAUUUAUU signal sequence in its 3′ UTR. These observations allow us to predict that mPer1 mRNA is less stable than luc mRNA, which supports our second and third mechanistic models. We still cannot rule out the absence of an inhibitory element in the mPer1-luc transgene (model 1); however, the data and sequence analyses better fit the models that account for mRNA stability.
Several issues beyond inherent mRNA stability must be addressed to explain the mPer1 and luc light-induction profile differences fully. Specifically, does light activate a pathway that further accelerates the rate of mPer1 mRNA degradation after long-term light exposure (model 2), is transcriptional activation at the mPer1 promoter blocked after long-term light exposure (model 3), or can both mechanisms apply? We used a theoretical approach in an effort to discriminate between models 2 and 3 in explaining the differential accumulation kinetics of mPer1 and luc. Because both models depend on mPer1 and luc having vastly different rates of accumulation and/or decay, we attempted to determine a function that fit the results for mPer1 and luc with appropriate time constants. We assumed that mRNA levels are determined by the difference between two exponential functions (for accumulation and degradation of mRNA molecules). These analyses indicated that the half-life of luc is longer than that of mPer1 (1.49 versus 0.93 h, respectively), but they failed to find a significant difference between the parameters for mPer1 and luc. Although other functions could be tried, future attempts to model the more rapid degradation of mPer1 mRNA in the light will require more frequent measurements of mRNA levels.
Determination of the precise activity of mPER1 remains a topic of intense investigation. Current evidence strongly suggests that the primary role for mPER1 lies in the light entrainment pathway; light induces mPer1 expression during the night, the presence of mPer1 antisense oligonucleotides inhibits the phase-shifting effects of light (47), and mPER1 levels remain high 4–9 h after a light pulse, long after control mPER1 abundance has fallen to trough values (8). Recently, targeted mutations of the mPer1 locus, named mPer1ldc and mPer1Brdm1, demonstrate the requirement for mPER1 activity within the oscillator mechanism as well (48, 49). Both mutations result in short circadian period length, and the mPer1ldc mutation leads to an eventual loss of circadian rhythms in constant darkness (48, 49). The mPer1ldc mutation also results in decreased peak mCRY1 protein levels in the SCN during the first day in constant darkness, whereas mCry1 mRNA abundance is not altered (48). These results are consistent with a model in which mPER1 stabilizes mCRY1 (and possibly mCRY2) independently of environmental conditions, and the increase in mPer1 mRNA and protein abundance in response to light leads to a sustained presence of mCRY proteins and a resultant phase shift (8, 48). In this model, mPER1 abundance must be controlled tightly to prevent inappropriate stabilization mCRY inhibitor activity. Posttranslational phosphorylation of mPER1 by casein kinase-1ɛ has been proposed to elicit mPER1 degradation (50); here, accelerated decay of mPer1 mRNA suggests the presence of a posttranscriptional regulatory pathway as well. This additional level of regulation suggests a highly refined temporal control of mPER1 abundance in both constant conditions and in response to light. We expect the continued study of mPer1 through targeted mutations, luc and green fluorescent protein reporters, and inducible constructs to further clarify these models.
Acknowledgments
We thank D. King for isolation of mPer1-containing bacterial artificial chromosome clones and K. Shimomura for helpful discussions. J.S.T. is an Investigator in the Howard Hughes Medical Institute. This work was supported by the National Science Foundation Center for Biological Timing and R37 MH39592 (to J.S.T.).
Abbreviations
- SCN
suprachiasmatic nucleus/nuclei
- P1L
mPer1-luc transgenic line
- UTR
untranslated region
- ZT
Zeitgeber time
- CT
circadian time
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Dunlap J C. Cell. 1999;96:271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 2.Wilsbacher L D, Takahashi J S. Curr Opin Genet Dev. 1998;8:595–602. doi: 10.1016/s0959-437x(98)80017-8. [DOI] [PubMed] [Google Scholar]
- 3.King D P, Takahashi J S. Annu Rev Neurosci. 2000;23:713–742. doi: 10.1146/annurev.neuro.23.1.713. [DOI] [PubMed] [Google Scholar]
- 4.Wager-Smith K, Kay S A. Nat Genet. 2000;26:23–27. doi: 10.1038/79134. [DOI] [PubMed] [Google Scholar]
- 5.Miyamoto Y, Sancar A. Proc Natl Acad Sci USA. 1998;95:6097–6102. doi: 10.1073/pnas.95.11.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Z S, Albrecht U, Zhuchenko O, Bailey J, Eichele G, Lee C C. Cell. 1997;90:1003–1011. doi: 10.1016/s0092-8674(00)80366-9. [DOI] [PubMed] [Google Scholar]
- 7.Tei H, Okamura H, Shigeyoshi Y, Fukuhara C, Ozawa R, Hirose M, Sakaki Y. Nature (London) 1997;389:512–516. doi: 10.1038/39086. [DOI] [PubMed] [Google Scholar]
- 8.Field M D, Maywood E S, O'Brien J A, Weaver D R, Reppert S M, Hastings M H. Neuron. 2000;25:437–447. doi: 10.1016/s0896-6273(00)80906-x. [DOI] [PubMed] [Google Scholar]
- 9.Hastings M H, Field M D, Maywood E S, Weaver D R, Reppert S M. J Neurosci. 1999;19:RC11. doi: 10.1523/JNEUROSCI.19-12-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kume K, Zylka M J, Sriram S, Shearman L P, Weaver D R, Jin X, Maywood E S, Hastings M H, Reppert S M. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 11.Okamura H, Miyake S, Sumi Y, Yamaguchi S, Yasui A, Muijtjens M, Hoeijmakers J H, van der Horst G T. Science. 1999;286:2531–2534. doi: 10.1126/science.286.5449.2531. [DOI] [PubMed] [Google Scholar]
- 12.Shearman L P, Zylka M J, Weaver D R, Kolakowski L F J, Reppert S M. Neuron. 1997;19:1261–1269. doi: 10.1016/s0896-6273(00)80417-1. [DOI] [PubMed] [Google Scholar]
- 13.Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros J J, Dunlap J C, Okamura H. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- 14.Vitaterna M H, Selby C P, Todo T, Niwa H, Thompson C, Fruechte E M, Hitomi K, Thresher R J, Ishikawa T, Miyazaki J, Takahashi J S, Sancar A. Proc Natl Acad Sci USA. 1999;96:12114–12119. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gekakis N, Staknis D, Nguyen H B, Davis F C, Wilsbacher L D, King D P, Takahashi J S, Weitz C J. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 16.Jin X, Shearman L P, Weaver D R, Zylka M J, de Vries G J, Reppert S M. Cell. 1999;96:57–68. doi: 10.1016/s0092-8674(00)80959-9. [DOI] [PubMed] [Google Scholar]
- 17.Sangoram A M, Saez L, Antoch M P, Gekakis N, Staknis D, Whiteley A, Fruechte E M, Vitaterna M H, Shimomura K, King D P, Young M W, Weitz C J, Takahashi J S. Neuron. 1998;21:1101–1113. doi: 10.1016/s0896-6273(00)80627-3. [DOI] [PubMed] [Google Scholar]
- 18.Griffin E A J, Staknis D, Weitz C J. Science. 1999;286:768–771. doi: 10.1126/science.286.5440.768. [DOI] [PubMed] [Google Scholar]
- 19.Kuhlman S J, Quintero J E, McMahon D G. Neuroreport. 2000;11:1479–1482. [PubMed] [Google Scholar]
- 20.Yamaguchi S, Mitsui S, Miyake S, Yan L, Onishi H, Yagita K, Suzuki M, Shibata S, Kobayashi M, Okamura H. Curr Biol. 2000;10:873–876. doi: 10.1016/s0960-9822(00)00602-3. [DOI] [PubMed] [Google Scholar]
- 21.Yamazaki S, Numano R, Michikazu A, Hida A, Takahashi R, Ueda M, Block G D, Sakaki Y, Menaker M, Tei H. Science. 2000;288:682–685. doi: 10.1126/science.288.5466.682. [DOI] [PubMed] [Google Scholar]
- 22.Antoch M P, Song E-J, Chang A-M, Vitaterna M H, Zhao Y, Wilsbacher L D, Sangoram A M, King D P, Pinto L H, Takahashi J S. Cell. 1997;89:655–667. doi: 10.1016/s0092-8674(00)80246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vitaterna M H, King D P, Chang A-M, Kornhauser J M, Lowrey P L, McDonald J D, Dove W F, Pinto L H, Turek F W, Takahashi J S. Science. 1994;264:719–725. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quandt K, Frech K, Karas H, Wingender E, Werner T. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ginty D D, Kornhauser J M, Thompson M A, Bading H, Mayo K E, Takahashi J S, Greenberg M E. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 26.Kornhauser J M, Mayo K E, Takahashi J S. Behav Genet. 1996;26:221–240. doi: 10.1007/BF02359382. [DOI] [PubMed] [Google Scholar]
- 27.Xia Z, Dudek H, Miranti C K, Greenberg M E. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albrecht U, Sun Z S, Eichele G, Lee C C. Cell. 1997;91:1055–1064. doi: 10.1016/s0092-8674(00)80495-x. [DOI] [PubMed] [Google Scholar]
- 29.Kondo T, Mori T, Lebedeva N V, Aoki S, Ishiura M, Golden S S. Science. 1997;275:224–227. doi: 10.1126/science.275.5297.224. [DOI] [PubMed] [Google Scholar]
- 30.Millar A J, Carre I A, Strayer C A, Chua N H, Kay S A. Science. 1995;267:1161–1163. doi: 10.1126/science.7855595. [DOI] [PubMed] [Google Scholar]
- 31.Plautz J D, Kaneko M, Hall J C, Kay S A. Science. 1997;278:1632–1635. doi: 10.1126/science.278.5343.1632. [DOI] [PubMed] [Google Scholar]
- 32.Stanewsky R, Frisch B, Brandes C, Hamblen-Coyle M J, Rosbash M, Hall J C. J Neurosci. 1997;17:676–696. doi: 10.1523/JNEUROSCI.17-02-00676.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanewsky R, Kaneko M, Emery P, Beretta B, Wager-Smith K, Kay S A, Rosbash M, Hall J C. Cell. 1998;95:681–692. doi: 10.1016/s0092-8674(00)81638-4. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi S, Kobayashi M, Mitsui S, Ishida Y, van der Horst G T, Suzuki M, Shibata S, Okamura H. Nature (London) 2001;409:684. doi: 10.1038/35055628. [DOI] [PubMed] [Google Scholar]
- 35.Sachs A B. Cell. 1993;74:413–421. doi: 10.1016/0092-8674(93)80043-e. [DOI] [PubMed] [Google Scholar]
- 36.Greenberg M E, Greene L A, Ziff E B. J Biol Chem. 1985;260:14101–14110. [PubMed] [Google Scholar]
- 37.Muller R, Bravo R, Burckhardt J, Curran T. Nature (London) 1984;312:716–720. doi: 10.1038/312716a0. [DOI] [PubMed] [Google Scholar]
- 38.Chen C Y, Chen T M, Shyu A B. Mol Cell Biol. 1994;14:416–426. doi: 10.1128/mcb.14.1.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shyu A B, Greenberg M E, Belasco J G. Genes Dev. 1989;3:60–72. doi: 10.1101/gad.3.1.60. [DOI] [PubMed] [Google Scholar]
- 40.Shyu A B, Belasco J G, Greenberg M E. Genes Dev. 1991;5:221–231. doi: 10.1101/gad.5.2.221. [DOI] [PubMed] [Google Scholar]
- 41.Wellington C L, Greenberg M E, Belasco J G. Mol Cell Biol. 1993;13:5034–5042. doi: 10.1128/mcb.13.8.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson T, Treisman R. Nature (London) 1988;336:396–399. doi: 10.1038/336396a0. [DOI] [PubMed] [Google Scholar]
- 43.Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Proc Natl Acad Sci USA. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw G, Kamen R. Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- 45.Zubiaga A M, Belasco J G, Greenberg M E. Mol Cell Biol. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.So W V, Rosbash M. EMBO J. 1997;16:7146–7155. doi: 10.1093/emboj/16.23.7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akiyama M, Kouzu Y, Takahashi S, Wakamatsu H, Moriya T, Maetani M, Watanabe S, Tei H, Sakaki Y, Shibata S. J Neurosci. 1999;19:1115–1121. doi: 10.1523/JNEUROSCI.19-03-01115.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bae K, Jin X, Maywood E S, Hastings M H, Reppert S M, Weaver D R. Neuron. 2001;30:525–536. doi: 10.1016/s0896-6273(01)00302-6. [DOI] [PubMed] [Google Scholar]
- 49.Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, Li Q, Sun Z S, Eichele G, Bradley A, Lee C C. Cell. 2001;105:683–694. doi: 10.1016/s0092-8674(01)00380-4. [DOI] [PubMed] [Google Scholar]
- 50.Lowrey P L, Shimomura K, Antoch M A, Yamakazi S, Zemenides P D, Ralph M R, Menaker M, Takahashi J S. Science. 2000;288:483–491. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]