

Abstract
Reactive oxygen intermediates (ROI) are strongly associated with plant defense responses. The origin of these ROI has been controversial. Arabidopsis respiratory burst oxidase homologues (rboh genes) have been proposed to play a role in ROI generation. We analyzed lines carrying dSpm insertions in the highly expressed AtrbohD and AtrbohF genes. Both are required for full ROI production observed during incompatible interactions with the bacterial pathogen Pseudomonas syringae pv. tomato DC3000(avrRpm1) and the oomycete parasite Peronospora parasitica. We also observed reduced cell death, visualized by trypan blue stain and reduced electrolyte leakage, in the Atrboh mutants after DC3000(avrRpm1) inoculation. However, enhanced cell death is observed after infection of mutant lines with P. parasitica. Paradoxically, although atrbohD mutation eliminated the majority of total ROI production, atrbohF mutation exhibited the strongest effect on cell death.
Early production of
reactive oxygen intermediates (ROI) is a hallmark of the plant defense
response. Many studies document the detection of O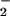 and/or its dismutation product,
H2O2, during incompatible
interactions between resistant plants and avirulent pathogens (1–4).
During the defense response, ROI can inhibit the pathogen by
strengthening cell walls via oxidative cross-linking of cell-wall
glycoproteins (5) or by directly killing the pathogen (3). ROI also
could act as signals to induce further defenses (6–9), including the
initiation of the hypersensitive response (HR). Nitric oxide (NO),
another reactive molecule that works synergistically with ROI in
driving mammalian cell death in macrophages (10), also has emerged as
an important mediator of plant defense response and cell death
signaling in plants (11, 12).
and/or its dismutation product,
H2O2, during incompatible
interactions between resistant plants and avirulent pathogens (1–4).
During the defense response, ROI can inhibit the pathogen by
strengthening cell walls via oxidative cross-linking of cell-wall
glycoproteins (5) or by directly killing the pathogen (3). ROI also
could act as signals to induce further defenses (6–9), including the
initiation of the hypersensitive response (HR). Nitric oxide (NO),
another reactive molecule that works synergistically with ROI in
driving mammalian cell death in macrophages (10), also has emerged as
an important mediator of plant defense response and cell death
signaling in plants (11, 12).
The likely source of ROI is an NADPH oxidase, originally postulated to be membrane-bound and to use molecular oxygen to make superoxide, based partly on inhibition studies using diphenylene iodonium (1). This NADPH oxidase is thought to be at least partially similar to the one present in the mammalian phagocytes (13, 14). In activated macrophages, the respiratory burst NADPH oxidase (RBO) is responsible for generation ROI (14). Mutations in gp91phox, encoding the catalytic subunit of the NADPH oxidase, result in chronic granulomatous disease, an immunological disorder in which macrophages are unable to stop the spread of infection (15). Several plant rboh genes, homologous to gp91phox, were identified, although they carry a 300-aa N-terminal extension compared with the mammalian proteins (16–19). Rac homologues in rice have been implicated in pathogen-induced cell death occurring in this plant (20), and Rac is required for assembly of an active respiratory burst NADPH oxidase in animals (14). However, no homologues to the p47 or p67 regulators of the mammalian NADPH oxidase were found in the Arabidopsis genome (21). These findings suggest that a superoxide-generating NADPH oxidase does exist in plants, although the plant NADPH oxidase is most likely regulated differently than the one present in mammalian macrophages. Alternative mechanisms of ROI production in plants also have been proposed invoking cell-wall-bound peroxidases as the main ROI source (22, 23).
Using a dSpm insertion mutagenesis system (24), we isolated mutants in eight Atrboh genes. We demonstrate that atrbohD and atrbohF mutations largely eliminate ROI accumulation during disease-resistance reactions of Arabidopsis to avirulent Pseudomonas syringae and Peronospora parasitica (Pp). Hence, an NADPH oxidase is responsible for ROI accumulation during some defense responses in Arabidopsis.
Materials and Methods
Identification of the Atrboh Mutants.
Insertions in AtrbohD and F were identified through a PCR screen on genomic DNA extracted from pools of Col-0 plants containing dSpm transposon insertions (24). Primers used include dSpm11 and dSpm1 from the transposon (24) and specific primers from each Atrboh gene: D122 (ATGAAAATGAGACGAGGCAATTC), D92b (GGATACTGATCATAGGCGTGGCTCCA), F171 (CTTCCGATATCCTTCAACCAACTC), and F172 (GAGATTGCCTTTATACTATAAGTG).
Insertions in each gene were confirmed by sequencing PCR products spanning the insertion. All of the lines identified contained a single transposon and were derived from gluforinate ammonium (BASTA)-resistant heterozygote parents. Plants were grown in a chamber under a 9-h photoperiod, 24°C day and 20°C night temperatures, 60% relative humidity, and 250 μeinsteins/m2/s.
Test with Pathogens.
Bacterial strains used in this study were P. syringae pv. tomato (Pst) DC3000, DC3000(avrRpm1), and DC3000(avrRpt2). Four-week-old plants were inoculated in a standard manner (25). atrbohD/F plants that did not display necrotic lesions were selected before infection. Various Pp isolates, Emco5, Cala2, Emwa1, Noco2, or Ahco1, were sprayed on 11-day-old seedlings (26).
Cell Death Measurements.
Trypan blue stain, used to visualize dying cells, was performed as described (27). The protocol for electrolyte leakage was adapted from Dellagi et al. (28). Four-week-old plants were injected with bacteria in 10 mM MgCl2. Ten minutes after injection, 7.5-mm-diameter leaf discs were collected from the injected area and washed extensively with water for 50 min, and then four discs were placed in a tube with 6 ml of water. Conductivity measurements (3–4 replicates for each treatment) were taken from the tubes over time by using an Orion (Boston) conductivity meter, model 130. The units of this measurement are μS/cm, where cm refers to the distance between electrodes. No increase in the conductivity was observed when plants were injected with MgCl2 or virulent DC3000.
Detection of ROI.
To visualize H2O2 in situ, 3,3′-diaminobenzidine (DAB) staining was performed on Arabidopsis seedlings sprayed with Pp spores as described (29). For bacterial experiments with DC3000(avrRPm1), leaves were collected 2 h after injection of the bacteria and vacuum-infiltrated with the DAB solution. Leaves then were placed in a plastic box under high humidity until brown precipitate was observed (5–6 h) and then fixed with a solution of 3:1:1 ethanol/lactic acid/glycerol. Catalase effectively eliminated the DAB stain. Application of glucose/glucose oxidase or H2O2 directly to leaves was used to verify that atrboh mutants are not impaired in the detection of ROI by this method. Quantification of the staining was performed with quantiscan (Biosoft, Milltown, NJ), using 20 leaves similar to the ones presented in Fig. 2A, coming from two different experiments. The index of staining was calculated for each leaf injected as the average of the index of brown pixels measured in three points inside the injected area minus the average of three points in the opposite side of each leaf.
Figure 2.

Reduced ROI accumulation and cell death in the atrboh mutants after inoculation of avirulent bacteria. (A) In situ detection of peroxides by using DAB staining on wt Col-0 and atrboh mutant leaves 5 h postinoculation with DC3000(avrRpm1) at 2.5 × 107 cfu/ml. (B) Quantitative analysis of DAB on leaves 6 h postinoculation with DC3000(avrRpm1) at 2.5 × 107 cfu/ml, DC3000 (same inoculum concentration), or 10 mM MgCl2. (Bars = SD.) (C) Detail of leaves stained with DAB 5 h postinoculation with DC3000(avrRpm1) at 2.5 × 107 cfu/ml. Genotypes are labeled at the top, left to right: Col-0, atrbohD, atrbohF, and atrbohD/F. (D) Detail of leaves stained with trypan blue 5 h postinoculation with DC3000(avrRpm1) at 2.5 × 107 cfu/ml. (E) Detail of leaves stained with trypan blue 12 h postinoculation with lower dose of DC3000(avrRpm1), 106 cfu/ml. (Bar = 25 μm.) All images in C–E have the same magnification.
Results
Identification of Mutants in AtrbohD and AtrbohF.
AtrbohD and AtrbohF are the highest expressed Atrboh genes in leaves (18), the tissue infected by our test pathogens. Thus, we focused on mutants in these two genes to test the role of NADPH oxidase in ROI production. We identified atrbohD and atrbohF mutants through PCR-based screening of pooled DNA from Arabidopsis plants containing random dSpm transposons in their genome (24). Insertion D3 is located in the fifth exon of AtrbohD after codon P535 (after nucleotide T1605 from the ATG). Insertions F3 and F4 are located in the first exon of AtrbohF, in codon Q177 (after nucleotide A530) and I221 (after nucleotide A661). F5 is in the second intron of AtrbohF, 11 nt downstream of the splice donor. Lines homozygous for the respective insertions were identified by PCR after gluforinate ammonium selection in the next generation. All three independent atrbohF mutant lines gave similar phenotypes with pathogens; therefore, we used only insertion F3 for these studies.
To verify that these mutant lines are null alleles, we performed RNA gel blot analyses and reverse transcription–PCR with specific primers on total RNA extracted from 10-day-old seedlings. These analyses revealed no normal full-length transcript, some aberrant transcripts, and chimeras between the transposon and the atrboh mRNA that presumably would give rise to nonfunctional proteins. atrbohD and atrbohF mutants are morphologically normal, although they look slightly smaller than wild type (wt) (Fig. 1B). This phenotype is enhanced in the atrbohD/F double mutant. Four-week-old atrbohD/F plants display some necrotic lesions and callose deposition (Fig. 1 C and D). Some of these plants stop growing and die before setting seeds. Importantly, in the experiments described below, we used morphologically normal plants at ages well before this phenotype developed.
Figure 1.

Arabidopsis atrbohD and atrbohF mutants. (A) Schematic representation of the transposon insertions in the Atrboh D and F genes. Boxes represent the exons of the genes, and red triangles mark the dSpm transposon insertions. (B) Representative 4-week-old rosettes. (C) Trypan blue staining of an atrbohD/F double mutant leaf, displaying spontaneous necrosis. (D) Detail of an atrbohD/F leaf stained with aniline blue to show callose deposition. Note that infection experiments are performed on young plants that do not exhibit ectopic cell death. [Bar = 1 cm (B) and 1 mm (D).]
Reduction of ROI Accumulation and Diminished Cell Death Symptoms in the atrboh Mutants After Inoculation with Avirulent P. syringae.
We examined the contribution of AtrbohD and AtrbohF to ROI production during responses to pathogenic bacteria. DAB polymerizes on contact with H2O2 in a reaction requiring peroxidase. Thus, H2O2 is visualized in situ as a reddish-brown precipitate (29). We observed a strong, brown precipitate in wt plants starting 3–4 h postinjection of the avirulent strain DC3000(avrRpm1) (Fig. 2 A and C), in accordance with previous observations of RPM1 function, by using CeCl3 staining (30). No stain was observed after injection of virulent DC3000 or MgCl2 (data not shown).
We conducted similar analyses with the atrbohD, atrbohF, and atrbohD/F double mutants. Although there was no change in DAB staining intensity in the atrbohF mutant compared with wt, staining is greatly reduced in the mutant atrbohD and in the double mutant atrbohD/F (Fig. 2 A and C). Similar results were obtained with DC3000(avrRpt2) (which produces a delayed HR compared with DC3000(avrRpm1); data not shown). Quantification of the DAB stain demonstrates that atrbohD and atrbohD/F double mutants display levels of DAB precipitate comparable to control plants inoculated with virulent DC3000 or MgCl2 (Fig. 2B). Trypan blue staining performed in additional leaves from the same experiment indicates that the DAB-staining procedure does not interfere with the progression of the HR (see below). Thus, Arabidopsis AtrbohD is required for most of the ROI observed after inoculation with avirulent Pst, whereas AtrbohF contributes little to this oxidative burst.
We wanted to study the requirement of Atrboh-generated ROI for HR and disease resistance. Although ROI production is strongly correlated with the early defense response, temporally preceding either HR or cessation of pathogen growth (31), it is unclear whether the oxidative burst is required for HR and/or stopping pathogen growth. Inoculation of avirulent DC3000(avrRpm1) or virulent DC3000 bacteria under conditions in which the former initiates a rapid hypersensitive reaction (≈2.5 × 107 cfu/ml) showed no clear differences in trypan blue staining between wt and atrboh mutants (Fig. 2D and data not shown). However, the atrboh mutants displayed less trypan blue stain than the wt after injection of an inoculum (106 cfu/ml) of DC3000(avrRpm1) that more closely reflects natural infection pressure. In particular, the atrbohD/F double mutant displayed reduced localized cell death compared with Col-0 (Fig. 2E). Measurement of in planta growth of both avirulent and virulent bacterial strains revealed no significant differences between the wt and the atrboh mutants (data not shown).
We monitored electrolyte leakage to quantify cell death during the HR (28). Leaf discs excised from control wt leaves infiltrated with DC3000(avrRpm1) exhibited significantly increased ion leakage compared with MgCl2 or DC3000 controls 4–6 h after injection (Fig. 3A). This corresponds to a time when leaves begin to exhibit macroscopic HR. The atrbohD/F double mutant displayed significantly lower ion leakage in repeated experiments (Fig. 3A). Mutations in atrbohF and atrbohD act redundantly in this assay. atrbohF mutants exhibit reproducibly greater diminution of ion leakage than atrbohD (Fig. 3B). Collectively, these data suggest that AtrbohD is responsible for most of the ROI produced in response to inoculation of avirulent Pst. Conversely, AtrbohF plays a key role in HR, particularly at low-dose inoculation. By contrast, ROI apparently are dispensable for mediating at least the RPM1-dependent signals that limit bacterial growth.
Figure 3.

Reduced electrolyte leakage in atrboh mutants after inoculation with avirulent bacteria. (A) Conductivity (μS/cm) of solution containing leaf discs from either wt Col-0 or atrbohD/F mutant inoculated with avirulent bacteria DC3000(avrRpm1) at 107 cfu/ml, virulent bacteria DC3000 at 107 cfu/ml, or 10 mM MgCl2. (B) Detailed differences in conductivity between Col-0 and the atrboh mutants during the incompatible interaction DC3000(avrRpm1) at 107 cfu/ml. Each value represents the mean and SD of three replicates (compatible interaction and MgCl2) or four replicates (incompatible interactions) per experiment. The experiment was repeated three times with similar results.
Atrboh Genes Modulate the Response to Pp Emco5 Infection.
We evaluated whether the atrboh mutants have an effect on the resistance response against the oomycete parasite Pp. Pp isolate Emco5 is fully virulent on Col-0 cotyledons, but triggers resistance, conditioned by a single locus, in the emerging true leaves (J. M. McDowell, S. Williams, and J.L.D., unpublished data). This “adult resistance” is associated with HR trailing the growing hyphae (trailing necrosis). DAB stain reveals peroxide production in the wt around the growing Emco5 hyphae in a pattern that resembles the appearance of HR revealed by trypan blue staining (Fig. 4). Thus, this adult resistance is weaker than the resistance mediated by the majority of R genes directed against Pp, because the latter typically are associated with discrete HR.
Figure 4.

Reduced peroxide accumulation and enhanced cell death in the atrboh mutants after inoculation with Pp isolate Emco5. (A) DAB staining of leaves 3 days after inoculation with Pp isolate Emco5. Arrows indicate HR sites in atrbohD/F. (B) Trypan blue stain of leaves from the same experiment 3 days after inoculation with Pp isolate Emco5. The experiment was repeated four times; ≈10 leaves per genotype per experiment analyzed. (Bar = 50 μm for all.)
Peroxide production, but not the typical trailing necrosis, triggered by Pp Emco5 is greatly reduced in atrbohD mutants (Fig. 4). Strikingly, both DAB staining and HR are enhanced, and focused into discrete HR lesions, in atrbohF plants (Fig. 4). Peroxide production is eliminated in the atrbohD/F double mutant but HR is also enhanced. The enhanced HR surrounds the emerging hyphae and prevents their growth in nearly all cases. This enhanced response also results in less sporangiophore formation in true leaves of the atrbohF and atrbohD/F double mutants compared with wt (Fig. 5). Consequently, at least in this interaction, assaying the weak adult R function directed toward Pp Emco5, atrbohD, and atrbohF results in separable phenotypes. The atrbohD mutation eliminates most of the peroxide produced, whereas atrbohF actually allows enhanced cell HR and improved resistance toward the parasite.
Figure 5.

Reduced sporangiophore formation in the atrboh mutants. The histogram represents the percentage of first leaves of seedlings (genotypes at bottom) that display >20, 11–20, 1–10, or 0 sporangiophores 7 days postinoculation with Pp isolate Emco5. The data are the mean and SD from three different experiments representing more than 200 total leaves evaluated for each genotype.
We also infected the Atrboh mutants with two additional avirulent isolates of Pp, Emwa1 and Cala2, that are recognized by the Col-0 RPP4 and RPP2 genes, respectively. These interactions result in typical HR limited to a discrete group of cells (32). atrbohD and the atrbohD/F double mutants exhibited reduced peroxide production in these interactions (data not shown). However, depletion of ROI in these experiments had no effect on either cell death or resistance. Additionally, we observed no DAB staining and no effect on disease progression after infection with virulent Pp isolates Noco2 and Ahco1 in either the Col-0 or the atrboh mutants (data not shown). This demonstrates that our data are not a result of general induction of defense response.
Discussion
We used reverse genetics in Arabidopsis to define functions for two Atrboh genes in the production of ROI during the defense response. We observed depletion of DAB staining, a standard marker of ROI accumulation, in these mutants after challenge with avirulent Pst or Pp. The AtrbohD gene is required for most of the ROI observed after inoculation with avirulent Pst, whereas AtrbohF makes a more limited contribution. In contrast, the atrboh mutants exhibit enhanced HR and less sporangiophore formation in response to the weakly avirulent Pp Emco5. Interestingly, although atrbohF exhibits minor diminution of ROI production, it expresses strongly enhanced cell death phenotypes. Finally, we demonstrate that the atrbohD/F double mutant exhibits even further reduced HR against Pst but enhances a weak resistance response against Pp. Thus, our most important findings are: (i) extracellular ROI production in Arabidopsis requires Atrboh function, (ii) AtrbohD and AtrbohF share functions in ROI generation but have separable functions in HR regulation, and (iii) atrbohF mutants have a limited effect on ROI production compared with atrbohD but a much greater effect on Pp-induced cell death.
We provide genetic evidence that AtrbohD and AtrbohF, encoding probable components of a plant NADPH oxidase, are responsible for the ROI produced in two widely used systems for the analysis of plant defense responses. Controversy regarding the origin of ROI in plant defense has existed since Doke (1) first documented the production of superoxide in potato tubers infected with Phytophthora infestans. Subsequent definition of similarities to the oxidative burst in mammalian macrophages suggested that an NADPH oxidase was responsible for this ROI production (13). However, H2O2, and not superoxide, is the ROI detected in most plant–pathogen interactions (2, 3). These data suggested either that H2O2 is the proximal burst product or that rapid dismutation converts superoxide to H2O2. Other studies demonstrated in several systems that superoxide is, in fact, the proximal burst product and suggested that H2O2 is not the key signal for HR or defense-response control in these systems (33, 34). Sagi and Fluhr (35), using a novel activity gel assay, recently confirmed that a putative plant plasma membrane NADPH oxidase can produce superoxide.
Cell wall-bound peroxidases were proposed as an alternate source for ROI–H2O2 generation (36). Induced gene expression and enzymatic activity concomitant with the burst also implicated oxalate oxidases in ROI production during powdery mildew–barley interactions (23). However, these studies are based on correlations derived from gene expression and protein accumulation. Additionally, some of these conclusions rest on pharmacological studies that require careful interpretation and specificity controls. There is, to date, no direct genetic evidence supporting peroxidase or oxalate oxidase as sources of the ROI produced after infection.
Although H2O2 is the
main ROI detected in many plant pathogen systems and cell culture
systems (2, 3), our demonstration that an NADPH oxidase subunit is
required for ROI production confirms Doke's original suggestion that
O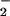 is the first ROI produced (at least in
interactions with avirulent Pst or Pp). Thus, our
results support previous findings identifying O
is the first ROI produced (at least in
interactions with avirulent Pst or Pp). Thus, our
results support previous findings identifying O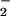 as
the key regulatory molecule (31, 33, 37). However, we failed to detect
O
as
the key regulatory molecule (31, 33, 37). However, we failed to detect
O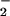 by nitroblue tetrazolium stain or cytochrome
c reduction after infection (data not shown). In fact, we
observed less nitroblue tetrazolium precipitate directly in leaf panels
injected with avirulent bacteria than in the surrounding areas (data
not shown). This might be a result of rapid superoxide dismutase
activity that increases after pathogen inoculation (38, 39). This
suggestion begs the question of how membrane-impermeable
O
by nitroblue tetrazolium stain or cytochrome
c reduction after infection (data not shown). In fact, we
observed less nitroblue tetrazolium precipitate directly in leaf panels
injected with avirulent bacteria than in the surrounding areas (data
not shown). This might be a result of rapid superoxide dismutase
activity that increases after pathogen inoculation (38, 39). This
suggestion begs the question of how membrane-impermeable
O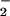 is dismutated in the absence of demonstrable
extracellular superoxide dismutase activity. Alternatively, the
O
is dismutated in the absence of demonstrable
extracellular superoxide dismutase activity. Alternatively, the
O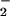 is dismutated rapidly in a superoxide
dismutase-independent, nonenzymatic manner. Its rate of nonenzymatic
dismutation is close to 105
M−1s−1, whereas its
reaction rate with nitroblue tetrazolium is 6 ×
104
M−1s−1 (40).
is dismutated rapidly in a superoxide
dismutase-independent, nonenzymatic manner. Its rate of nonenzymatic
dismutation is close to 105
M−1s−1, whereas its
reaction rate with nitroblue tetrazolium is 6 ×
104
M−1s−1 (40).
Our analysis of the atrbohD and atrbohF mutants strongly suggests that these genes act together to produce ROI (AtrbohD) and to control cell death (AtrbohF) in response to avirulent Pst DC3000(avrRpm1) because the double mutant HR reduction phenotype is stronger than atrbohF. However, HR appears not to be required for limiting bacterial growth. Other studies indicated separation between cell death and resistance to pathogens. For example, dnd1 mutant (defense, no death) displays normal gene for gene resistance to bacterial pathogens although the HR appears inhibited (41).
The HR provoked by Pst DC3000(avrRpm1) is not suppressed completely in the atrboh mutants. Both trypan blue stain and electrolytic leakage measurements indicate residual HR in the atrbohD/F double mutant that might be explained by residual ROI below our limit of detection (Fig. 2 A–C). Alternatively, other mechanisms may contribute to the cell death induced during interaction with avirulent bacteria. Trypan blue stain after inoculation of 106 cfu/ml DC3000(avrRpm1) identifies localized lesions in the atrbohD/F double mutant, compared with more spreading staining in the wt at each infection point (Fig. 2E). Therefore, the proximally produced ROI may act not as the initial trigger for HR in the directly infected cells but, rather, as a local signal for HR in nearby cells.
ROI may play a role in the establishment of systemic acquired resistance (SAR), a defense system that acts in distal parts of an infected plant (42). Salicylic acid (SA) is required for this establishment of SAR, because SAR is compromised in nahG plants that have reduced SA (43, 44). However, SA itself is not the translocated signal that mediates SAR (45, 46). SA and ROI metabolism are interconnected because ROI accumulation is potentiated by very small doses of SA (6, 47) and ROI induce SA accumulation (48). H2O2 has been proposed as a systemic signal (9). However, Dorey et al. (49) indicate that H2O2 is neither necessary nor sufficient to drive the expression of defense markers in areas surrounding infection sites. We induced SAR by inoculation of DC3000(avrRpt2) and assayed distal leaves for both protection against virulent Pp isolates and expression of the SAR-related gene PR-1. None of the atrboh mutants were compromised in their ability to mount an SAR response (data not shown). Thus, even in plants devoid of an oxidative burst, SAR still can be induced.
Surprisingly, we identified two different roles in HR control for Atrboh-generated ROI. Although the atrboh mutants, especially the atrbohD/F double mutant, display less HR after inoculation with avirulent Pst, they exhibit enhanced HR after inoculation with Pp Emco5. This unexpected phenotype was revealed only when we assayed the function of a weak R gene (J. M. McDowell, S. Williams, and J.L.D., unpublished data). This weak recognition does not completely block sporulation. The atrboh mutants display stronger cell death around the growing hyphae that prevents sporulation. Note that this is not simply enhancement of basal defense because it does not occur in response to infection with either of two virulent Pp isolates. We suggest that HR may have a clear mechanistic relation to resistance in interaction with the Pp, as has been demonstrated for the interaction of powdery mildew and barley (50). Our failure to define an effect of atrboh mutants on stronger R-mediated HR responses could reflect the fact that, despite ROI depletion, sufficient R signaling was generated. This concept is in line with findings of Bendahmane et al. (51), demonstrating that HR occurrence and magnitude can be correlated directly to R and Avr protein levels.
How can suppression of atrboh-dependent ROI lead to
enhanced cell death? One possible scenario is provided by Delledonne
et al. (38). In mammalian phagocytes, superoxide reacts with
NO generating peroxynitrite (ONOO−), a very
reactive molecule with many biological targets (52). Delledonne
et al. (38) suggest that the bacterial-induced HR in soybean
suspension-cultured cells requires a fine poise between ROI and NO.
However, HR does not appear to be mediated directly by
ONOO−. NO and O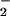 accumulate
independently after pathogen recognition (12). When the two are in
balance, ONOO− can be formed, which is, in fact,
not lethal in plants (38). However, increased superoxide dismutase
activity, or spontaneous dismutation, drives O
accumulate
independently after pathogen recognition (12). When the two are in
balance, ONOO− can be formed, which is, in fact,
not lethal in plants (38). However, increased superoxide dismutase
activity, or spontaneous dismutation, drives O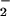 into
H2O2, potentially forcing
an NO/O
into
H2O2, potentially forcing
an NO/O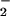 imbalance. As a consequence, free NO
accumulates and, together with
H2O2, initiates HR.
imbalance. As a consequence, free NO
accumulates and, together with
H2O2, initiates HR.
Thus, NO and H2O2 are
the elements directly involved in inducing cell death. Yet,
O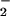 is the proximal ROI produced, and its levels and
rates of conversion to either ONOO− or
H2O2 determine the outcome.
In agreement with these studies, the effect of NO in potato as a
protectant against ROI-mediated cytotoxic processes indicates a fine
balance between NO/ROI to produce cell death and a role of
ONOO− as a harmless sink for these reactive
species (53). NO is a likely mediator of HR, but its activity is
contingent on relative ROI levels. Reduction of ROI levels in the
atrboh mutants would imply reduction of both positive
(H2O2) and negative signals
(O
is the proximal ROI produced, and its levels and
rates of conversion to either ONOO− or
H2O2 determine the outcome.
In agreement with these studies, the effect of NO in potato as a
protectant against ROI-mediated cytotoxic processes indicates a fine
balance between NO/ROI to produce cell death and a role of
ONOO− as a harmless sink for these reactive
species (53). NO is a likely mediator of HR, but its activity is
contingent on relative ROI levels. Reduction of ROI levels in the
atrboh mutants would imply reduction of both positive
(H2O2) and negative signals
(O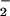 , in its role as a scavenger of NO) for the HR.
The alteration of the fine balance between ROI and NO may explain the
opposite effect observed between Pst and Pp
responses. Alteration in the levels of NO also may be the origin of the
spontaneous necrosis displayed by old atrbohD/F plants
(Fig. 1B).
, in its role as a scavenger of NO) for the HR.
The alteration of the fine balance between ROI and NO may explain the
opposite effect observed between Pst and Pp
responses. Alteration in the levels of NO also may be the origin of the
spontaneous necrosis displayed by old atrbohD/F plants
(Fig. 1B).
Concluding Remarks
The identification of the insertion mutations in AtrbohD and F enabled a stringent test of their role in plant defense. We demonstrate that these proteins are responsible for the ROI production observed in some interactions with avirulent pathogens. The AtrbohD contribution to ROI production in leaves is greater than AtrbohF. However, atrbohF mutants display a stronger effect on cell death, which indicates a qualitative (spacial or temporal) difference in the ROI produced by each Atrboh. Depletion of ROI production in these lines has opposite effects on cell death: HR is reduced during interaction with DC3000(avrRpm1) and enhanced after Pp Emco5. This suggests a different role for ROI in cell death signaling in resistance to these pathogens. We propose that the enhanced cell death phenotype that these mutants display in the Emco5 interaction may be produced through an effect of this ROI depletion on the levels of other signaling components of the defense–cell death response, particularly NO.
Acknowledgments
We thank K. Patel and S. Marillonnet for help during the isolation of the atrboh mutants. We thank R. Subramanian and T. Eulgem for critical reading of the manuscript. This research was funded by National Institutes of Health Grant 1-R01-GM057171-01, National Science Foundation Grant IBN-0077887 (to J.L.D.), and the Gatsby Foundation (to J.D.G.J.).
Abbreviations
- ROI
reactive oxygen intermediates
- Pst
Pseudomonas syringae pv. tomato
- Pp
Peronospora parasitica
- HR
hypersensitive response
- ONOO−
peroxynitrite
- wt
wild type
- SA
salicylic acid
- SAR
systemic acquired resistance
- DAB
3,3′-diaminobenzidine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Doke N. Physiol Plant Pathol. 1983;23:359–367. [Google Scholar]
- 2.Apostol I, Heinstein P F, Low P S. Plant Physiol. 1989;99:109–116. doi: 10.1104/pp.90.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine A, Tenhaken R, Dixon R, Lamb C J. Cell. 1994;79:583–593. doi: 10.1016/0092-8674(94)90544-4. [DOI] [PubMed] [Google Scholar]
- 4.Nürnberger T, Nennstiel D, Jabs T, Sacks W R, Hahlbrock K, Scheel D. Cell. 1994;78:449–460. doi: 10.1016/0092-8674(94)90423-5. [DOI] [PubMed] [Google Scholar]
- 5.Bradley D, Kjellbom P, Lamb C. Cell. 1992;70:21–30. doi: 10.1016/0092-8674(92)90530-p. [DOI] [PubMed] [Google Scholar]
- 6.Shirasu K, Nakajima H, Rajasekhar V K, Dixon R A, Lamb C J. Plant Cell. 1997;9:261–270. doi: 10.1105/tpc.9.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammond-Kosack K E, Jones J D G. Plant Cell. 1996;8:1773–1791. doi: 10.1105/tpc.8.10.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamb C, Dixon R A. Annu Rev Physiol Plant Mol Biol. 1997;48:251–275. doi: 10.1146/annurev.arplant.48.1.251. [DOI] [PubMed] [Google Scholar]
- 9.Alvarez M E, Pennell R I, Meijer P-J, Ishikawa A, Dixon R A, Lamb C. Cell. 1998;92:773–784. doi: 10.1016/s0092-8674(00)81405-1. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt H H H, Walter U. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 11.Delledonne M, Xia Y, Dixon R A, Lamb C J. Nature (London) 1998;394:585–588. doi: 10.1038/29087. [DOI] [PubMed] [Google Scholar]
- 12.Durner J, Wendehenne D, Klessig D F. Proc Natl Acad Sci USA. 1998;95:10328–10333. doi: 10.1073/pnas.95.17.10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Low P S, Merida J R. Physiol Plant. 1996;96:533–542. [Google Scholar]
- 14.Segal A W, Abo A. Trends Biochem Sci. 1993;18:48–52. doi: 10.1016/0968-0004(93)90051-n. [DOI] [PubMed] [Google Scholar]
- 15.Roos D, Deboer M, Kuribayashi F, Meischl C, Weening R S, Segal A W, Ahlin A, Nemet K, Hossle J P, Bernatowska-Matuszkiewicz E, et al. Blood. 1996;87:1663–1681. [PubMed] [Google Scholar]
- 16.Groom Q J, Torres M A, Fordham-Skelton A P, Hammond-Kosack K E, Robinson N J, Jones J D G. Plant J. 1996;10:515–522. doi: 10.1046/j.1365-313x.1996.10030515.x. [DOI] [PubMed] [Google Scholar]
- 17.Keller T, Damude H G, Werner D, Doerner P, Dixon R A, Lamb C. Plant Cell. 1998;10:255–266. doi: 10.1105/tpc.10.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torres M-A, Onouchi H, Hamada S, Machida C, Hammond-Kosack K E, Jones J D G. Plant J. 1998;14:365–373. doi: 10.1046/j.1365-313x.1998.00136.x. [DOI] [PubMed] [Google Scholar]
- 19.Amicucci E, Gaschler K, Ward J M. Plant Biol. 1999;1:524–528. [Google Scholar]
- 20.Kawasaki T, Henmi K, Ono E, Hataleuama S, Iwano M, Satoh H, Shimamoto K. Proc Natl Acad Sci USA. 1999;96:10922–10926. doi: 10.1073/pnas.96.19.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dangl J D, Jones J D G. Nature (London) 2000;411:826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- 22.Bolwell G P, Davies D R, Gerrish C, Auh C-K, Murphy T M. Plant Physiol. 1999;116:1379–1385. doi: 10.1104/pp.116.4.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou F, Zhang Z, Gregersen P L, Mikkelsen J D, de Neergaard E, Collinge D B, Thordal-Christensen H. Plant Physiol. 1998;117:33–41. doi: 10.1104/pp.117.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tissier A F, Marillonnet S, Klimyuk V, Patel K, Torres M A, Murphy G, Jones S D G. Plant Cell. 1999;11:1841–1852. doi: 10.1105/tpc.11.10.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debener T, Lehnackers H, Arnold M, Dangl J L. Plant J. 1991;1:289–302. doi: 10.1046/j.1365-313X.1991.t01-7-00999.x. [DOI] [PubMed] [Google Scholar]
- 26.Dangl J L, Holub E B, Debener T, Lehnackers H, Ritter C, Crute I R. In: Methods in Arabidopsis Research. Koncz C, Chua N-H, Schell J, editors. London: World Scientific; 1992. pp. 393–418. [Google Scholar]
- 27.Koch E, Slusarenko A J. Plant Cell. 1990;2:437–445. doi: 10.1105/tpc.2.5.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dellagi A, Brisset M N, Paulin J P, Expert D. Mol Plant–Microbe Interact. 1998;11:734–742. doi: 10.1094/MPMI.1998.11.8.734. [DOI] [PubMed] [Google Scholar]
- 29.Thordal-Christensen H, Zhang Z, Wei Y, Collinge D B. Plant J. 1997;11:1187–1194. [Google Scholar]
- 30.Grant M, Brown I, Adams S, Knight M, Ainslie A, Mansfield J. Plant J. 2000;24:441–450. doi: 10.1046/j.1365-313x.2000.00804.x. [DOI] [PubMed] [Google Scholar]
- 31.Scheel D. Curr Opin Plant Biol. 1998;1:305–310. doi: 10.1016/1369-5266(88)80051-7. [DOI] [PubMed] [Google Scholar]
- 32.Holub E B, Beynon J L, Crute I R. Mol Plant–Microbe Interact. 1994;7:223–239. doi: 10.1094/mpmi-8-0916. [DOI] [PubMed] [Google Scholar]
- 33.Jabs T, Colling C, Tschöpe M, Hahlbrock K, Scheel D. Proc Natl Acad Sci USA. 1997;94:4800–4805. doi: 10.1073/pnas.94.9.4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glazener J A, Orlandi E W, Baker C J. Plant Physiol. 1996;110:759–763. doi: 10.1104/pp.110.3.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sagi M, Fluhr R. Plant Physiol. 2001;126:1281–1290. doi: 10.1104/pp.126.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolwell G P, Butt V S, Davies D R, Zimmerlin A. Free Radical Res. 1995;23:517–532. doi: 10.3109/10715769509065273. [DOI] [PubMed] [Google Scholar]
- 37.Jabs T, Dietrich R A, Dangl J L. Science. 1996;273:1853–1856. doi: 10.1126/science.273.5283.1853. [DOI] [PubMed] [Google Scholar]
- 38.Delledonne M, Zeier J, Marocco A, Lamb C. Proc Natl Acad Sci USA. 2001;98:13454–13459. doi: 10.1073/pnas.231178298. . (First Published October 23, 2001; 10.1073/pnas.231178298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kliebenstein D J, Dietrich R A, Martin A C, Last R L, Dangl J L. Mol Plant–Microbe Interact. 1999;12:1022–1026. doi: 10.1094/MPMI.1999.12.11.1022. [DOI] [PubMed] [Google Scholar]
- 40.Rice-Evans C, Halliwell B, Lunt G G. Free Radicals and Oxidative Stress: Environment, Drugs and Food Additives. London: The Biochemical Society; 1995. [Google Scholar]
- 41.Yu I-C, Parker J, Bent A F. Proc Natl Acad Sci USA. 1998;95:7819–7824. doi: 10.1073/pnas.95.13.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryals J L, Neuenschwander U H, Willits M C, Molina A, Steiner H-Y, Hunt M D. Plant Cell. 1996;8:1809–1819. doi: 10.1105/tpc.8.10.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaffney T, Friedrich L, Vernooij B, Negrotto D, Nye G, Uknes S, Ward E, Ryals J. Science. 1993;261:754–756. doi: 10.1126/science.261.5122.754. [DOI] [PubMed] [Google Scholar]
- 44.Bi Y-M, Kenton P, Mur L, Darby R, Draper J. Plant J. 1995;8:235–246. doi: 10.1046/j.1365-313x.1995.08020235.x. [DOI] [PubMed] [Google Scholar]
- 45.Vernooij B, Friedrich L, Morse A, Reist R, Kolditz-Jawhar R, Ward E, Uknes S, Kessmann H, Ryals J. Plant Cell. 1994;6:959–965. doi: 10.1105/tpc.6.7.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pallas J A, Paiva N L, Lamb C J, Dixon R A. Plant J. 1996;10:281–294. [Google Scholar]
- 47.Draper J. Trends Plant Sci. 1997;2:162–165. [Google Scholar]
- 48.Chamnongpol S, Willekens H, Moeder W, Langebartels C, Sanderman H J, Van Montagu M, Inze D, Van Camp W. Proc Natl Acad Sci USA. 1998;95:5818–5823. doi: 10.1073/pnas.95.10.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorey S, Kopp M, Geoffroy P, Fritig B, Kauffmann S. Plant Physiol. 1999;121:163–173. doi: 10.1104/pp.121.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freialdenhoven A, Scherag B, Hollricher K, Collinge D, Christensen H-T, Schulze-Lefert P. Plant Cell. 1994;6:983–994. doi: 10.1105/tpc.6.7.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bendahmane A, Kanyuka K, Baulcombe D C. Plant Cell. 1999;11:781–791. doi: 10.1105/tpc.11.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Groves J T. Curr Opin Chem Biol. 1999;3:226–235. doi: 10.1016/S1367-5931(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 53.Beligni M V, Lamattina L. Nitric Oxide. 1999;3:199–208. doi: 10.1006/niox.1999.0222. [DOI] [PubMed] [Google Scholar]