

Abstract
Regulation of activity in neuronal circuits is poorly understood. Alterations in regulatory mechanisms may underlie the transition from interictal to ictal activity. Epileptogenesis is thought to arise from hyperexcitability in populations of neurons, resulting from alterations in connectivity and synaptic physiology. Recent studies suggest that excitability in local circuits of neurons also can be changed by short-term plasticity induced by repetitive stimulation and actions of neuromodulators. It is suggested that these factors could be elements underlying excitability changes that contribute to the periodic nature of seizures.
A defining feature of epilepsy is the periodic nature of seizures. The interval between seizures varies both within and between individuals. Seizure frequency can be influenced or modified by stressors, such as sleep deprivation, drug or alcohol abuse or both, or a host of less well-defined factors (1). Alterations in connectivity and synaptic physiology in local neural circuits are thought to underlie development of intractable epilepsy (2) and to increase seizure propensity in the immature brain (3). The periodic nature of seizures suggests that some mechanisms must be capable of regulating the excitability in neural circuits, thereby potentially altering seizure initiation. Multiple mechanisms exist that are capable of regulating circuits and excitability. For example, excitability can be influenced by factors intrinsic to the epileptic circuit, such as short-term changes in synaptic plasticity (depression or facilitation), or via extrinsic influences projecting to the hyperexcitable circuit, such as dopaminergic inputs to a local cortical network.
Short-term Synaptic Plasticity and Regulation of Excitability
Recurrent connections between both γ-aminobutyric acid (GABA)ergic interneurons and pyramidal cells are a common feature of cortical circuits. The recurrent excitatory axon collaterals of pyramidal cells provide positive feedback, which can amplify inputs to a circuit and lead to epileptiform discharges. A key element in determining the behavior of cortical circuits is the balance between inhibition and excitation. It has long been known that modest reductions in inhibition can result in epileptiform activity (4). What now is becoming apparent is that during repetitive activation, both excitatory and inhibitory synapses show marked frequency-dependent, short-term plasticity. Surprisingly, considerable heterogeneity in short-term plasticity has been observed. Alterations in synaptic facilitation or depression or both could be an important factor in regulating excitability in circuits involved in epileptogenesis.
Short-term plasticity recently has been examined in synaptically connected pairs of neocortical pyramidal cells and pairs of interneurons (5). Sustained activity was elicited by exciting presynaptic cells at a frequency of 20 Hz. Both excitatory postsynaptic potentials (EPSPs) and inhibitory postsynaptic potentials (IPSPs) showed depression during prolonged stimulation. Compared with inhibitory responses, excitatory synaptic responses showed a much larger depression and recovered more slowly. Short bursts of action potentials (similar to those observed during epileptiform discharges) produced comparable differences in depression. The ability of inhibitory synapses to sustain their outputs during repetitive activation may contribute to maintenance of stability in neuronal circuits and control of excitability of principal cells. Enhanced depression of IPSPs or decreases in EPSP depression could enhance circuit excitability, resulting in epileptiform activity.
These interactions are likely to be very complex. Different cell populations (e.g., interneurons versus pyramidal cells) may respond differently to repetitive or prolonged stimulation. A given cell can show facilitation or depression, depending on the nature of the input (e.g., thalamocortical vs. intracortical) (6). The frequency dependence of short-term plasticity also shows regional variability and may contribute to differences in seizure susceptibility. Short-term plasticity is generally viewed as a presynaptic phenomenon. Possible presynaptic mechanisms include changes in the readily releasable pool of vesicles and activation of presynaptic receptors. GABAB and kainate receptors are known to be localized to presynaptic nerve terminals, where they can have homo- and heterosynaptic influences (7,8). In addition, it is likely that these receptors are subject to up- and downregulation. Changes in the transmitter phenotype of certain cells after seizures (9) could result in different patterns of short-term plasticity. Finally, alterations in short-term plasticity mechanisms by a variety of stressors could modify the dynamics in cortical circuits and contribute to the induction of seizures.
Monoaminergic Regulation of Circuit Excitability
Dopamine (DA) is an endogenous neuromodulator in cortical circuits and is known to be important for normal brain function. Immunohistochemical studies in rat and primate cortex show that both inhibitory interneurons and excitatory pyramidal cells are targets for DA innervation (10). It also has been suggested that pyramidal neurons and interneurons may express different DA-receptor subtypes (11). This receptor heterogeneity provides a basis for differential DA modulation of cortical neurons. Therefore dopaminergic activation may shift the balance between excitation and inhibition in neuronal circuits, which may be related to clinical and experimental observations suggesting a potential link between DA and epilepsy (12,13).
An important factor in epileptogenesis is the degree to which neurons in a circuit become synchronized and whether this activity spreads or propagates though the brain. Recent studies showed that DA increases the amplitude of excitatory postsynaptic currents (EPSCs) in layer II/III neocortical pyramidal cells (14). Subsequent work by the same investigators examined whether DA enhancement of EPSCs alters excitability in neocortical networks, by using optical imaging with voltage-sensitive dyes to monitor reliably the initiation, distribution, and spread of activity in the neocortex in vitro (15). Modern optical methods allow reproducible detection of dye signals in single trials without serious photobleaching or toxicity, making it feasible to ask the question whether DA modulation affects the spatiotemporal pattern of activation in local circuits.
In this study, neocortical slices were stained with the voltage-dependent dye RH414 (30 μM). Activity-dependent changes in fluorescence were detected by using a Neuroplex 464 diode array (see Fig. 1; the pial surface is indicated by the white bar in the first frame). With a ×5 objective, an area of cortex with an approximate width and height of 3.6 mm was imaged. Decreases in fluorescence were plotted as an upward deflection and are associated with a depolarization. In the presence of bicuculline, which blocks GABAA receptor–mediated inhibition, weak intracortical stimulation was used to activate neocortical circuits. Figure 1A shows that such stimulation produced localized activation of a small region of cortex under control conditions. After activation of DA receptors with the D1 receptor agonist SKF 38393, the same stimulation resulted in a more extensive and persistent activation of cortical circuits, as shown in Figure 1B. Activity from two individual diodes is shown the lower portions of the figure. These traces depict changes in fluorescent intensity as a function of time and reflect depolarization of neuronal elements. It can be seen that DA-receptor activation enhanced and prolonged the responses. This finding suggests that the modest (∼20%) increase of individual EPSCs induced by DA (14) is exaggerated as activity spreads through the cortical mantle. In other experiments, D1-receptor activation also resulted in lowering of the threshold for evoking epileptiform activity.
Figure 1.
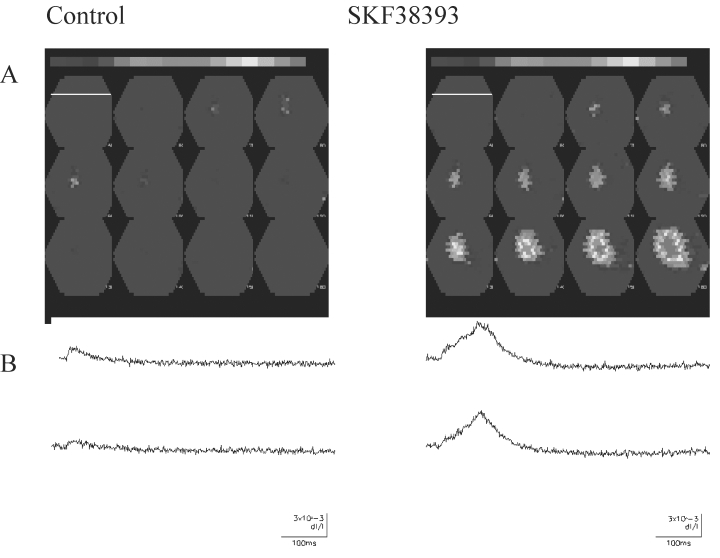
Dopamine enhancement of circuit activity in rat neocortex. A: Recordings under control conditions. Upper panel, Multiple frames obtained at 6-msec intervals. Frame 1 (Control) is in the upper left hand corner, and the time sequence is from left to right. White line, in the first frame shows the location of the pial surface. Stimulation occurred between the first two frames. In frames 5–8, activity-dependent changes are observed. Lower panel, Fluorescence changes (as a function of time) from individual diodes. B: Similar recordings obtained after bath application of the D1-receptor agonist SKF 38393. The area activated was always larger after DA-receptor activation. Changes also were more persistent. The amplitude and duration of evoked responses in the individual diodes also were increased in the presence of SKF 38393.
It is apparent that neuromodulators can significantly regulate excitability in neuronal circuits. Enhancements seen at the single-cell level are magnified as activity propagates through multiple elements in a local neuronal network. Alterations in extrinsic neuromodulatory inputs to a hyperexcitable circuit could be an important factor in controlling seizure initiation or frequency or both. The exact effects of DA or other modulators are hard to predict. Regional and cell-specific differences in receptor-subtype expression, existence of positive or negative feedback circuits to alter DA cell excitability, and differing effector mechanisms are all complicating factors. Understanding regulation of circuits and excitability remains an exciting challenge.
Acknowledgments
I thank C. Gonzalez-Islas and S. Bandyopadhyay for their work on the dopamine studies. This work was supported by NINDS grants NS18145 and NS22373.
References
- 1.Engel J., Jr . Seizure and epilepsy. Philadelphia: FA Davis; 1989. [Google Scholar]
- 2.Sayin U, Osting S, Hagen J, Rutecki P, Sutula T. Spontaneous seizures and loss of axo-axonic and axo-somatic inhibition induced by repeated brief seizures in kindled rats. J Neurosci. 2003;23:2759–2768. doi: 10.1523/JNEUROSCI.23-07-02759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swann JW, Hablitz JJ. Cellular abnormalities and synaptic plasticity in seizure disorders of the immature nervous system. Ment Retard Dev D R. 2000;6:258–267. doi: 10.1002/1098-2779(2000)6:4<258::AID-MRDD5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 4.Chagnac-Amitai Y, Connors BW. Horizontal spread of synchronized activity in neocortex and its control by GABA-mediated inhibition. J Neurophysiol. 1989;61:747–758. doi: 10.1152/jn.1989.61.4.747. [DOI] [PubMed] [Google Scholar]
- 5.Galarreta M, Hestrin S. Frequency-dependent synaptic depression and the balance of excitation and inhibition in the neocortex. Nat Neurosci. 1998;1:587–594. doi: 10.1038/2822. [DOI] [PubMed] [Google Scholar]
- 6.Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 7.MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- 8.Huettner JE. Kainate receptors and synaptic transmission. Prog Neurobiol. 2003;70:387–407. doi: 10.1016/s0301-0082(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 9.Sloviter RS. Excitatory dentate granule cells normally contain GAD and GABA, but does that make them GABAergic, and do seizures shift granule cell function in the inhibitory direction? Epilepsy Curr. 2003;3:3–5. doi: 10.1046/j.1535-7597.2003.03101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sesack SR, Snyder CL, Lewis DA. Axon terminals immunolabeled for dopamine or tyrosine hydroxylase synapse on GABA-immunoreactive dendrites in rat and monkey cortex. J Comp Neurol. 1995;363:264–280. doi: 10.1002/cne.903630208. [DOI] [PubMed] [Google Scholar]
- 11.Mrzljak L, Bergson C, Pappy M, Huff R, Levenson R, Goldman-Rakic PS. Localization of dopamine D4 receptors in GABAergic neurons of the primate brain. Physiol Rev. 1996;78:189–225. doi: 10.1038/381245a0. [DOI] [PubMed] [Google Scholar]
- 12.Starr MS. The role of dopamine in epilepsy. Synapse. 1996;22:159–194. doi: 10.1002/(SICI)1098-2396(199602)22:2<159::AID-SYN8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 13.Trimble MR. Biological psychiatry. 2nd ed. West Sussex: John Wiley & Sons; 1996. [Google Scholar]
- 14.Gonzalez-Islas C, Hablitz JJ. Dopamine enhances EPSCs in layer II-III pyramidal neurons in rat prefrontal cortex. J Neurosci. 2003;23:867–875. doi: 10.1523/JNEUROSCI.23-03-00867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hablitz JJ, Gonzalez-Islas C. Dopamine alters threshold for initiation of epileptiform activity in rat neocortex. Epilepsia. 2002;43(suppl 7):129. [Google Scholar]