

Abstract
MapMan is a user-driven tool that displays large genomics datasets onto diagrams of metabolic pathways or other processes. Here, we present new developments, including improvements of the gene assignments and the user interface, a strategy to visualize multilayered datasets, the incorporation of statistics packages, and extensions of the software to incorporate more biological information including visualization of coresponding genes and horizontal searches for similar global responses across large numbers of arrays.
Expression arrays (De Risi et al., 1997; Celis et al., 2000; Michaut et al., 2003; Scheible et al., 2004) and metabolite profiling (Fiehn et al., 2000; Stitt and Fernie, 2003) provide a comprehensive overview of system responses. However, their interpretation depends on the availability of appropriate bioinformatics tools. While many tools exist to support statistical analysis of profiling data, relatively few tools integrate profiling datasets with preexisting biological knowledge. Examples include GenMAPP (http://www.GenMAPP.org), PathwayAssist (http://www.ariadnegenomics.com), Pathway Processor (Grosu et al., 2002), BioMiner (http://www.zbi.uni-saarland.de/chair/projects/BioMiner/index.shtml), and the Saccharomyces genome database (Christie et al., 2004). GenMAPP, Pathway Processor, and the Saccharomyces genome database include many yeast (Saccharomyces cerevisiae) pathway diagrams and provide yeast researchers with powerful tools to visualize their experiments. This is especially true for Pathway Processor, which also supports statistical analysis. However, these tools are of limited utility for plant research because they are based on microbial or animal systems, contain irrelevant categories, lack plant-specific pathways and processes, and lack predefined plant pathways. The plant-specific tool Aracyc (Mueller et al., 2003) is easy to use and intuitive, but suffers from the limitation of being designed as an online tool and from offering only prebuilt pathways.
In addition to stand-alone studies, large public domain databases (e.g. AtGenExpress) that integrate datasets from hundreds of profiling studies are being established. These allow more sophisticated questions to be posed. Tools have been developed to query the responses of individual genes horizontally across hundreds of physiological or environmental treatments or developmental series (Rhee et al., 2003; Craigon et al., 2004; Zimmermann et al., 2004). The next level is to investigate which groups of genes are expressed coordinately (e.g. Metagenealyse; Daub et al., 2003). Databases have been created that contain precomputed coresponse values for large sets of experiments, for example, the comprehensive systems biology database CSB.DB (Steinhauser et al., 2004). Coresponding genes may encode proteins that are involved in the same pathway, are members of the same complex, or are connected by other functional constraints such as metabolic coupling (Lee et al., 2004). Many tools are being developed to search for pathway enrichments within a given set of genes (e.g. Wang et al., 2004). It is already apparent that in yeast, transcripts of genes involved in a particular pathway often respond similarly (Brown et al., 2000). In Arabidopsis (Arabidopsis thaliana), sugar depletion (Thimm et al., 2004) or nitrogen addition (Scheible et al., 2004) lead to coordinate repression and induction, respectively, of large numbers of genes that are required for inorganic nitrogen assimilation, amino acid synthesis, amino acid activation, and protein synthesis. In other situations a more complex picture emerges. This can occur for many reasons: There may be gene families for one or more of the steps whose members are differentially expressed (for examples, see Wang et al., 2003; Scheible et al., 2004; Thimm et al., 2004), pathways or segments of pathways may have multiple and only partly overlapping functions, functional categories may not have been sufficiently resolved, or there may quite simply be errors in gene assignments.
We recently presented a visualization tool called MapMan (Thimm et al., 2004; the application can be used as a Web-based application or downloaded from http://gabi.rzpd.de/projects/MapMan/) that allows the users to display genomics datasets onto pictorial diagrams. MapMan consists of a Scavenger module that collects and classifies the measured parameters into hierarchical functional categories, and an ImageAnnotator module that uses these classifications to organize and display data on diagrams of the user's choice. Each gene is represented by a discrete signal, allowing individual responses to be identified. Signals for genes that are involved in a particular process are grouped spatially, making it possible to discern general trends that would be less apparent from lists of individual genes. The modular structure allows users to display many different sorts of data and to create new functional categories and diagrams as they learn more about the systems they are studying.
This article presents developments in MapMan including (1) improvements in the Scavenger module, which has been updated using The Institute for Genomic Research (TIGR) release 5 Arabidopsis annotation and expanded to incorporate more functional categories and genes; (2) improvements in the user interface of the ImageAnnotator module, including more rapid and flexible loading of files, more convenient inspection of gene annotations, and a search function; (3) integration of tools for statistical treatment of replicated arrays, allowing the display of results on the basis of their statistical significance rather than the extent of the changes; (4) development of a module that assesses the statistical significance of differences in the response of sets of genes assigned to different biological functions; and (5) the development of strategies that allow simultaneous handling and visualization of different kinds of data (e.g. transcripts and metabolites). Further, to allow the versatility of MapMan to be combined with the wealth of information available in the large-array databases in the public domain (6) an online tool has been developed that links MapMan with CSB.DB and displays sets of coresponding genes on MapMan maps, and (7) new types of mapping files and maps have been developed that allow users to rapidly screen in silico for similarities between the changes of expression in an experiment of their choice and those in many other biological responses.
RESULTS
Overall Structure of MapMan
The overall design of MapMan is described in Thimm et al. (2004) and at http://gabi.rzpd.de/projects/MapMan/. Briefly, a Scavenger module classifies the measured parameters (e.g. the genes represented on the ATH1 array) into hierarchical functional categories (BINS, subBINS, individual enzymes, etc.). It generates mapping files that contain, for each measured parameter, a unique identifier (e.g. the Affymetrix identifier, the Arabidopsis Genome Initiative code), a text annotation, and the numeric code of the functional category (or categories) to which the parameter has been assigned. The ImageAnnotator module uses these mapping files to map the experimental data into functional categories (BINS) and display them onto diagrams or maps. A screen shot of the user interface is given in Figure 1, showing a browser that the user can use to call up maps, link them with the appropriate mapping file, and then open up and display experimental data files. The diagrams, or maps, can be downloaded from Web sites, scanned in from textbooks, or custom-made by the user. They are stored as files in a BMP, PNG, JPG, or other format. To prepare a map for data display, the user mouse-clicks on a chosen location of the map to open a pop-up dialog box, and enters the numerical code of the BIN or subBIN that is to be displayed there. This step is repeated for all of the BINS or subBINS that are to be displayed on the map. The resulting assignments are stored as an XML file, which is linked to and automatically opened with the map image. The experimental data files can be accessed via the browser. Experimental data files contain the unique identifier and an experimental value (e.g. the change in expression between the treatment and a control sample on a log2 scale) for each measured parameter. After opening an experimental file by mouse click, the data are automatically visualized on the map image. The direction of the change of expression is reflected in the color (items that increase and decrease are colored blue and red, respectively), and the extent of the change is given by the intensity of the color (genes showing no or little change are white, and an increasingly large change is reflected by a darker and darker color on a user-adjustable scale). Items that are flagged as being unreliably measured (e.g. “not present” transcripts in the MAS Affymetrix software) are colored gray (see Fig. 1 for an example of the user interface).
Figure 1.

A screen shot of the user interface of the ImageAnnotator module. The ImageAnnotator module organizes maps, mapping files, and experiment files in a browsable hierarchy tree (left side). The bottom part of the window can display logging messages (data not shown) and statistics. The main part of the window is reserved for the display of the actual map file with superimposed experimental data. The data are displayed at sites on the map that have been defined previously by the user as described in the text. Each parameter is displayed as a discrete signal using an adjustable false color code. The scale and size of the spots can be chosen by the user by clicking on the icon “Options.” Here, a combined experiment is displayed showing the changes of metabolites and transcripts in Arabidopsis seedlings after 2 d of nitrogen deprivation. Metabolites are in the darker fields, whereas the transcripts encoding for enzymes responsible for a reaction are located adjacent to the respective arrows.
Updating the Mapping Files Using TIGR5 and Addition of More Highly Resolved Maps of Pathways and Processes
The usefulness of MapMan depends in large part on the accuracy and completeness with which the measured parameters (e.g. genes) are assigned to hierarchical categories. This immense ongoing task is hampered by the current quality of annotations and our own limited expertise. The original mapping file for genes was based on the TIGR2 annotation. It has been updated to the current TIGR5 release, using a combination of a text mining approach (J. Fluck, M. Hofmann, J. Selbig, M. Stitt, and O. Thimm, unpublished data) and manual curation of nearly 10,000 genes. Since the first presentation of MapMan, input about biological pathways or gene annotations that have been provided by many experts (see “Acknowledgments”; for details, visit http://gabi.rzpd.de/projects/MapMan/Collaborations.shtml) have been used to correct and improve the assignments.
Manual annotation has further resolved many BINS and subBINS, in some cases down to individual gene families. For example, in addition to Suc and starch metabolism, glycolysis, and the TCA cycle, further pathways including the Calvin cycle, trehalose metabolism, some parts of cell wall synthesis, and, in secondary metabolism, genes for the individual enzymes of the monolignol synthesis are individually annotated and corresponding maps provided. The structure of amino acid metabolism has been reorganized to better reflect the actual biological pathways and to decrease redundancy. An improved breakdown is provided of genes involved in protein synthesis, the ubiquitin pathway for protein degradation, and transcription factors and has started for hormone metabolism. Mapping files can be opened online to inspect their structure and breakdown by clicking on the menu. A complete overview of available maps is provided in “MapMan Help.”
Previously, each breakdown of a pathway to the enzyme level was supplied as a separate mapping file. This required the user to select the correct mapping file for each map. The new version simplifies this procedure by placing all parameters pertaining to Affymetrix arrays in one single mapping file. Moreover, a new Arabidopsis gene index code-based mapping file has been added to allow MapMan to be used with other microarray systems.
Affymetrix arrays track changes of gene expression by measuring hybridization to a probe set of short oligonucleotides. Continuing updating of the Arabidopsis annotation could lead to some of the probe sets becoming unspecific or inappropriate. The oligonucleotides present on the ATH1 chip were compared by a BLAST search (Altschul et al., 1990) against TIGR5 CDS sequences. Only 89.5% of the _at-labeled spots had a completely unique hit. The resulting reallocation can be downloaded from (http://gabi.rzpd.de/services/Downloads.shtml) and was used for the new mapping file. Reasons for the reallocations could include updated genome information, as well as the higher precision obtained by BLASTing with the oligonucleotides present in the probe set rather than the sequence from which they were designed. The possibility of cross hybridizations is provided in the information about the gene, which the user can call up by a mouse-over action.
In addition, a metabolite mapping file including all metabolites routinely measured on gas chromatography-mass spectrometry, liquid chromatography-mass spectrometry, HPLC, and enzyme-based platforms and including synonyms and identifiers for each metabolite is close to completion.
Software and User Interface Improvements in the ImageAnnotator
Improvements have been made in the software and the user interface to improve the versatility. For the original version of MapMan, Microsoft Excel (Redmond, WA) format was used for the data files. The new version also supports tab-delimited text files. This not only allows faster import of data, but also makes the user independent of the Microsoft platform. This makes it easier to combine the visualization capacity of the ImageAnnotator with specialized analyses including statistical software packages such as R (R Development Core Team, 2004), which can be used to generate custom files for display in the ImageAnnotator module (see below for examples). Second, the mouse-over function that calls up the annotation and information about the gene online has been improved to temporarily store information while the mouse is being used for further actions. Third, link out has been introduced that allows the user to right click on spots in the ImageAnnotator module and open a Web browser window online. This displays information about the gene from the GABI Primary Database Web site. From there it is then possible to directly query various information resources such as The Arabidopsis Information Resource (TAIR; Rhee et al., 2003) for a particular gene associated with an Affymetrix probe set. This is useful when interesting candidate genes have been identified and one wants to learn more about them.
A new search-and-highlighting function has been introduced that allows the user to search for keywords from the description in the mapping file and rapidly pinpoint items of interest online. There is a text field on the toolbar where one can insert text (*, ? as wildcards) or regular expressions (e.g. RE://at3g\d+). All items displayed on the map that is currently in use, which are identified by the search function, can in this way be highlighted in a user-definable way. This allows rapid online location of processes or genes of interest.
The search-and-highlighting function introduces a further important functionality into MapMan. It allows the user to apply other attributes or classifications horizontally, which complement the hierarchical classification defined in the BINS and subBINS. In principle, any sort of attribute can be incorporated in text annotation of the genes in the mapping file, and the search-and-highlighting function then used online to identify all the genes that share a particular attribute. This adds a large degree of flexibility. This would allow, for example, all genes assigned to a particular tissue (e.g. root) or subcellular structure (e.g. plastid-located) to be simultaneously identified, and their changes of expression inspected. Other examples that can be inspected are all genes that respond to a particular response (e.g. all genes induced by more than x-fold in low sugar could be flagged by “low sugar induced” in the mapping file) or all genes for which a particular resource is available (e.g. all genes for which a knockout mutant is available could be flagged by “KO available” in the mapping file), and then could be easily highlighted by searching for the requisite term. The modular structure of MapMan and the use of tab-delimited files will make it easy for the user to add such terms to their mapping files at will. Such extensions of the mapping files will be added to MapMan in the next months.
Visualizing Statistical Analyses of Replicate Arrays in MapMan
Used in its simplest mode, MapMan compares a treatment with a control treatment. The change of expression ratio of each gene is calculated as log fold change to generate the experimental file, which is then visualized (Thimm et al., 2004). However, the modular structure of MapMan allows many other types of experimental data files to be imported and visualized. Examples include layover plots, which provide a qualitative overview of the similarities between two different experiments (see Thimm et al., 2004), and derivative data files, in which a more complex analysis of the data has been performed (O. Blaesing and M. Stitt, unpublished data; MapMan was used to visualize the amplitudes of the diurnal changes of gene expression or the weightings of individual genes after principle components analysis).
MapMan can also be used to visualize the results of a standard statistical analysis of replicated arrays as transformed P values. An instructive approach is to display all nonsignificant changes as white spots by setting the cutoff value in the experiment file to zero. More significant P values are then transformed to generate an increasingly intense color in the ImageAnnotator as the P value decreases. This can be done with the following formula:
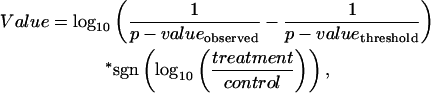 |
substituting NA values by zero. The first term provides a statistical analysis of the significance of the change, and the second term distinguishes between induction and repression. When the resulting values are visualized in MapMan, the significance of the change is reflected in the intensity and the direction by the color. As an example we chose the above given dataset (O. Blaesing and M. Stitt, unpublished data) and computed a P value using Student's t test for end of day versus end of night.
Figure 2 compares the same set of data (biological triplicates of Arabidopsis rosettes of 5-week-old plants growing in a 12-h-light/12-h-dark diurnal cycle harvested at the end of the night and at the end of the light period) showing (Fig. 2A) the ratio of the change calculated from the average of the three experiments and (Fig. 2B) the P values calculated from the individual values in the three experiments. The scales used for display in MapMan can be selected by the user. The scale can be adjusted in such a way that fold change values uninteresting for the biologist are not visualized. The qualitative results are very similar in both display modes. However, there are genes for which a change was barely visible in Figure 2A, but a clear change is revealed after statistical analysis because although the change is small, the values are well replicated in all three experiments.
Figure 2.

A part of the response of Arabidopsis to the day diurnal cycle. A, A part of the diurnal cycle was visualized by calculating the log ratios for the average transcript abundance measured with an Affymetrix chip at the end of the day and the end of the night. The resulting file was then loaded into the ImageAnnotator module and visualized on a general map (metabolism overview). B, For the same experiment, a Student's t test was performed to test for difference at a significance level of 1%. The resulting P values were transformed to fit into the ImageAnnotator scale (see text for details).
A more comprehensive way to view the results of a statistical analysis is currently in preparation. These experimental data files can easily be extended by adding the results of a statistical analysis of the data. A second search and highlight that allows the experimental data files to be queried will be added. This will allow statistically significant results to be flagged.
Statistical Analysis of Differences in the Response of Genes Assigned to Different BINS or SubBINS
Changes in gene expression often lead to coordinated changes in the expression of genes involved in a particular function (see introduction). MapMan has been extended to facilitate identification and statistical analysis of responses that affect a large proportion of the genes in a particular functional category. To do this, a statistical module has been included that performs a Wilcoxon rank sum test with the members of individual BINS seen on the screen versus all other items of the same type. This rank sum test provides a robust procedure to reveal whether the average response of a BIN is different from the response of all the other BINS. The results are dynamically calculated upon loading of an experiment file and displayed in a separate log window, which can be called up by the user.
Table I shows the window that would be called up for Figure 2A. This reveals that, for categories of genes in metabolism, the most significant shared change between the end of the night and the end of the day occurs in BIN 1.1 (photosystem.light reaction). The extent of the changes of individual genes is small (Fig. 2A) and relatively few of the changes are significant at the level of the individual genes (Fig. 2B), but the response becomes extremely significant when the response of the total BIN1.1 is analyzed (Table I).
Table I.
Results pasted from a MapMan session
The microarray experiment end of day versus end of night was visualized in the ImageAnnotator module. The results were taken from the statistical tab of the ImageAnnotator module, which are calculated on the fly and sorted by probability. Here, the BIN, the number of elements in that particular BIN, and the corresponding probability for that BIN being identical to the overall distribution are shown.
| BIN | Elements | Probability |
|---|---|---|
| 1.1 | 127 | 5.68E−12 |
| 4 | 58 | 2.67E−5 |
| 5 | 12 | 4.20E−5 |
Display of Multilevel Data Sets in MapMan
Changes of transcripts need to be combined with information about changes of proteins, enzyme activities, metabolites, and fluxes to allow their physiological significance to be evaluated (see e.g. Gibon et al., 2004; Scheible et al., 2004). To aid the display of multilevel genomics data, a new feature has been added into the mapping files and the ImageAnnotator to allow each parameter to be identified as a transcript, an enzyme (activity), a protein (level), or a metabolite (level). This allows these different kinds of data to be combined and displayed together on one map and using one single mapping file. The advantage of this approach is shown in Figure 1, where the changes of metabolites and transcripts in nitrogen-starved seedlings (Scheible et al., 2004) are displayed together. Metabolites are distinguished by displaying them against a darker background, while transcripts encoding enzymes are located adjacent to the arrows linking the metabolites. In this example, most amino acids are low and organic acids increased, and this is associated with induction of genes encoding for enzymes in the lower part of glycolysis, the TCA cycle, one isoform of Glu dehydrogenase but with no coordinated changes in amino acid biosynthesis or degradation. As multiparallel, multiplatform measurements become more widespread, this feature will provide a useful tool to visualize these datasets.
Visualizing Coresponses in MapMan Reveals Interacting Pathways
As outlined in the introduction, tools have been developed in the last months to identify sets of coresponding genes to be identified by horizontal queries in large databases of expression arrays. The output of coresponse analyses is typically a long list of genes, which it is difficult to overview in tabular form. The flexible structure of MapMan makes it possible to visualize which sets of genes corespond with a given query transcript. To facilitate this, CSB.DB (Steinhauser et al., 2004; http://csbdb.mpimp-golm.mpg.de) has been extended to automatically convert a coresponse table into a MapMan readable file, which then can be immediately downloaded or directly displayed in the online version of MapMan. An analogous display to the normal MapMan is preserved, allowing for easy identification of processes which might be changed: coresponding genes are blue, genes behaving reciprocally are red, and the color intensity indicates the tightness of the agreement.
As an example, CSB.DB was queried with a gene from the Calvin cycle (GAPA; Fig. 3). All the genes in the light reactions and almost all genes assigned to the Calvin cycle are deep blue, as are sets of genes required for chloroplast biogenesis including tetrapyrrole biosynthesis, carotene synthesis via the DXS pathway, and plastid (but not cytosolic) ribosomal proteins. Despite the qualitatively homogenous response in these BINS, some genes are less tightly related than others. For example, plastid genome-encoded genes for light reaction components show a weaker relation than nuclear-encoded components (data not shown). In some cases, individual members of BINS show a very high coresponse. Examples include the chloroplast envelope triose phosphate transporter and cytosolic FBPase, which are the first two key steps in photosynthetic Suc synthesis; two amino transferases annotated to use glyoxylate, which are presumably primarily involved in photorespiration; the plastid NADP malate dehydrogenase; individual genes involved in RNA synthesis and amino acid activation (of which many are already annotated as plastid-located); genes involved in protein import into the plastid (data not shown in Fig. 3); genes assigned to fatty acid desaturation and phospholipids synthesis that are involved in the synthesis of thylakoid lipids; and a small number of transcription factors. These genes are all known to be involved in photosynthesis or plastid biogenesis, making it attractive to hypothesize that other genes with a similarly high coresponse coefficient (e.g. a small number of transcription factors; data not shown) contribute in a specific way to these processes. Other BINS contain a high proportion of genes that show a weak coresponse, for example, starch synthesis and degradation, indicating that these pathways are coregulated with genes involved in photosynthesis, and also by other independent inputs. This is expected because starch is synthesized and degraded in leaves but also in heterotrophic tissues. On the other hand, sets of genes that are exclusively associated with respiratory processes, like those from the TCA cycle and mitochondrial electron transport chain, are mainly colored red, indicating they change independently of and sometimes even reciprocally to genes involved in photosynthesis.
Figure 3.

Coresponse visualized with the ImageAnnotator module. A coresponse query was performed using the CSB database with GAPA (At3g26650) using the M0271 matrix and standard settings. The resulting output in MapMan-readable format was visualized in the ImageAnnotator module. The most interesting sections of the metabolism overview, transport, secondary metabolism, and protein synthesis maps are reproduced here.
Response Maps to Allow Comparison with Many Other Biological Responses
The maps discussed so far aid interpretation by visualizing changes of expression to learn how genes that are involved in different metabolic (e.g. a pathway or sector of metabolism) or cellular processes (e.g. RNA synthesis, protein synthesis, protein degradation, transcription factors) respond. Another important question that can be asked is whether there is any similarity between a new response and the hundreds of responses that have been seen and characterized in earlier experiments. Such information could provide important clues about underlying regulatory mechanisms and network structure. For example, comparison of the global changes of expression in a mutant with the responses to different environmental or physiological challenges might provide information about the metabolic or cellular processes that are altered in the mutant. It could also reveal if unexpected side effects are complicating an experiment. Comparison of the global response across many arrays is best done using classification approaches (see e.g. Statnikov et al., 2005). However, often proper statistical methods are not available, and there are not enough replicates for these approaches.
MapMan has been extended to allow a facile identification of similarities between the response in a new experiment and the responses in large numbers of earlier experiments. To do this, a new type of mapping file and map has been introduced. These response maps group sets of genes whose expression is strongly increased or decreased in many different organs, developmental stages, or experimental treatments. The user can visually inspect the response of these sets of genes in a new experiment by uploading the corresponding experimental file. If the response resembles an earlier experiment (treatment “x”), many of the genes that are grouped as “induced by x” should be induced in the new experiment, and many of the genes that are grouped as “repressed by x” should be repressed.
A combination of strategies was used to retrieve sets of genes that are induced and repressed by different challenges from public microarray experiments (AtGenExpress) and our own data pool. The basic approach was to extract the 100 most up-regulated and the 100 most down-regulated genes in each treatment. In addition, treatment-specific genes were extracted by identifying outliers (Kadota et al., 2003). Although the latter approach facilitates nonparametric statistics, most of the genes that it retrieved had only low deviations in our experiments. Only the most responsive genes were included in the actual maps. A list of the maps and displayed conditions is available in the MapMan help. Perl scripts that allow users to automatically generate response maps to further challenges can be obtained via e-mail (usadel@mpimp-golm.mpg.de).
The use of response maps is illustrated by comparing the publicly available responses of pho1 and pho3 (Fig. 4). Both were isolated in screens that were designed to identify mutants that were altered in Pi levels or Pi sensing. The pho1 mutant (Poirier et al., 1991) has low Pi levels in the shoot and high Pi levels in the root. PHO1 encodes a member of a new family of putative Pi transporters and is located in the vascular system (Hamburger et al., 2002). This information, in combination with the pho1 phenotype, implicates PHO1 in the loading of Pi into the xylem. A dataset in Figure 4A displays the changes of gene expression in the shoot of the pho1 compared to wild-type plants (Hammond et al., 2004, downloaded from Nottingham Arabidopsis Stock Centre (NASC) arrays; see also Craigon et al., 2004) on a response map that collects sets of genes that are induced or repressed by depletion and subsequent readdition of carbon, nitrate, phosphate, or sulfate (the sets of genes were retrieved from Scheible et al., 2004, and other in-house nutrient response treatments). The response of pho1 is very similar to the response in phosphate-deficient plants and opposite to the response after Pi readdition. A completely different picture emerged for pho3 (Lloyd and Zakhleniuk, 2004; data downloaded from NASC arrays). This mutant was originally isolated in a screen based on loss of the ability to induce acid phosphatase in conditions of phosphate depletion (Zakhleniuk et al., 2001). The changes of gene expression in pho3 (Fig. 4B) showed little similarity to the responses in Pi starvation and especially Pi readdition, but instead showed similarities to the response during nitrogen depletion or after carbon readdition. It has recently been reported that PHO3 encodes a Suc transporter (SUC2; unpublished data referenced in Lloyd and Zakhleniuk, 2004). Hence, it is not difficult to understand that there is little similarity to the changes during Pi depletion and readdition. One prediction from Figure 3 is that sugars accumulate in the leaf of pho3, as would indeed be expected if the genetic lesion is a mutation in a phloem Suc transporter. The results also predict that nitrogen uptake or assimilation is seriously disturbed in pho3. One possible explanation is that low levels of sugars in the root inhibit nitrate uptake. It is known that low sugars repress nitrate transporters including NRT2.1 (Lejay et al., 1999) and inhibit nitrate uptake (A. Gojon, personal communication).
Figure 4.

Response of the pho1 and pho3 mutants visualized on a nutrient response map. For both pho1 and pho3 mutants, the response to the wild type was calculated as a log ratio. The results were displayed on a response map showing the hundred most up- or down-regulated genes of Arabidopsis in response to different starvation conditions. In the case where a similar response is encountered, the individual spots in an area should have the same color as the guide bars in the same column.
Access to MapMan
MapMan is available as a downloadable tool for users who wish to inspect their own data. Upon user invocation, the current ImageAnnotator module will automatically query the MapMan homepage for new updates (e.g. mapping files, updated and new maps). It will also remain possible to download updates by hand. MapMan is also provided as a Web version in combination with approximately 200 downloaded arrays from the public domain. The Web version is not designed to allow a user to explore his or her own data, but it instead provides a tool that will allow the user to explore data sets in the public domain without having to download the MapMan software package and all the individual array datasets. It may allow users to preselect interesting datasets from microarray repositories such as TAIR or NASC, which can then be downloaded for more detailed analysis. This feature may become increasingly useful as more and more Affymetrix experiments are being produced by large consortiums such as AtGenExpress.
FUTURE PERSPECTIVES
MapMan is a versatile and flexible but easy-to-use tool that will be continually updated with new biological information and expanded to increase its ease of use and its applications. The gene assignments and files will be made available on request to users who wish to modify and extend them. As MapMan will rely on expert input to continue improvements, we hope that these changes will be returned to us to allow them to be incorporated into the central files. The previous Scavenger Module relied on Excel sheets in conjunction with Microsoft Access. The underlying Scavenger Module now has been rewritten to interface an enterprise class database with a flexible Java interface, which will allow multiple experts and/or users to directly access the Scavenger module (D. Weicht, B. Usadel, O. Thimm, M. Piques, M. Stitt, and J. Selbig, unpublished data). The underlying gene assignments are useful not only for analysis of data from the model system Arabidopsis, but also for organizing and interpreting expression data from other species. Gene assignment files have already been made available to other groups to allow their adaptation to several crop plants.
The applications will also be expanded, including further modules into the ImageAnnotator module to allow more flexible handling and loading of the experimental data sets; simple, standard statistical tests; the exploration strategies to link changes of expression with multidimensional hierarchies of gene assignment using the search-and-highlight function; the display and the integration of different sorts of datasets; and the extension and refinement of ways to combine the versatility of MapMan with the power of horizontal searches across large public domain databases. Finally, to allow better integration of array data from diverse sources, standardized descriptions about the material and experimental design will be required. This might be facilitated by bioinformatics tools that allow biologists to define their experimental design in a controlled vocabulary and translate this information into a machine-readable format.
Terms Used in Article
Terms used in the article are as follows.
BIN, A container for items such as transcripts, metabolites, or proteins. The BINS are structured hierarchically and contain one or several levels of subBINS. Each BIN and subBIN contains transcripts (enzymes/metabolites) which, based on their annotation, can be (tentatively) assigned to a particular biological process. Some BINS are also included that organize genes whose biological function is not known according to their membership of large gene families.
Mapping File, A file that classifies items (e.g. transcripts) into the hierarchical BIN structure. Currently three Arabidopsis files are supplied with MapMan.
Map, A diagram or photorealistic picture showing biological processes or pathways onto which array data can be visualized. It consists of (1) the actual diagram and (2) an accompanying XML file, which stores information about which (sub)BINS are to be displayed at what locations on the diagram. Each map and the associated XML files must be used in conjunction with an appropriate mapping file. The XML files are supplied by the MapMan team but can be modified by the user. When users wish to create their own new maps, they will need to also create the accompanying XML file.
Experiment file, A file that harbors the results of a microarray, metabolic profile, etc. experiment giving log ratios of treatment versus control. Each experiment can be viewed on any map provided the measured items are displayed on the map.
Acknowledgments
We thank Oliver Fiehn for aid in gas chromatography-mass spectrometry profiling; Melanie Höhne and Manuela Günter for excellent support in plant growth, harvesting, extraction, and analysis; Matthias Steinfath for providing expertise in application and approximation of the Wilcoxon test; Stefanos Petrakis for skilled programming and many useful suggestions; Iris Bertram for her support in graphical design; and the people from TAIR for their cooperation. We are indebted to Dr. John Hammond for depositing his unpublished microarray data in the NASC database. Moreover, we are indebted to Frederic Beisson, Lieven de Veylder, Chris Helliwell, Matthew Hannah, Claire Hutchinson, Joe Kieber, Ute Krämer, Yan-Xia Liu, John Lunn, Steven Maere, John Ohlrogge, Wolf-Rüdiger Scheible, Jen Sheen, Alison Smith, Joost van Dongen, Klaas van Wijk, Dirk Warnecke, and Rita Zrenner for their precious expert input on various pathways.
This work was supported by Bildungsministerium für Bildung und Forschung (BMBF). The original ImageAnnotator was developed within the BMBF-funded project Genomanalyse im biologischen System Pflanze (GABI)-Primary Database (project no. 0312272), and the TranscriptScavenger and MetaboliteScavenger as well as the experimental studies reported in this article were supported by the BMBF-funded project GABI Verbund Arabidopsis III Gauntlets, Carbon and Nutrient Signaling: Test Systems, and Metabolite and Transcript Profiles (project no. 0312277A). The developments reported in this article were largely supported by the BMBF-funded project GABI-MapMan (project nos. 313112 and 313110). This work was also supported by the European Community (contract no. QLK1–CT–2001–01080 to N.P.-R.).
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M Jr, Haussler D (2000) Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci USA 97: 262–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis JE, Kruhoffer M, Gromove I, Frederiksen C, Ostergaard M, Thykjaer T, Gromova P, Yu J, Palsdottir H, Mangnusson N, et al (2000) Gene expression profiling: monitoring transcription and translation products using DNA microarrays and proteomics. FEBS Lett 480: 2–16 [DOI] [PubMed] [Google Scholar]
- Christie KR, Weng S, Balakrishnan R, Costanzo MC, Dolinski K, Dwight SS, Engel SR, Feierbach B, Fisk DG, Hirschman JE, et al (2004) Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res 32: D311–D314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craigon DJ, James N, Okyere J, Higgins J, Jotham J, May S (2004) NASCArrays: a repository for microarray data generated by NASC's transcriptomics service. Nucleic Acids Res 32: D575–D577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub CO, Kloska S, Selbig J (2003) MetaGeneAlyse: analysis of integrated transcriptional and metabolite data. Bioinformatics 19: 2332–2333 [DOI] [PubMed] [Google Scholar]
- De Risi JL, Iyer VR, Brown PO (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278: 680–686 [DOI] [PubMed] [Google Scholar]
- Fiehn O, Kopka J, Dörmann P, Altmann T, Trethewey RN, Willmitzer L (2000) Metabolite profiling for plant functional genomics. Nat Biotechnol 18: 1157–1161 [DOI] [PubMed] [Google Scholar]
- Gibon Y, Blaesing OE, Hannemann J, Carillo P, Hohne M, Hendriks JH, Palacios N, Cross J, Selbig J, Stitt M (2004) A Robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell 16: 3304–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosu P, Townsend JP, Hartl DL, Cavalieri D (2002) Pathway Processor: a tool for integrating whole-genome expression results into metabolic networks. Genome Res 12: 1121–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger D, Rezzonico E, MacDonald-Comber Petétot J, Somerville C, Poirier Y (2002) Identification and characterization of the Arabidopsis PHO1 gene involved in phosphate loading to the xylem. Plant Cell 14: 889–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JP, Bennett MJ, Broadley MR, White PJ (2004) Differential gene expression patterns in the phosphate deficient mutant, pho1. NASCarrays. http://affymetrix.arabidopsis.info/narrays/experimentpage.pl?experimentid=102 (February 1, 2005)
- Kadota K, Nishimura S, Bono H, Nakamura S, Hayashizaki Y, Okazaki Y, Takahashi K (2003) Detection of genes with tissue-specific expression patterns using Akaike's information criterion procedure. Physiol Genomics 12: 251–259 [DOI] [PubMed] [Google Scholar]
- Lee I, Date SV, Adai AT, Marcotte EM (2004) A probabilistic functional network of yeast genes. Science 306: 1555–1558 [DOI] [PubMed] [Google Scholar]
- Lejay L, Tillard P, Lepetit M, Olive F, Filleur S, Daniel-Vedele F, Gojon A (1999) Molecular and functional regulation of two NO3- uptake systems by N- and C-status of Arabidopsis plants. Plant J 18: 509–519 [DOI] [PubMed] [Google Scholar]
- Lloyd JC, Zakhleniuk OV (2004) Responses of primary and secondary metabolism to sugar accumulation revealed by microarray expression analysis of the Arabidopsis mutant, pho3. J Exp Bot 55: 1221–1230 [DOI] [PubMed] [Google Scholar]
- Michaut L, Flister S, Neeb M, White KP, Certa U, Gehring WJ (2003) Analysis of the eye developmental pathway in Drosophila using DNA microarrays. Proc Natl Acad Sci USA 100: 4024–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller LA, Zhang P, Rhee SY (2003) AraCyc: a biochemical pathway database for Arabidopsis. Plant Physiol 132: 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier Y, Thoma S, Somerville C, Schiefelbein J (1991) A mutant of Arabidopsis deficient in xylem loading of phosphate. Plant Physiol 97: 1087–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2004) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org (February 1, 2005)
- Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M, et al (2003) The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res 31: 224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheible WR, Morcuende R, Czechowski T, Fritz C, Osuna D, Palacios-Rojas N, Schindelasch D, Thimm O, Udvardi MK, Stitt M (2004) Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol 136: 2483–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statnikov A, Aliferis CF, Tsamardinos I, Hardin D, Levy S (2005) A comprehensive evaluation of multicategory classification methods for microarray gene expression cancer diagnosis. Bioinformatics 21: 631–643 [DOI] [PubMed] [Google Scholar]
- Steinhauser D, Usadel B, Luedemann A, Thimm O, Kopka J (2004) CSB.DB: a comprehensive systems-biology database. Bioinformatics 20: 3647–3651 [DOI] [PubMed] [Google Scholar]
- Stitt M, Fernie AR (2003) From measurements of metabolites to metabolomics: an ‘on the fly’ perspective illustrated by recent studies of carbon-nitrogen interactions. Curr Opin Biotechnol 14: 136–144 [DOI] [PubMed] [Google Scholar]
- Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37: 914–939 [DOI] [PubMed] [Google Scholar]
- Wang R, Okamoto M, Xing X, Crawford NM (2003) Microarray analysis of the nitrate response in Arabidopsis roots and shoots reveals over 1,000 rapidly responding genes and new linkages to glucose, trehalose-6-phosphate, iron, and sulfate metabolism. Plant Physiol 132: 556–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Tischner R, Gutierrez RA, Hoffman M, Xing X, Chen M, Coruzzi G, Crawford NM (2004) Genomic analysis of the nitrate response using a nitrate reductase-null mutant of Arabidopsis. Plant Physiol 136: 2512–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhleniuk OV, Raines CA, Lloyd JC (2001) pho3: a phosphorus-deficient mutant of Arabidopsis thaliana (L.) Heynh. Planta 212: 529–534 [DOI] [PubMed] [Google Scholar]
- Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W (2004) GENEVESTIGATOR: Arabidopsis microarray database and analysis toolbox. Plant Physiol 136: 2621–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]