

Abstract
The secreted Mycobacterium tuberculosis complex proteins CFP-10 and ESAT-6 have recently been shown to play an essential role in tuberculosis pathogenesis. We have determined the solution structure of the tight, 1:1 complex formed by CFP-10 and ESAT-6, and employed fluorescence microscopy to demonstrate specific binding of the complex to the surface of macrophage and monocyte cells. A striking feature of the complex is the long flexible arm formed by the C-terminus of CFP-10, which was found to be essential for binding to the surface of cells. The surface features of the CFP-10·ESAT-6 complex, together with observed binding to specific host cells, strongly suggest a key signalling role for the complex, in which binding to cell surface receptors leads to modulation of host cell behaviour to the advantage of the pathogen.
Keywords: CFP-10, ESAT-6, pathogenesis, tuberculosis, virulence
Introduction
Tuberculosis kills 2–3 million people annually (World Health Organisation, 2004), yet we still have little understanding of the molecular basis of pathogenesis. Comparisons of the genomes of virulent Mycobacterium tuberculosis and Mycobacterium bovis with attenuated BCG vaccine strains identified a deletion (RD1) in all BCG strains that plays a key role in virulence (Mahairas et al, 1996; Behr et al, 1999; Brosch et al, 2001; Pym et al, 2002). This region contains nine protein-coding genes (Rv3871–3879c) and subsequent work showed that inactivation of just two of these (Rv3874 and Rv3875), coding for the secreted proteins CFP-10 (100 residues) and ESAT-6 (95 residues), results in dramatically reduced virulence (Behr et al, 1999; Wards et al, 2000; Pym et al, 2002; Hsu et al, 2003; Stanley et al, 2003). These proteins clearly play an essential role in tuberculosis pathogenesis, but show no homology to any proteins of known structure or function.
CFP-10 and ESAT-6 are members of a large family of mycobacterial proteins, including 22 found in M. tuberculosis, which are encoded by genes arranged in pairs in the genome (Renshaw et al, 2002). In the case of CFP-10 and ESAT-6, the genes have been shown to be coordinately regulated (Berthet et al, 1998) and both proteins are secreted despite lacking a conventional signal sequence (Pym et al, 2003). Recent work indicates that secretion of the proteins is an active process involving a membrane protein complex formed from the products of several flanking genes (Gey van Pittius et al, 2001; Hsu et al, 2003; Sassetti and Rubin, 2003; Stanley et al, 2003; Guinn et al, 2004). Several other CFP-10/ESAT-6 family members are also known to be secreted, including the products of Rv0287/Rv0288 and Rv3019c/Rv3020c (Alderson et al, 2000; Rosenkrands et al, 2000; Skjøt et al, 2000, 2002), which we have recently shown form tight complexes, as observed for CFP-10 and ESAT-6 (Renshaw et al, 2002; Lightbody et al, 2004). The Mycobacterium leprae genome contains only 1604 functional protein genes compared to 4006 for M. tuberculosis and has been proposed to represent the minimal gene set for a pathogenic mycobacterium (Cole et al, 2001). Strikingly, of the 11 pairs of CFP-10/ESAT-6 family proteins found in M. tuberculosis, only orthologues of CFP-10 and ESAT-6 are individually conserved in M. leprae (ML0050c and ML0049c, respectively), which further emphasises their importance in the lifecycle of mycobacterial pathogens (Renshaw et al, 2002).
Recently, we showed that CFP-10 and ESAT-6 form a tight (Kd⩽1.1 × 10−8 M), 1:1 complex (Renshaw et al, 2002) and here we report the solution structure of the complex, together with fluorescence microscopy data showing specific binding of the complex to the surface of primary macrophages, the main cell type infected by M. tuberculosis. The structural features of the CFP-10·ESAT-6 complex, together with clear evidence for specific binding of the complex to the surface of host cells, strongly imply a signalling role for the CFP-10·ESAT-6 complex, in which binding to cell surface receptors may lead to modulation of host cell behaviour.
Results and discussion
We have determined the solution structure of the CFP-10·ESAT-6 complex to high precision, which is clearly evident from the overlay of the protein backbone shown for the family of 28 satisfactorily converged structures in Figure 1. This is also reflected in low root mean squared deviation values to the mean structure for both the backbone and all heavy atoms (0.49±0.13 and 0.93±0.12 Å, respectively) for the well-defined regions of the complex (residues 6–85 in CFP-10 and ESAT-6). The family of converged structures contain no distance or van der Waals violations greater than 0.5 Å and no dihedral angle violations greater than 5o, with an average value for the CYANA target function of 7.41±0.85 Å2 (Herrmann et al, 2002). The sums of the violations for upper distance limits, lower distance limits, van der Waals contacts and torsion angle constraints were 34.8±2.44 Å, 2.1±0.28 Å, 13.3±1.29 Å and 81.3±12.54°, respectively. Similarly, maximum violations for the converged structures were 0.35±0.05 Å, 0.25±0.03 Å, 0.27±0.06 Å and 3.95±0.45°, respectively. Analysis of the backbone dihedral angles for the family of converged structures using PROCHECK (Laskowski et al, 1996) revealed that 82% of the residues adopt backbone conformations found within the most favoured regions of a Ramachandran plot and that 17% lie within the additional allowed regions, with no residues consistently found in disallowed regions.
Figure 1.
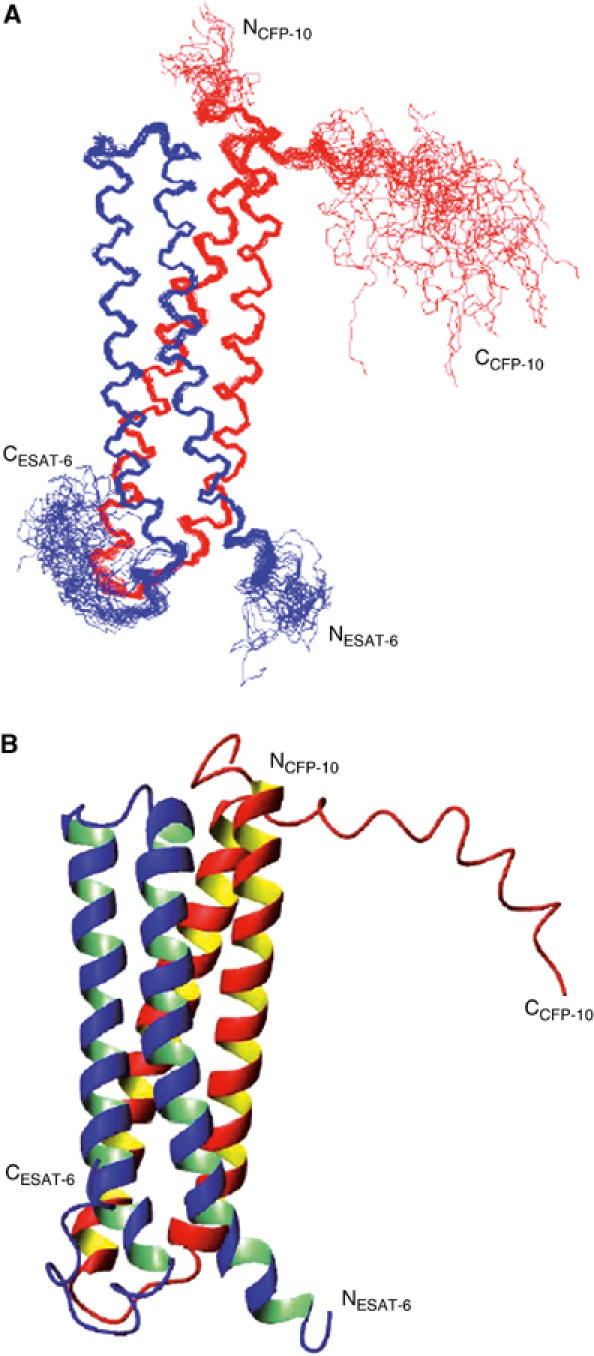
Solution structure of the CFP-10·ESAT-6 complex. (A) A best-fit superposition of the protein backbone for the family of 28 converged structures obtained, with CFP-10 shown in red and ESAT-6 in blue. The long flexible C-terminal arms of both proteins are clearly visible, as is the propensity to helical structure in this region of CFP-10. (B) A ribbon representation of the backbone topology of the CFP-10·ESAT-6 complex based on the converged structure closest to the mean, which illustrates the two helix–turn–helix hairpin structures formed by the individual proteins. The orientation of the complex is identical to that shown in panel A, with CFP-10 in red and ESAT-6 in blue. The helical propensity of residues 85–95 in the flexible C-terminus of CFP-10 can be clearly seen in the top right of the figure.
The well-defined core of the CFP-10·ESAT-6 complex consists of two similar helix–turn–helix hairpin structures formed from the individual proteins, which have an extensive hydrophobic contact surface and lie antiparallel to each other to form a four-helix bundle (Figure 1). A striking feature of the complex is the disordered N- and particularly C-termini of both proteins (residues 2–5 and 86–100 in CFP-10 and 1–3 and 86–95 in ESAT-6), which form long flexible arms at both ends of the four-helix bundle core. The two long helices in the hairpin structures are formed from residues Ala8–Gln40 and Ala47–Ala79 in CFP-10, and from Phe8–Trp43 and Glu49–Ala79 in ESAT-6. The helices in CFP-10 are completely α-helical, whereas in ESAT-6 both long helices terminate with a single turn of 310 helix and ESAT-6 also contains a short 310 helix close to the N-terminus (Gln4–Trp6). Chemical shift and NOE data indicate that part of the exposed C-terminal region of CFP-10 (Arg85–Ser95) has a distinct propensity to adopt a helical conformation, which is clearly evident in Figure 1. This region of CFP-10 may be involved in interactions with a host cell target protein (discussed below), resulting in stabilisation of the helical conformation.
The CFP-10·ESAT-6 complex has recently been proposed to have host cell lysis activity mediated via the formation of pores in cell membranes (Hsu et al, 2003); however, analysis of the electrostatic surface of the complex strongly argues against a pore forming role. The surface of the complex has a very uniform distribution of positive and negative charge, with no hint of a significant hydrophobic patch (Figure 2), which is clearly inconsistent with a membrane spanning pore. In addition, the complex is soluble to over 2 mM in aqueous solution with no sign of aggregation, which is certainly not typical behaviour for a pore forming protein. The surface of the complex is also devoid of any striking acidic or basic patches, which in the latter case suggests that the complex is not involved in interactions with nucleic acids. Similarly, there are no significant clefts in the surface of the structure indicative of an enzyme active site, which suggests a noncatalytic role for the complex. Overall, the surface features of the CFP-10·ESAT-6 complex seem most consistent with a function based on specific binding to one or more target proteins, perhaps playing a key role in pathogen–host cell signalling.
Figure 2.
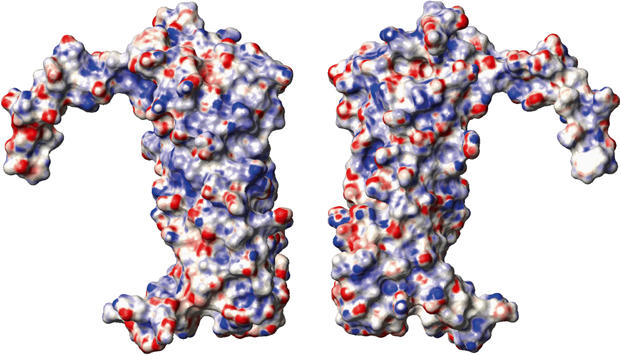
Space-filled views of the surface of the CFP-10·ESAT-6 complex based on the converged structure closest fit to the mean. The right-hand view depicts the complex in the same orientation as in Figure 1, while the left-hand view is rotated by 180o about the vertical axis to show the opposite face. The surface is coloured according to electrostatic potential, with areas of significant negative charge in red, significant positive charge in blue and neutral as white. The electrostatic potential was calculated using MOLMOL (Koradi et al, 1996), with the threshold for depicting significant areas of charge chosen to obtain a neutral representation for the fully exposed aromatic ring of Phe100 in CFP-10.
The extensive contact surface between CFP-10 and ESAT-6 is essentially hydrophobic in nature and comprises about 25% (∼1800 Å2) of the total surface area of both proteins. In the case of CFP-10, 29 residues account for nearly 90% of the interface (Lys5, Thr6, Leu11, Glu14, Asn17, Phe18, Ile21, Leu25, Gln28, Val32, Thr35, Leu39, Gln42, Trp43, Arg44, Ala46, Ala47, Ala50, Ala54, Phe58, Ala61, Lys64, Gln65, Glu68, Glu71, Ile72, Asn75, Ile76 and Ala79) and for ESAT-6 just 26 residues form approximately 85% of the contact surface (Ile11, Ala14, Ile18, Asn21, Ile25, Leu28, Leu29, Glu31, Gly32 Ser35, Lys38, Leu39, Ala41, Ala42, Trp43, Lys57, Trp58, Thr61, Glu64, Leu65, Ala68, Leu69, Leu72, Thr75, Ile76 and Met83; see Figure 3). The tight interaction between the two proteins in the complex appears to be primarily based on extensive and favourable van der Waals contacts; however, two salt bridges between CFP-10 and ESAT-6 (Glu14–Lys38 and Glu71–Lys57) appear to stabilise interactions between the N-terminal end of helix-1 in CFP-10 and the C-terminal end of the corresponding helix in ESAT-6, and between the C-terminal region of helix-2 in CFP-10 and the N-terminal region of the equivalent helix in ESAT-6, respectively. The positions of the residues forming the two salt bridges are indicated on the multiple sequence alignment for CFP-10- and ESAT-6-related proteins from M. tuberculosis shown in Figure 3 and their lack of conservation indicates that these specific salt bridges are not a general feature of the complexes formed by CFP-10/ESAT-6 family proteins, nor a predictor of which family members will form complexes. Analysis of the multiple sequence alignment (Figure 3) reveals that over half of the interface residues are conserved to type in at least two-thirds of the sequences. This, together with predicted helical structures for all members of the M. tuberculosis CFP-10/ESAT-6 family and known complex formation for several genome pairs, strongly suggests that all pairs of these proteins will form similar, four-helix bundle containing complexes. However, careful consideration of both the structural and sequence conservation data provides no clear rules for predicting which nongenome paired members of the family will form tight complexes, beyond the importance of close sequence similarity discussed previously (Lightbody et al, 2004).
Figure 3.
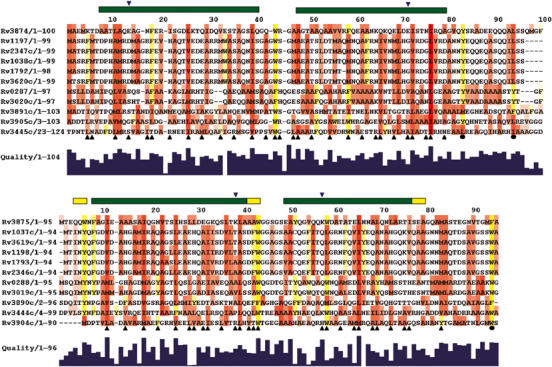
Conservation of the amino-acid sequences of the CFP-10- (top) and ESAT-6- (bottom) related proteins from M. tuberculosis. Aliphatic residues with hydrophobic side chains (Leu, Ile, Val, Met and Ala) are highlighted in red and aromatic residues (Phe, Tyr and Trp) in yellow. The extent of sequence conservation is indicated by the histogram quality score and for highlighted residues the greater the conservation, the more intense the colour. Residues forming the interface between CFP-10 and ESAT-6 are indicated by upright black triangular symbols and conserved hydrophobic residues in the C-terminal arms of the proteins by closed circles. Similarly, the locations of four residues forming two intermolecular salt bridges are indicated by down-turned blue triangles. The positions of helices in the structure of the complex are shown by bars above the sequences (α in green and 310 in yellow). The sequences were aligned using ClustalW, with a standard Blosum30 scoring matrix, a gap opening penalty of 10 and a gap extension penalty of 1 (Thompson et al, 1997).
The multiple sequence alignments shown in Figure 3 reveal that there are a number of hydrophobic and aromatic residues located in the C-terminal regions of CFP-10 (Tyr83 and Leu94) and ESAT-6 (Phe94), which are conserved across the whole family of M. tuberculosis-related proteins. These residues are found on the surface of the CFP-10·ESAT-6 complex and play no structural role, implying some functional significance. The C-terminal regions of CFP-10 and ESAT-6 are also as well conserved between M. tuberculosis and M. leprae as the overall proteins (overall 69% amino-acid sequence homology for CFP-10/ML0050c and 62% for ESAT-6/ML0049c) despite no structural role in the complex, which again implies functional pressure to conserve these regions. It therefore seems possible that the conserved flexible arms in the CFP-10·ESAT-6 complex form part of the interaction site with a target protein.
As discussed above, the structure of the complex strongly suggests that its function is mediated via binding to specific target proteins. The complex has recently been shown to be actively secreted from M. tuberculosis and M. bovis bacilli (Hsu et al, 2003; Pym et al, 2003; Stanley et al, 2003; Guinn et al, 2004), and the expression of both proteins was significantly downregulated by bacteria internalised in macrophages (Schnappinger et al, 2003), raising the possibility that target proteins may be found on the surface of host cells. To test this hypothesis, we have covalently labelled the N-termini of both proteins in the complex with a fluorophore (Alexa Fluor 546) and used fluorescence microscopy to look for specific binding to a variety of cell types, including primary monocytes and macrophages, U937 and MonoMac 6 (MM6) monocyte cell lines, and the fibroblast cell lines COS-1 and NIH-3T3.
The primary monocytes and macrophages, together with both monocyte cell lines, consistently showed intense fluorescence at the cell surface after incubation with the labelled complex, which in a significant proportion of cells was further focused in patches reminiscent of the ‘cap-like' structures associated with cell surface receptors (Kwiatkowska and Sobota, 1999). In contrast, no significant fluorescence labelling was seen for the two fibroblast cell lines. This is illustrated by the representative images shown in Figure 4. Analogous experiments were also carried out with Alexa Fluor 546-labelled MPB70 (another major secreted protein of M. tuberculosis and M. bovis) and MM6 cells. In this case, no significant labelling of the surface of MM6 cells was detected, which strongly argues that the fluorescence localisation observed with the labelled CFP-10·ESAT-6 complex is not mediated by the fluorophore. Similarly, the cell type-specific labelling observed for the CFP-10·ESAT-6 complex suggests that the localisation results from tight binding to a cell surface receptor (probably a protein) expressed in monocytic cells, the main cell type infected by M. tuberculosis, rather than a nonspecific interaction between the complex or fluorophore and cell surface. This point was further investigated using U937 cells, which were incubated with fluorescently labelled CFP-10·ESAT-6 complex in the presence of a 20-fold molar excess of unlabelled complex. The intensity of the fluorescence associated with the surface of U937 cells under these conditions was very significantly reduced (Figure 5), which clearly indicates that binding is mediated by the protein complex and not the attached fluorophore.
Figure 4.
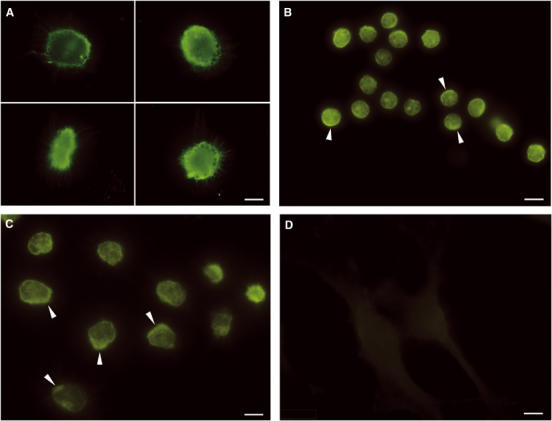
Binding of fluorescently labelled CFP-10·ESAT-6 complex to the surface of monocyte lineage cells. (A–C) (A, C, 100 ms exposure; B, 200 ms exposure) The typical fluorescence localisation observed after incubating primary macrophages, primary monocytes and the monocyte cell line U937 with 1 μM fluorescently labelled CFP-10·ESAT-6. The fluorescence labelling is focused at the cell surface for all three monocytic cells and is often seen concentrated in patches (indicated by arrows), resembling the ‘cap-like' structures associated with receptor-mediated signalling. In contrast, no significant fluorescence labelling was detected for several fibroblast cell lines, as illustrated by the representative images shown for NIH3T3 cells in (D) (200 ms exposure). The size bars shown correspond to 5 μm.
Figure 5.
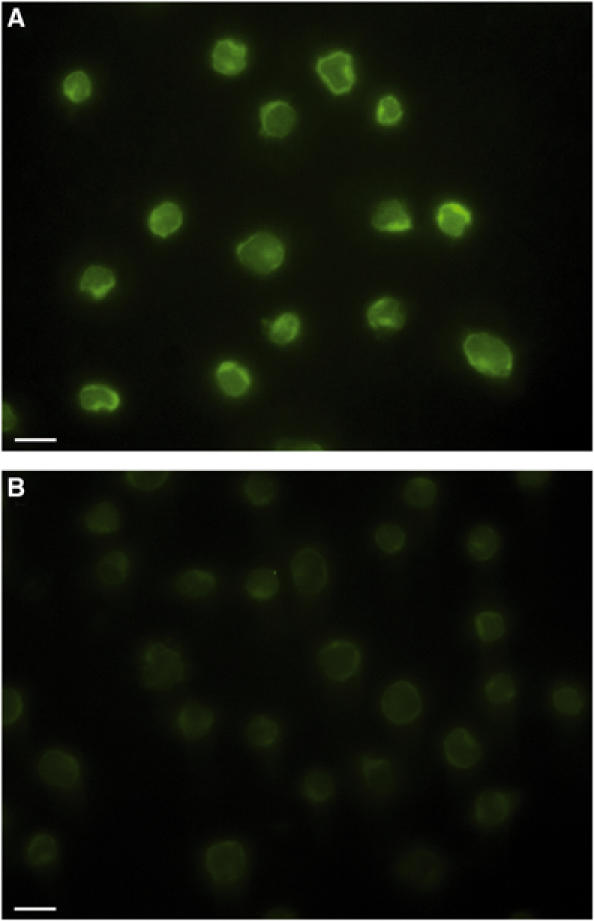
Blocking of fluorescently labelled CFP-10·ESAT-6 complex binding to U937 cells by competition with unlabelled complex. (A) Typical fluorescence images obtained for U937 cells after incubating with 1 μM Alexa Fluor 546-labelled CFP-10·ESAT-6 complex for 15 min at 4°C. (B) The dramatic effect of adding a 20-fold molar excess of unlabelled complex. Clearly, the fluorescence labelling of U937 cells in the presence of an excess of unlabelled CFP-10·ESAT-6 complex is several orders of magnitude lower, which indicates that binding is mediated via the protein complex and not the attached fluorophore. The size bars shown correspond to 10 μm.
In order to test the hypothesis that the binding of the CFP-10·ESAT-6 complex to the surface of target host cells involves the flexible C-termini of either CFP-10 or ESAT-6, complexes formed from truncated CFP-10 (residues 1–86) bound to full-length ESAT-6 and full-length CFP-10 bound to truncated ESAT-6 (residues 1–84) were similarly labelled with Alexa Fluor 546 and incubated with U937 monocytes. The fluorescence labelling observed for cells incubated with the full-length CFP-10·truncated ESAT-6 complex was indistinguishable from that observed for the intact CFP-10·ESAT-6 complex. In contrast, the fluorescence labelling observed for U937 cells incubated with truncated CFP-10 bound to full-length ESAT-6 was dramatically reduced (Figure 6). These findings clearly confirm that binding of fluorescently labelled CFP-10·ESAT-6 complex to the surface of U937 cells is mediated by the protein complex and also show that the flexible C-terminal arm of CFP-10 forms an essential part of the cell surface receptor binding site. It is also worth noting that during time course experiments not reported in detail here, both primary and MM6 cells exposed to the labelled CFP-10·ESAT-6 complex for periods of at least several hours showed no evidence of lysis, supporting the conclusion that the complex is not associated with cytolytic activity.
Figure 6.
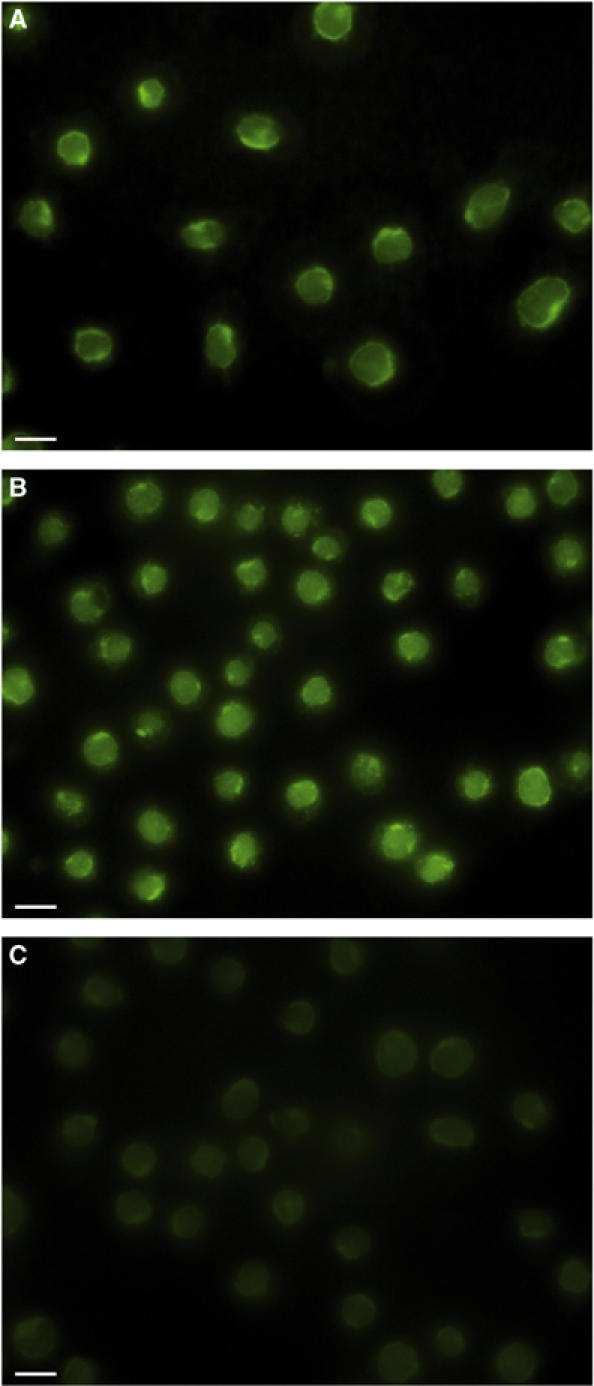
Dramatically reduced binding of the CFP-10·ESAT-6 complex containing C-terminally truncated CFP-10 to U937 cells. Panel (A) illustrates the typical fluorescence localisation observed for U937 cells incubated with full-length labelled complex, while panels (B) and (C) show comparable images obtained with labelled complexes containing C-terminally truncated ESAT-6 and CFP-10, respectively. Removal of the exposed C-terminus of ESAT-6 appears to have no significant effect on the ability of the complex to bind to U937 cells. In contrast, the fluorescence associated with U937 cells incubated with the complex containing C-terminally truncated CFP-10 is several orders of magnitude lower than that observed for the intact complex, which indicates that the flexible C-terminus of CFP-10 forms an essential component of the binding site for the cell surface receptor. The size bars shown correspond to 10 μm.
The work reported here implies a possible signalling role for the CFP-10·ESAT-6 complex, in which binding to cell surface receptors leads to modulation of host cell behaviour, and clearly represents a major advance in our understanding of the essential role of the CFP-10·ESAT-6 complex in tuberculosis pathogenesis. During final preparation of this manuscript, it was reported that secreted RD1 virulence determinants are required for macrophage aggregation and the subsequent formation of granulomas in zebrafish infected with Mycobacterium marinum (Volkman et al, 2004). This finding clearly supports our conclusion that the CFP-10·ESAT-6 complex acts as a signalling molecule and further work is now ongoing to identify the host cell target proteins for the complex.
Materials and methods
Protein preparation
The nonlabelled and uniformly 15N- and 15N/13C-labelled CFP-10 and ESAT-6 were prepared as described previously (Renshaw et al, 2002, 2004). In addition, 13C/1H HMQC-NOESY spectra were acquired from samples of the complex in which only the nonaromatic residues were uniformly 15N/13C labelled. This was achieved by the preparation of both proteins from Escherichia coli grown in labelled minimal media supplemented with 50 mg/l of L-histidine, L-tyrosine, L-phenylalanine and L-tryptophan (Carr et al, 2003). The mixed complexes of labelled CFP-10 bound to nonlabelled ESAT-6 and vice versa were produced by mixing equimolar solutions of the purified proteins at room temperature in 25 mM NaH2PO4, 100 mM NaCl and 0.02% (w/v) NaN3, pH 6.5, with the individual proteins at a concentration of 5–15 μM. The complex was concentrated by ultrafiltration to give 0.35 ml NMR samples containing 0.9–1.5 mM CFP-10·ESAT-6 complex in either a 90% H2O/10% D2O or 100% D2O buffer as appropriate.
Protein corresponding to a truncated variant of CFP-10 lacking the final 14 C-terminal residues (Asp87–Phe100) was prepared from a pET28a-based E. coli expression vector, which was produced using a PCR-based approach, essentially as described previously (Renshaw et al, 2002). Purification of the expressed protein was carried out in two stages using a 10 ml Q-Sepharose column (Renshaw et al, 2002), with truncated CFP-10 eluted from the column in the 50 mM NaCl step at pH 8.0 and in the 20 mM NaCl step at pH 5.8.
C-terminally truncated ESAT-6 corresponding to residues 1–84 is produced as a by-product during purification of the full-length protein. The two species are separated by anion exchange (Renshaw et al, 2002), with the truncated species eluted from the column in the 100 mM NaCl step.
NMR spectroscopy
NMR spectra were acquired at 35°C on either an 800 MHz Varian Inova or a 600 MHz Bruker Avance spectrometer. The 2D and 3D spectra recorded to obtain essentially complete sequence-specific backbone and side-chain assignments for CFP-10 and ESAT-6 in the complex, and to obtain conformational constraints for structural calculations were as follows: 1H TOCSY and NOESY; 15N/1H HSQC, TOCSY-HSQC and NOESY-HSQC; 13C/1H HCCH-TOCSY and HMQC-NOESY; and 15N/13C/1H HNCACB, CBCA(CO)NH and HBHA(CBCACO)NH, as described previously (Renshaw et al, 2004).
The 3D NMR data were processed using NMRPipe (Delaglio et al, 1995), with linear prediction used to extend the effective acquisition times by up to 1.5-fold in F1 and F2 and mild resolution enhancement applied in all dimensions using a shifted sine-squared function. Apart from the omission of linear prediction, the 2D spectra were similarly processed using Varian or Bruker software. All the spectra were analysed using the program XEASY (Bartels et al, 1995).
Structural calculations
The family of converged CFP-10·ESAT-6 complex structures was calculated in a two-stage process using the program CYANA (Herrmann et al, 2002). Initially, the combined automated NOE assignment and structure determination protocol (CANDID) was used to automatically assign the NOE crosspeaks identified in 3D 15N- and 13C-edited NOESY spectra of the complex and to produce preliminary structures. Subsequently, several cycles of simulated annealing combined with redundant dihedral angle constraints (REDAC) to increase convergence were used to produce the final converged CFP-10·ESAT-6 complex structures (Muskett et al, 1998; Lemercinier et al, 2001; Carr et al, 2003). The input for the CANDID stage primarily consisted of essentially complete 15N, 13C and 1H resonance assignments for the nonexchangeable groups in the CFP-10·ESAT-6 complex and four manually picked NOE peak lists obtained from 3D 15N- and 13C-edited NOESY spectra of complexes in which only one protein was labelled. In the 15N-edited spectra, 1165 NOE peaks were identified with labelled CFP-10 and 1237 with labelled ESAT-6, and in the 13C-edited spectra 1962 NOEs with CFP-10 and 2580 with ESAT-6 were identified. In addition, the CANDID stage included φ and ϕ dihedral angle constraints for 95 residues in CFP-10 and 89 in ESAT-6, which were obtained from the 13C and 1H chemical shifts of backbone resonances using TALOS (Cornilescu et al, 1999). The CANDID calculations were carried out using the default parameter settings in CYANA 1.0.6 apart from slightly increasing the chemical shift tolerances to 0.03 ppm for 1H and 0.4 ppm for 15N and 13C.
The final converged CFP-10·ESAT-6 complex structures were produced from 100 random starting conformations using a torsion angle-based simulated annealing protocol combined with six cycles of REDAC (Muskett et al, 1998; Lemercinier et al, 2001; Carr et al, 2003). The calculations were mainly based on 3315 nonredundant, NOE-derived upper distance limits, assigned to unique pairs of protons using CANDID and corresponding to over 90% of the NOE peaks identified. However, constraints were also included for φ and ϕ dihedral angles in 184 residues and for hydrogen bonds formed by 37 residues with slowly exchanging backbone amide signals and where the hydrogen bond acceptor was unambiguous in preliminary structures (residues 22–26, 28–33, 58, 59, 61, 62, 65, 66, 68, 69 and 72 in CFP-10, and 28–33, 35, 36, 39, 62, 63 and 65–70 in ESAT-6). Slowly exchanging backbone amides in the complex were identified from a series of 15N/1H HSQC spectra recorded over a period of several hours after dissolving samples of the complex in D2O. The final family of CFP-10·ESAT-6 complex structures obtained were analysed using the programs CYANA, PROCHECK and MOLMOL, which included standard combined distance and orientation-based searches for hydrogen bonds and salt bridges (Koradi et al, 1996; Laskowski et al, 1996; Herrmann et al, 2002). Coordinates for the family of converged CFP-10·ESAT-6 complex structures, together with the NMR constraints, have been deposited in the Protein Data Bank under accession number 1wa8.
Fluorescence microscopy
Samples of complexes corresponding to full-length CFP-10 bound to full-length ESAT-6, truncated CFP-10 bound to full-length ESAT-6 and full-length CFP-10 bound to truncated ESAT-6 were labelled with the fluorophore Alexa Fluor 546 (Molecular Probes) by incubating a 10-fold molar excess of the succinimidyl ester derivative of the dye with the respective complexes in a 25 mM NaH2PO4 and 100 mM NaCl, pH 7.5, buffer at room temperature overnight. At pH 7.5, the reactive succinimidyl ester group on the fluorophore is able to react with the N-terminal amino group of the two proteins, but not with charged lysine side-chain amino groups. Excess dye was removed by dialysis and the extent of labelling (typically 1.5–1.9:1) determined from the absorbance of the labelled complex at 280 and 556 nm, as per the supplier's instructions.
Primary monocyte, monocyte-derived macrophages, NIH-3T3 and COS-1 cells were grown directly on glass coverslips in appropriate media. The MonoMac 6 and U937 monocyte cell lines were initially grown in suspension and then allowed to adhere to glass coverslips precoated with 160 μg/ml poly-L-lysine for 20 min at 37°C. To assay for potential binding of the full-length CFP-10·ESAT-6 complex to the surface of specific cell types, cells adhered to coverslips were incubated with 1 μM Alexa Fluor 546-labelled complex for 15 min in PBS at either room temperature or 4°C. Nonbound complex was removed by two PBS washes prior to fixing of the cells with 4% (w/v) paraformaldehyde and permeabilisation with 0.2% (v/v) Triton X-100. The coverslips were mounted onto slides using ProLong antifade reagent (Molecular Probes) and stored at room temperature in the dark until dry. Fluorescence microscopy was carried out using a Nikon TE300 inverted microscope and the images recorded with a Hamamatsu CCD camera.
Similarly, U937 monocyte cells were incubated with 1 μM samples of Alexa Fluor 546-labelled combinations of truncated and full-length complexes for 15 min at 4°C, to minimise cell wall fluidity and possible receptor cycling, prior to being washed, fixed and imaged as described above. The blocking experiments were also carried out with U937 cells, which were incubated with a solution containing 1 μM labelled full-length complex and a 20-fold molar excess of unlabelled complex for 15 min at 4°C.
Acknowledgments
This work was initially supported by the award of a PhD studentship to Philip Renshaw from the Biotechnology and Biological Sciences Research Council and the Veterinary Laboratories Agency. Recent support has been provided by a project grant from the Wellcome Trust (066047). Kirsty Lightbody is supported by a PhD studentship from the Department for Environment, Food and Rural Affairs. Mark Carr is a member of the Mycobacterium tuberculosis Structural Genomics Consortium.
References
- Alderson MR, Bement T, Day CH, Zhu L, Molesh D, Skeiky YAW, Coler RN, Lewinson DM, Reed SG, Dillon DC (2000) Expression cloning of an immunodominant family of Mycobacterium tuberculosis antigens using human CD4+ T cells. J Exp Med 191: 551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C, Xia T-H, Billeter M, Güntert P, Wüthrich K (1995) The program XEASY for computer-supported nmr spectral analysis of biological macromolecules. J Biomol NMR 5: 1–10 [DOI] [PubMed] [Google Scholar]
- Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, Small PM (1999) Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284: 1520–1523 [DOI] [PubMed] [Google Scholar]
- Berthet F-X, Ramussen PB, Rosenkrands I, Andersen P, Gicquel B (1998) A Mycobacterium tuberculosis operon encoding ESAT-6 and a novel low-molecular-mass culture filtrate protein (CFP-10). Microbiology 144: 3195–3203 [DOI] [PubMed] [Google Scholar]
- Brosch R, Pym AS, Gordon SV, Cole ST (2001) The evolution of mycobacterial pathogenicity: clues from comparative genomics. Trends Microbiol 9: 452–458 [DOI] [PubMed] [Google Scholar]
- Carr MD, Bloemink MJ, Dentten E, Whelan AO, Gordon SV, Kelly G, Frenkiel TA, Hewinson RG, Williamson RA (2003) Solution structure of the Mycobacterium tuberculosis complex protein MPB70. From tuberculosis pathogenesis to inherited human corneal disease. J Biol Chem 278: 43736–43743 [DOI] [PubMed] [Google Scholar]
- Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honoré N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holyroyd S, Hornsby T, Jagels K, Lacroix C, McLean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandrearn M-A, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG (2001) Massive gene decay in the leprosy bacillus. Nature 409: 1007–1011 [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13: 289–302 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Gey van Pittius NC, Gamieldien J, Hide W, Brown GD, Siezen RJ, Beyers AD (2001) The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol 2:Research. 0044.1–0044.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR (2004) Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol 51: 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann T, Güntert P, Wüthrich K (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319: 209–227 [DOI] [PubMed] [Google Scholar]
- Hsu T, Hinigley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR (2003) The primary mechanism of attenuation of Bacillus Calmette-Guérin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA 100: 12420–12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14: 51–55 [DOI] [PubMed] [Google Scholar]
- Kwiatkowska K, Sobota A (1999) Tyryosine phosphorylation/dephosphorylation controls capping of Fcγ receptor II in U937 Cells. Cell Motil Cytoskeleton 42: 298–314 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477–486 [DOI] [PubMed] [Google Scholar]
- Lemercinier X, Muskett FW, Cheeseman B, McIntosh PB, Thim L, Carr MD (2001) High resolution solution structure of human intestinal trefoil factor and functional insights from detailed structural comparisons with the other members of the trefoil family of cell motility factors. Biochemistry 40: 9552–9559 [DOI] [PubMed] [Google Scholar]
- Lightbody KL, Renshaw PS, Collins ML, Wright RL, Hunt DM, Gordon SV, Hewinson RG, Buxton RS, Williamson RA, Carr MD (2004) Characterisation of complex formation between members of the Mycobacterium tuberculosis complex CFP-10/ESAT-6 protein family; towards an understanding of the rules governing complex formation and thereby functional flexibility. FEMS Microbiol Lett 238: 255–262 [DOI] [PubMed] [Google Scholar]
- Mahairas GG, Sabo PJ, Hickey MJ, Sing DC, Stover CK (1996) Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol 178: 1274–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muskett FW, Frenkiel TA, Feeney J, Freedman RB, Carr MD, Williamson RA (1998) High resolution structure of the N-terminal domain of tissue inhibitor of metalloproteinases-2 and characterisation of its interaction site with matrix metalloproteinase-3. J Biol Chem 273: 21736–21743 [DOI] [PubMed] [Google Scholar]
- Pym AS, Brodin P, Brosch R, Huerre M, Cole ST (2002) Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 46: 709–717 [DOI] [PubMed] [Google Scholar]
- Pym AS, Brodin P, Majlessi L, Brosch R, Demangel C, Williams A, Griffiths KE, Marchal G, Leclerc C, Cole ST (2003) Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat Med 9: 533–539 [DOI] [PubMed] [Google Scholar]
- Renshaw PS, Panagiotidou P, Whelan A, Gordon SV, Hewinson RG, Williamson RA, Carr MD (2002) Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterisation of the structural properties of ESAT-6, CFP-10 and the ESAT-6.CFP-10 complex: implications for pathogenesis and virulence. J Biol Chem 277: 21598–21603 [DOI] [PubMed] [Google Scholar]
- Renshaw PS, Veverka V, Kelly G, Frenkiel TA, Williamson RA, Gordon SV, Hewinson RG, Carr MD (2004) Letter to the editor: sequence-specific assignment and secondary structure determination of the 195-residue complex formed by the Mycobacterium tuberculosis proteins CFP-10 and ESAT-6. J Biomol NMR 30: 225–226 [DOI] [PubMed] [Google Scholar]
- Rosenkrands I, Weldingh K, Jacobsen S, Hansen CV, Florio W, Gianetri I, Andersen P (2000) Mapping and identification of Mycobacteriuim tuberculosis proteins by two-dimensional gel electrophoresis, microsequencing and immunodetection. Electrophoresis 21: 935–948 [DOI] [PubMed] [Google Scholar]
- Sassetti CM, Rubin EJ (2003) Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA 100: 12989–12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med 198: 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjøt RLV, Brock I, Arend SM, Munk ME, Theisen M, Ottenhoff THM, Andersen P (2002) Epitope mapping of the immunodominant antigen TB10.4 and the two homologous proteins TB10.3 and Tb12.9, which constitute a subfamily of the esat-6 gene family. Infect Immun 70: 5446–5453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjøt RLV, Oettinger T, Rosenkrands I, Ravn P, Brock I, Jacobsen S, Andersen P (2000) Comparative evaluation of low-molecular-mass proteins from Mycobacterium tuberculosis identifies members of the ESAT-6 family as immunodominant T-cell antigens. Infect Immun 68: 214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SA, Raghaven S, Hwang WH, Cox JS (2003) Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialised secretion system. Proc Natl Acad Sci USA 100: 13001–13006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman HE, Clay H, Beery D, Chang JCW, Sherman DR, Ramakrishnan L (2004) Tuberculous granuloma formation is enhanced by a Mycobacterium virulence determinant. PLoS Biol 2: 1946–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wards BJ, de Lisle GW, Collins DM (2000) An esat6 knockout mutant of Mycobacterium bovis produced by recombination will contribute to the development of a live tuberculosis vaccine. Tubercle Lung Dis 80: 185–189 [DOI] [PubMed] [Google Scholar]
- World Health Organisation (WHO) Geneva (2004) Global tuberculosis control: WHO Report 2004. WHO/HTM/TB/2004.331