

Abstract
Bloom's syndrome is a hereditary cancer-predisposition disorder resulting from mutations in the BLM gene. In humans, BLM encodes one of five members of the RecQ helicase family. One function of BLM is to act in concert with topoisomerase IIIα (TOPO IIIα) to resolve recombination intermediates containing double Holliday junctions by a process called double Holliday junction dissolution, herein termed dissolution. Here, we show that dissolution is highly specific for BLM among human RecQ helicases and critically depends upon a functional HRDC domain in BLM. We show that the HRDC domain confers DNA structure specificity, and is required for the efficient binding to and unwinding of double Holliday junctions, but not for the unwinding of a simple partial duplex substrate. Furthermore, we show that lysine-1270 of BLM, which resides in the HRDC domain and is predicted to play a role in mediating interactions with DNA, is required for efficient dissolution.
Keywords: Bloom's syndrome, Holliday junction resolution, HRDC domain, RecQ DNA helicases, topoisomerase III
Introduction
Mutations in the BLM gene give rise to Bloom's syndrome (BS) in humans (Ellis et al, 1995). This rare genetic disorder is characterized by short stature, sunlight sensitivity, immunodeficiency and a predisposition to the development of many different types of malignancies (Bachrati and Hickson, 2003; Hickson, 2003). BLM encodes a DNA helicase that belongs to a highly conserved family, the prototypical member of which is encoded by the recQ gene of Escherichia coli (Figure 1A) (Bachrati and Hickson, 2003). Germline mutations in two other human RecQ family helicase genes also give rise to cancer-predisposition disorders: WRN mutations give rise to Werner's syndrome and RECQ4 mutations are associated with some cases of Rothmund–Thomson syndrome (Yu et al, 1996; Kitao et al, 1999).
Figure 1.

(A) Schematic representation of selected members of the RecQ helicase family. All proteins are from humans, except E. coli RecQ and S. cerevisiae Sgs1, and are aligned about the conserved helicase domain. Also shown are the relative positions of the exonuclease (Exo) and ssDNA annealing domains of WRN and RECQ5β, respectively, and the RQC (R) and HRDC domains. Nonconserved regions are shaded in gray. Relevant amino-acid residue positions of truncation mutants used in this study are indicated above BLM and RecQ. See text for details. (B) Alignment of the HRDC domain of BLM, WRN, Sgs1 and E. coli RecQ. Residues comprising α-helices in the three-dimensional structure of Sgs1 are indicated by boxes above. Identical or similar residues are shaded in gray. Shown below is the amino-acid substitution K1270V used in the generation of BLM(K1270V). See text for details. The figure was adapted from Liu et al (1999).
The existence of clinically distinct disorders associated with inactivation of any of three different RecQ family helicases indicates that the human RecQ family helicases have specialized functions in the cell (Oakley and Hickson, 2002; Bachrati and Hickson, 2003; Wang et al, 2003). In the budding yeast, Saccharomyces cerevisiae, mutation of the sole RecQ helicase gene, SGS1, results in elevated levels of genome-wide genetic recombination (Gangloff et al, 1994; Watt et al, 1996). This role in regulating homologous recombination levels appears to have been conserved in humans, at least in the case of BLM, since BS cells also show excessive levels of genome-wide homologous recombination. This is particularly apparent for recombination events between sister chromatids, giving rise to a characteristically elevated level of sister chromatid exchanges (SCEs) that is sufficiently specific for BS to be used in the diagnosis of the disorder (Chaganti et al, 1974). The hyper-recombination phenotype of certain RecQ helicase mutants is likely due to aberrant processing of recombination intermediates (Wu and Hickson, 2002b) since RecQ helicases from several organisms have been purified, and have been shown to act upon DNA intermediates that arise during the process of homologous recombination (Bachrati and Hickson, 2003).
RecQ helicases all contain a conserved helicase domain that defines the family (Bachrati and Hickson, 2003). There are, however, additional sequence motifs that flank the signature helicase domain (Figure 1A). In certain cases, additional enzymatic activities reside in these flanking domains. In the case of WRN, a 3′–5′ exonuclease domain is located in the N-terminal region, whereas the C-terminal domain of RECQ5β directs a DNA strand annealing function (Huang et al, 1998; Kamath-Loeb et al, 1998; Garcia et al, 2004) (Figure 1A). Other sequence motifs identified in RecQ helicases include the RQC domain, which is situated C-terminal to the helicase domain and appears to be found exclusively in some RecQ helicases, and the HRDC domain, which is found C-terminal to the RQC domain of some RecQ helicases and in RNaseD homologs (Figure 1A and B) (Bachrati and Hickson, 2003). The structures of the RQC and HRDC domains have been determined for RecQ and Sgs1, respectively (Liu et al, 1999; Bernstein et al, 2003). The RQC domain resembles a so-called winged-helix domain, which is a member of the helix–turn–helix superfamily, and plays a role in binding duplex DNA (Bernstein et al, 2003). Interestingly, in BLM and WRN, the RQC domain has been implicated also in the orchestration of protein–protein interactions (Brosh et al, 2001; von Kobbe et al, 2002; Bachrati and Hickson, 2003). The HRDC domain is thought to function as an auxiliary DNA-binding domain and, structurally, it resembles domains found in many DNA-metabolizing enzymes such as helicases, polymerases and recombinases (Morozov et al, 1997; Liu et al, 1999). How these conserved motifs impact on the biochemical properties of RecQ helicases and affect the in vivo function of these enzymes has not been determined.
In addition to the presence or absence of the aforementioned functional domains, the different cellular functions of the various vertebrate RecQ helicases likely depend on their ability to form specific interactions with different protein partners. BLM is proposed to act in a complex with replication protein A and the type IA topoisomerase, topoisomerase IIIα (hTOPO IIIα) (Brosh et al, 2000; Johnson et al, 2000; Wu et al, 2000; Meetei et al, 2003). This association with a type IA topoisomerase is evolutionarily conserved, since Sgs1p also acts together with Top3p, the only type IA topoisomerase expressed in budding yeast (Gangloff et al, 1994; Bennett and Wang, 2001; Fricke et al, 2001). top3 mutant cells have elevated levels of recombination (Wallis et al, 1989), which can be suppressed, at least partially, by inactivation of either Sgs1p or Rad52p, the latter of which is required for all forms of homologous recombination (Gangloff et al, 1994; Oakley et al, 2002; Shor et al, 2002). The conserved coupling of a RecQ helicase and a type IA topoisomerase, therefore, appears to have a central role in regulating recombination levels in the cell (Wu and Hickson, 2001). In particular, Sgs1p and BLM appear to suppress the formation of crossover products that arise from the resolution of homologous recombination intermediates containing Holliday junctions (Ira et al, 2003; Wu and Hickson, 2003). We have recently proposed a mechanism by which this is achieved by BLM. Together with hTOPO IIIα, BLM can catalyze double Holliday junction dissolution (DHJ), a reaction mechanism in which DHJs are resolved exclusively into non-crossover recombinant products (Wu and Hickson, 2003). Here, we have demonstrated that BLM cannot by substituted by other RecQ helicases in dissolution reactions containing hTOPO IIIα. Furthermore, we reveal the first biochemical function for the HRDC domain of a RecQ helicase through the demonstration of a requirement for this domain in the specific recognition, processing and dissolution of DHJs.
Results
Double Holliday junction dissolution is specific for BLM
Despite the similar biochemical properties of many RecQ helicases in vitro, and their ability to act upon a wide variety of DNA structures, an elevated frequency of SCEs is thought to be a feature unique to BLM-deficient cells among human RecQ helicase mutants. We set out, therefore, to examine the apparent contradiction between, on the one hand, the promiscuous and redundant biochemical activities of RecQ helicases in vitro and, on the other, the distinct cellular phenotypes of RecQ helicase mutants. To do this, we analyzed the ability of three other human RecQ helicases to catalyze dissolution in conjunction with hTOPO IIIα. Dissolution was assessed using an oligonucleotide-based molecule (DHJ) that we had used previously to assay for dissolution activity (Figure 2) (Fu et al, 1994; Wu and Hickson, 2003). Using this substrate, we found that WRN, RECQ1 and RECQ5β could not substitute for BLM in dissolution reactions with hTOPO IIIα, indicating that the reaction mechanism of dissolution displays a high degree of specificity for BLM (Figure 3).
Figure 2.
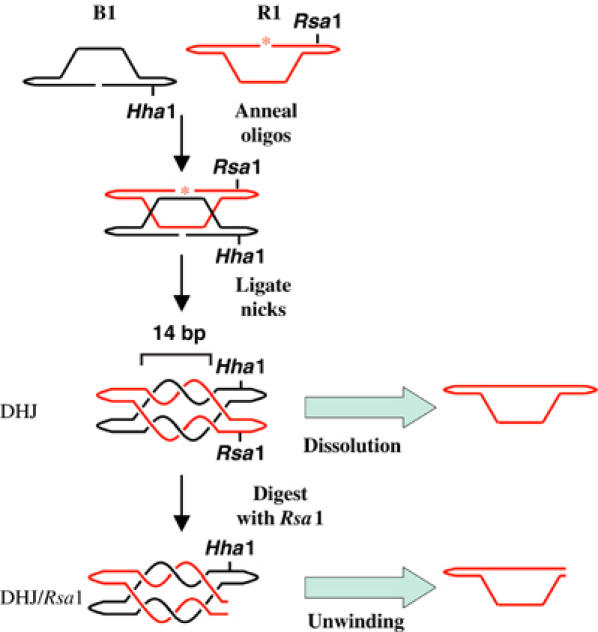
Outline of the strategy used in the generation of the DHJ-containing substrates analyzed in this study, DHJ and DHJ/Rsa1. DHJ comprises two oligonucleotides (B1 and R1), each of which contains two regions of complementary sequences giving rise to two 11 bp internal duplex arms. When incubated together, B1 and R1 anneal to form a molecule that contains two juxtaposed HJs separated by two 14 bp heteroduplex regions (Fu et al, 1994; Wu and Hickson, 2003). These heteroduplexes contain approximately 1.5 helical turns, which results in the catenation of B1 and R1 when both oligonucleotides are circularized by ligation. Topologically relevant helical turns are shown. Each oligonucleotide can be detected individually by the 5′ end-labeling of one of the oligonucleotides prior to annealing. In this scheme, oligonucleotide R1 has been labeled, as indicated by the asterisk. Blue arrows indicate the enzymatic activity each substrate was used to assess and the detectable product generated from each substrate. Dissolution of DHJ results in the decatenation of B1 and R1 releasing the two oligonucleotides as circular DNA species, each of which has a faster mobility on denaturing PAGE than the DHJ substrate (Wu and Hickson, 2003). Unwinding of DHJ/Rsa1 by BLM releases linear R1 and circular B1.
Figure 3.
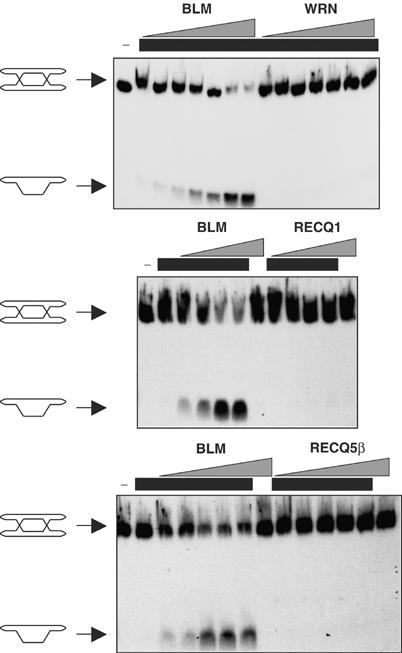
Dissolution is specifically catalyzed by BLM. Dissolution reactions containing a series of two-fold dilutions of BLM (upper panel, 5, 2.5, 1.25, 0.625, 0.3125, 0.15 and 0.075 nM; the middle and lower panels did not contain the three and two lowest concentrations, respectively), WRN (15, 7.5, 3.75, 1.87, 0.94, 0.47 and 0.24 nM), RECQ1 (100, 50, 25 and 12.5 nM) or RECQ5β (80, 40, 20, 10 and 5 nM), as indicated above the panels. Lanes containing hTOPO IIIα (250 nM) are indicated above by black bars. Positions of the DHJ substrate and the dissolution product are shown on the left. For simplicity, the intertwining of the strands of DHJ is omitted.
The N-terminal domain of BLM is not required to catalyze dissolution
The apparently specific ability of BLM to catalyze dissolution indicated that the BLM protein itself must possess biochemical functions that are not present in the other human RecQ helicases. To identify structural features of BLM that are required for catalysis of dissolution, we generated a series of truncated versions of BLM that all retained the catalytic core of BLM (Figure 1) and assessed the ability of these BLM variants to catalyze dissolution. Residues 1–133 of BLM contain an hTOPO IIIα-interaction domain (Hu et al, 2001). We therefore wanted to determine if this interaction domain was required for dissolution. Deletion of residues 1–212 of BLM, and thus removal of this hTOPO IIIα-interaction domain, did not affect the ability of BLM to catalyze dissolution (Figure 4A). Moreover, a truncated BLM protein generated by removal of the entire nonconserved N-terminal domain of BLM (residues 1–641) was still able to catalyze dissolution (data not shown). Dissolution of DHJ is a symmetrical reaction and generates two circular products in an ATP-dependent manner. To eliminate the possibility that both of the N-terminally truncated proteins catalyze a pseudodissolution reaction by, for instance, asymmetric nicking of the substrate in the unlabeled oligonucleotide, we monitored the fate of both oligonucleotides by performing dissolution reactions on the two forms of DHJ in which either oligonculeotide R1 or B1 was labeled. The reaction catalyzed by both of the N-terminal BLM truncation mutants on each DHJ substrate was indistinguishable from that carried out by full-length BLM with regard to ATP dependency and the structure of the reaction products, that is, two intact circular species (Figure 4A and B and data not shown). Together, these data indicate that the N-terminal domain of BLM, and thus a physical association of hTOPO IIIα with this portion of BLM, is not required for the catalysis of dissolution.
Figure 4.
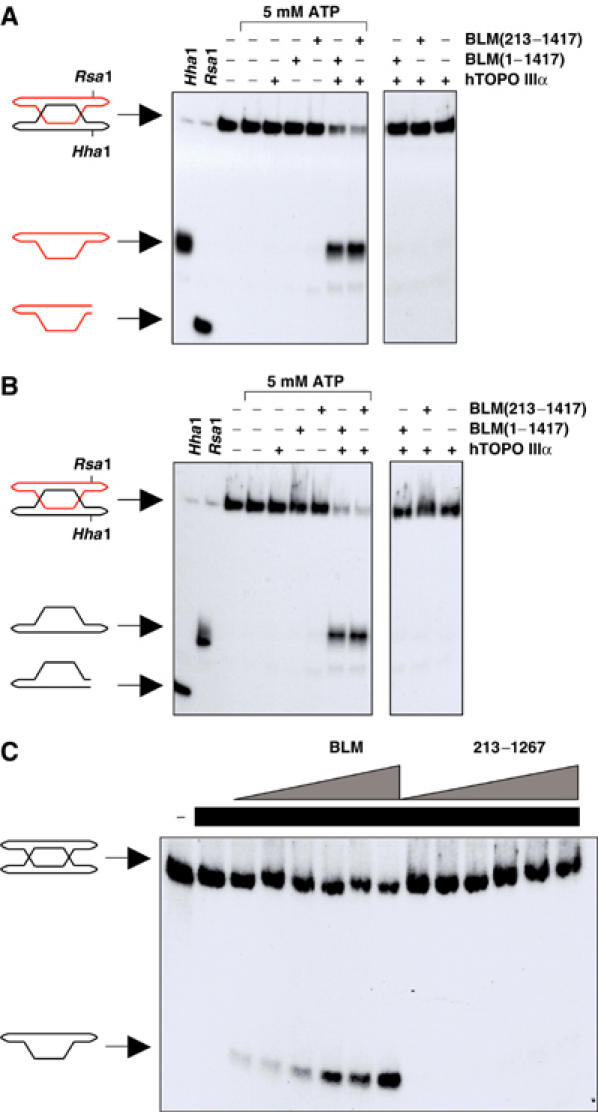
The C- but not the N-terminal domain of BLM is required for dissolution. (A) Dissolution reactions in the presence or absence of 5 mM ATP and containing hTOPO IIIα and full-length BLM(1–1417) or BLM(213–1417), as indicated. In the DHJ substrate, oligonucleotide R1 is labeled. Positions of the DHJ substrate, dissolution product and products of the indicated restriction digests, confirming the identity of the labeled oligonucleotide, are shown on the left. (B) Identical reactions to those shown in (A) were carried out except that a DHJ molecule was used in which oligonucleotide B1 was labeled. (C) Dissolution reactions containing a series of two-fold dilutions of BLM (highest concentration, 5 nM) or BLM(213–1267) (highest concentration, 7 nM). Lanes containing hTOPO IIIα (250 nM) are indicated by black bars. Positions of the DHJ substrate and dissolution product are shown on the left.
The C-terminal domain of BLM is essential for dissolution
The dispensable nature of the N-terminal domain of BLM for dissolution led us to examine the role of the C-terminal domain of BLM using a previously described, truncated version of BLM that consists of residues 213–1267, herein designated BLM(213–1267) (Wu and Hickson, 2002a). Despite being an active helicase, we found that BLM(213–1267), in contrast to BLM(213–1417), which contains the natural C-terminus of BLM, was defective in catalyzing dissolution (Figure 4C). This indicates that in addition to the helicase domain, residues 1268–1417 of BLM are required for dissolution (Figure 4).
The RecQ family HRDC domain is required for the binding to and unwinding of double Holliday junctions
The deletion of residues 1268–1417 to generate BLM(213–1267) resulted in truncation of the HRDC domain, which consists of residues 1210–1290 in BLM (Figure 1A and B). We reasoned, therefore, that the failure of BLM(213–1267) to catalyze dissolution might be reflected in a diminished ability to interact with and/or process DHJs as a result of the inactivation of the DNA-binding function of the BLM HRDC domain. To address this, we compared the ability of BLM and BLM(213–1267) to unwind, in the absence of hTOPO IIIα, a modified DHJ molecule (DHJ/Rsa1) in which DHJ had been digested with Rsa1 (Wu and Hickson, 2003). The linearization of oligonucleotide R1 by Rsa1 results in the topological unlinking of the two constituent oligonucleotides of DHJ and thus creates a substrate that can be disrupted by thermal denaturation or by a helicase activity (Figure 2) (Wu and Hickson, 2003). The intact DHJ substrate is resistant to conventional unwinding due to its covalently closed nature. While we found that both BLM and BLM(213–1267) were able to unwind DHJ/Rsa1, BLM(213–1267) displayed an approximately 10-fold reduction in helicase activity toward this substrate compared to that of full-length BLM (Figure 5). This impaired helicase activity was solely due to the loss of residues 1268–1417 of BLM, since BLM(213–1417), which lacks only the N-terminal 1–212 residues, displayed helicase activity on this substrate that was equivalent to that displayed by full-length BLM (Figure 5).
Figure 5.
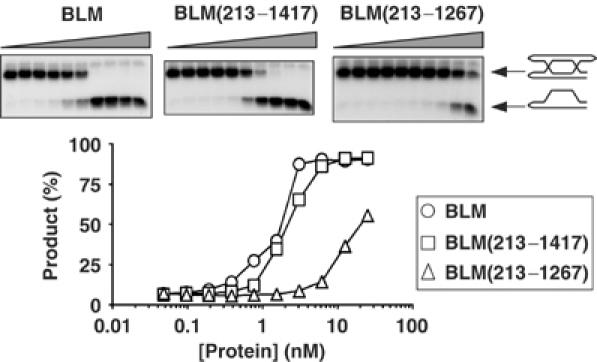
The C-terminal domain of BLM is required for efficient unwinding of DHJs. Helicase assays using DHJ/Rsa1 and a series of two-fold dilutions of BLM, BLM(213–1417) or BLM(213–1267) on DHJ/Rsa1, as indicated. The positions of DHJ/Rsa1 and the unwound product are shown on the right. The graph shows quantification of the level of unwound product generated by BLM (circles), BLM(213–1417) (squares) and BLM(213–1267) (triangles) on DHJ/Rsa1. Note that the scale on the horizontal axis is logarithmic.
One possible explanation for the reduced helicase activity displayed by BLM(213–1267) was that the truncated protein has a reduced affinity for DHJs. To analyze this, we employed two independent means to compare the binding affinities of BLM(213–1417) and BLM(213–1267) for the DHJ substrate: a filter-binding assay, in which we measured protein-dependent retention of DHJ on a nitrocellulose membrane, and an electrophoretic mobility shift assay (EMSA), in which potentially different protein/DHJ complexes were visualized as a result of their reduced electrophoretic mobility compared to protein-free DHJ. Using the filter-binding assay, we found that BLM(213–1267) had a significantly reduced ability to bind DHJ as compared to BLM(213–1417) (Figure 6A). Using EMSAs, BLM(213–1417) was found to generate two differentially retarded species and a small amount of material that could not be resolved and was retained in the gel wells (Figure 6B). The level of all retarded species was dramatically reduced when BLM(213–1267) replaced BLM(213–1417) in the EMSA analyses (Figure 6B), thus confirming the conclusion of the filter-binding assay.
Figure 6.
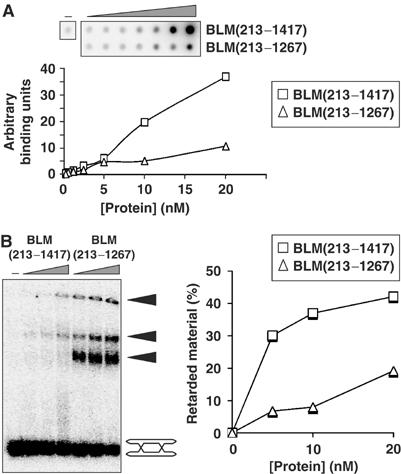
The C-terminal domain of BLM is required for efficient binding to DHJs. (A) DNA filter-binding assay using a series of two-fold dilutions of BLM(213–1417) or BLM(213–1267) on DHJ. The graph shows the quantification of the relative level of DNA bound to the filter in reactions containing either BLM(213–1417) (squares) or BLM(213–1267) (triangles). (B) DNA EMSA using a series of two-fold dilutions of BLM(213–1417) or BLM(213–1267) on DHJ, as indicated above the lanes. The positions of the unbound substrate and protein–DNA complexes (black arrowheads) are indicated on the right. The graph shows the quantification of the total amount of DNA bound by BLM(213–1417) (squares) or BLM(213–1267) (triangles).
Next, we addressed the question of whether the observed reduction in DNA-binding affinity and helicase activity of BLM(213–1267) was specific for DHJ structures. To do this, we compared the helicase activities of BLM, BLM(213–1417) and BLM(213–1267) on a conventional helicase substrate, a forked partial duplex. Interestingly, all three proteins displayed very similar levels of helicase activity on this substrate and, in contrast to the results with the DHJ substrate, BLM and BLM(213–1267) displayed similar binding affinities for the forked duplex substrate (Figure 7). Together, these data strongly suggest that the C-terminal domain of BLM contains a DNA structure-specific binding motif, and implicate residues 1268–1417 of BLM in the processing of DHJ structures.
Figure 7.
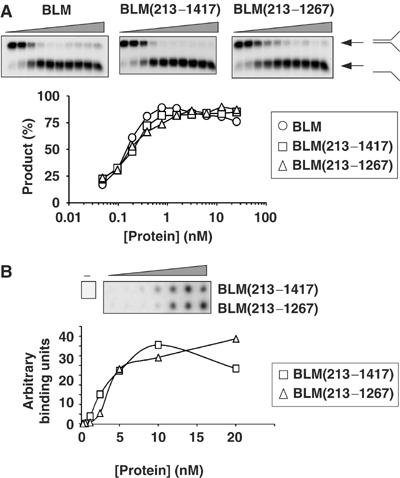
The C-terminal domain of BLM is not required for the unwinding or binding of forked duplexes. (A) Helicase assays using a series of two-fold dilutions of BLM, BLM(213–1417) and BLM(213–1267) on a forked partial duplex substrate. The positions of the forked duplex and the unwound product are shown on the right. The graph shows quantification of the amount of unwound product generated by BLM (circles), BLM(213–1417) (squares) or BLM(213–1267) (triangles). (B) DNA filter-binding assay using a series of two-fold dilutions of BLM or BLM(213–1267) on a forked partial duplex substrate. The graph shows quantification of the relative amount of DNA bound to the filter in reactions containing BLM(213–1417) (squares) or BLM(213–1267) (triangles).
To provide further support for the proposal that it is loss of the HRDC domain that is responsible for the reduced ability of BLM(213–1267) to unwind DHJ/Rsa1, a comparison was made between the activities of E. coli RecQ and a truncated version of this protein (residues 1–523) that lacks the entire HRDC domain, RecQ(1–523) (Figure 1A and B) (Bernstein and Keck, 2003). Both proteins were able to unwind DHJ/Rsa1 to some extent, but RecQ(1–523) had a much reduced ability to unwind this molecule compared to full-length RecQ (>10-fold reduction; Figure 8). As was the case with BLM, loss of the RecQ HRDC domain appeared to specifically affect the helicase activity of RecQ on the DHJ/Rsa1 substrate since it had no significant effect on its helicase activity on a forked DNA substrate (Figure 8). These data suggest a conserved function for the HRDC domain of RecQ family helicases in the specific processing of DHJs. The fact that the RecQ and BLM HRDC domains apparently have similar functions in facilitating the efficient unwinding of DHJ/Rsa1 raised the possibility that RecQ might also catalyze dissolution together with bacterial Top3. RecQ and Top3 have been shown to cooperate in the mediation of a strand passage activity that facilitates the catenation and decatenation of covalently closed plasmid DNAs (Harmon et al, 1999). However, we found that RecQ and Top3 could not catalyze dissolution under conditions that supported dissolution by BLM and hTOPO IIIα (data not shown).
Figure 8.
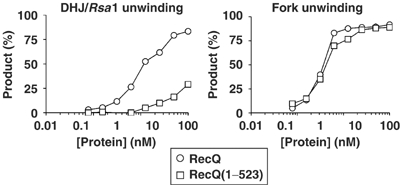
The E. coli RecQ HRDC domain is required for the unwinding of DHJ/Rsa1 but not of a forked duplex. Helicase assays using RecQ and RecQ(1–523) on either DHJ/Rsa1 (left) or a forked partial duplex (right) substrate. The graphs show the quantification of unwound product for each substrate for RecQ (circles) and RecQ(1–523) (squares). Note that the scale on the horizontal axis is logarithmic.
Lysine-1270 of BLM is required for efficient dissolution activity
Thus far, we conclude that the inability of BLM(213–1267) to catalyze dissolution is likely due to the absence of a functional HRDC domain, which had been inactivated by truncation. However, to demonstrate this directly and to eliminate the possibility that the truncation of the C-terminal 150 residues to generate BLM(213–1267) might have had long-range effects on other portions of the BLM protein and thereby led to inactivation of non-HRDC-mediated functions, we sought to abrogate the HRDC domain function in the context of the full-length protein. The similarity of the predicted secondary structure of the HRDC domain in RecQ helicases to motifs in unrelated DNA-metabolizing enzymes suggests that it is the conserved protein fold of the HRDC domain that is important for function. We set out, therefore, specifically to disrupt the conformation of the HRDC domain in BLM by site-directed mutagenesis. Initial attempts involved either the substitution of proline for the highly conserved alanine-1250 residue in order to destabilize α-helix 2 of the HRDC domain, or the creation of an internal deletion removing residues 1251–1262 to truncate α-helix 2 and remove α-helix 3 (Figure 1B). However, neither of these proteins could be expressed successfully, indicating that disruption of the fold of the HRDC domain likely reduces the solubility and/or stability of BLM. As an alternative, we chose to mutate highly conserved residues that, in the predicted three-dimensional structure of the BLM HRDC domain, reside on the protein surface. We reasoned that such residues were less likely to be essential for the overall structural integrity of the HRDC domain, but may be important for function through the mediation of intermolecular interactions. Lysine-1329 of Sgs1 has been shown to contact ssDNA, consistent with the role of the HRDC domain as an auxiliary DNA-binding domain (Liu et al, 1999). This residue is conserved among RecQ helicases and corresponds to residue 1270 in BLM (Figure 1B). We therefore mutated the BLM cDNA to convert lysine-1270 to valine in the full-length BLM protein to generate BLM(K1270V). BLM(K1270V) could be expressed and purified to give protein yields that were comparable to those seen with wild-type BLM (data not shown). Purified BLM(K1270V) was active as a helicase, and had a level of activity on a forked partial duplex substrate that was indistinguishable from that of wild-type BLM (Figure 9A). The substitution of lysine-1270 to valine, therefore, did not appear to have any gross effect on the overall stability or the helicase activity of BLM. However, BLM(K1270V) had a substantially reduced ability in comparison with wild-type BLM to support dissolution together with hTOPO IIIα (Figure 9B). Time-course experiments performed with a fixed concentration of protein indicated an approximately eight-fold reduction in the rate of dissolution for BLM(K1270V) (Figure 9B). Taken together, these data reveal that lysine-1270 of the HRDC domain in BLM is essential for the efficient dissolution of DHJ.
Figure 9.
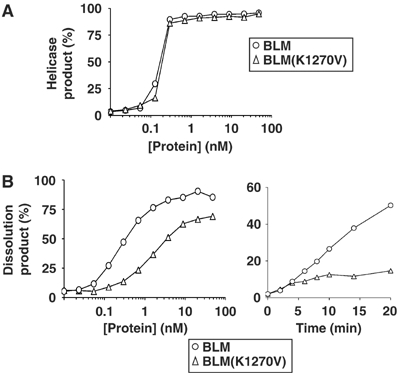
Lysine-1270 is required for efficient catalysis of dissolution. (A) Graph of relative helicase activity for BLM (circles) and BLM(K1270V) (triangles) on a forked partial duplex substrate. (B) Graph of relative dissolution activity catalyzed by BLM (circles) and BLM(K1270V) (triangles) as a function of either protein concentration (left) with a logarithmic scale on the horizontal axis or time in the presence of a fixed protein concentration of 0.8 nM (right).
Discussion
A cellular marker of BLM deficiency used in the diagnosis of BS is an abnormally high frequency of spontaneous SCEs. In normal cells, it has been proposed that SCEs are suppressed by the ability of BLM, together with hTOPO IIIα, to catalyze dissolution. This is an alternative mechanism to that of endonuclease-mediated resolution of HJs, such as that catalyzed by RuvC, Mus81 or the XRCC3/RAD51C complex (Chen et al, 2001; Osman et al, 2003; West, 2003; Liu et al, 2004). Here, we have shown that despite being one of five highly related human RecQ helicases, BLM could not be substituted by WRN, RECQ1 or RECQ5β in dissolution reactions containing hTOPO IIIα. Structurally, proteins of the RecQ helicase family are modular in nature. Several members, including BLM, contain, in addition to the signature helicase domain, a number of identifiable sequence motifs. Previously (and in this study), we have shown that BLM-mediated dissolution requires ATP hydrolysis, indicating a requirement for the helicase domain of BLM in this reaction (Wu and Hickson, 2003). In this study, we have identified the HRDC domain of BLM as also being required for dissolution. The structural similarity of the HRDC domain found in RecQ helicases to domains found in other DNA-metabolizing enzymes strongly indicates a role in mediating contacts with DNA. However, the RecQ helicase HRDC domain is clearly not required for intrinsic helicase activity since we demonstrated that variants of both BLM and RecQ that lack HRDC domains have a helicase activity on forked duplexes that is similar to that of their respective full-length proteins. Rather, we propose that the HRDC domain found in RecQ helicases performs a more specialized role in the recognition of alternate DNA structures, and, more specifically, that it is required for the efficient unwinding of DHJ structures. In particular, we have shown that the amino-acid substitution K1270V in BLM, which is predicted to alter the charge of the DNA-binding face of the HRDC domain, resulted in a diminished ability to mediate dissolution. This is the first specific biochemical function to have been ascribed to the HRDC domain. We did not attempt to complement the hyper-SCE phenotype of BS cells by expression of the BLM(K1270V) cDNA since we and others (Ellis et al, 1999; Hu et al, 2001; Davies et al, 2004) have made the observation that complementation with the wild-type BLM cDNA is often only partial and results in a broad range of SCE frequencies. Therefore, given that the BLM(K1270V) mutant is only partially defective for HRDC function and can still catalyze dissolution, to some extent, we think it is unlikely that any statistically significant difference between BLM(K1270V) and BLM to correct the elevated SCEs seen in BS cells will be demonstrable. However, Yankiwski et al (2001) have shown that expression of a mutant BLM cDNA, which contains an internal deletion that removes the whole of the HRDC domain and thus will be completely defective for HRDC function, is unable to complement the hyper-SCE phenotype of BS cells. This observation is consistent with the proposal that there is a direct link between the dissolution of DHJs and the suppression of SCEs in vivo.
While the HRDC domain is necessary for BLM to catalyze dissolution, its presence in other RecQ helicases is not sufficient to facilitate dissolution. One question that arises from this study, therefore, is does the HRDC domain function in a similar manner in those RecQ helicases where it is present or has it evolved different but related auxiliary functions that contribute to the functional diversification of RecQ helicases? Superficially, the BLM and RecQ HRDC domains appear to have a similar role, in vitro, in the specific recognition of DHJs. An HRDC domain is also present in WRN. However, unlike BS cells, Werner syndrome cells do not show obvious alterations in the levels of genome-wide recombination, but do have telomere maintenance defects (Wyllie et al, 2000; Crabbe et al, 2004). Indeed, in murine models of WS, many of the phenotypes of WS are only manifested in the presence of a second mutation that inactivates telomerase (Chang et al, 2004; Du et al, 2004). Therefore, one role for WRN may be in a recombination-dependent pathway that acts to maintain telomeres in the absence of telomerase. The HRDC domain of WRN may therefore have evolved a role in the specific recognition of recombination structures that arise at telomeres (Opresko et al, 2004). Structural comparisons of the RecQ and Sgs1 HRDC domains indicate that the domain is a highly versatile DNA-binding module that can support several different DNA-binding modes (Liu et al, 1999; DA Bernstein and JL Keck, unpublished data). Indeed, the distribution of surface charge of the HRDC domain, to which lysine-1270 of BLM contributes, is quite distinct between Sgs1, BLM and WRN, which is consistent with the notion that the HRDC domain may have evolved different roles in different RecQ helicases (Liu et al, 1999, DA Bernstein and JL Keck, unpublished data).
We have demonstrated that the entire N-terminal domain of BLM is dispensable for dissolution. This result was somewhat surprising since this portion of BLM contains an hTOPO IIIα-interaction domain that is essential in vivo for the interaction with hTOPO IIIα and for the suppression of SCEs (Hu et al, 2001). Moreover, a similar functional domain organization exists in Sgs1, where the N-terminal domain of Sgs1p is required for the interaction with Top3p (Gangloff et al, 1994; Bennett et al, 2000; Fricke et al, 2001). One possible explanation for these apparently contradictory in vitro and in vivo requirements for a physical association of BLM and hTOPO IIIα is that, in vivo, the physical association between hTOPO IIIα and the N-terminal domain of BLM may facilitate, not a biochemical activity, but rather the recruitment of hTOPO IIIα to its correct subcellular localization. This proposal is consistent with the mislocalization of hTOPO IIIα observed in BS cells (Wu et al, 2000).
RecQ and Top3, respectively, constitute the sole RecQ helicase family member and topoisomerase III enzyme in E. coli. The fact that these two enzymes were unable to catalyze DHJ dissolution suggests that the ancestral function of coupling the activities of a RecQ helicase and topoisomerase III may not be to catalyze dissolution. Indeed, in contrast to BS cells, RecQ mutants have decreased, not increased, levels of crossovers (Kusano et al, 1994). Dissolution results exclusively in non-crossover products and therefore suppresses the exchange of genetic material between molecules engaged in the process of homologous recombination. We propose that this mechanism to resolve recombination intermediates evolved with the advent of diploid organisms with large genomes containing repetitive sequences. In such organisms, the suppression of crossover events during recombinational repair is paramount for the prevention of mutagenic genomic rearrangements that can arise when the DNA molecules participating in recombinational repair are nonsisters. In particular, crossing-over in mammalian cells between sequences lying on homologous chromosomes can lead to loss of heterozygosity, a known driver of the tumorigenesis process. Notably, Blm-deficient mice show excessive loss of heterozygosity and accelerated tumor development (Luo et al, 2000).
In summary, we have shown that the RecQ helicase family HRDC domain confers DNA structure specificity and, in BLM, is required for the dissolution of DHJs. Further analyses to elucidate how the HRDC domain confers structural specificity and how it may function differentially in the various RecQ helicase family members will be crucial in our understanding of the role played by these enzymes in the maintenance of genome integrity and the suppression of tumorigenesis in man.
Materials and methods
Proteins
BLM, BLM (K1270V), BLM(213–1267) and BLM(213–1417) proteins were expressed in yeast and purified by nickel-chelate affinity chromatography using methods described previously (Karow et al, 1997). WRN, RECQ1, RECQ5β, RecQ and RecQ(1–523) were expressed and purified as described previously (Brosh et al, 1999; Bernstein and Keck, 2003; Bernstein et al, 2003; Cui et al, 2004; Garcia et al, 2004). The expression construct for BLM(K1270V) was generated by replacing in the yeast expression vector, pJK1, the HindIII/BamHI fragment, internal to the BLM cDNA, with a PCR-generated mutant fragment containing the desired single codon substitution. hTOPO IIIα was a kind gift from Drs Hélène Goulaouic and Jean-François Riou (Goulaouic et al, 1999).
DNA substrates
DHJ, DHJ/Rsa1 and partial duplex fork substrates were prepared and purified as described previously (Mohaghegh et al, 2001; Wu and Hickson, 2003).
Dissolution and unwinding assays
Reactions were carried out in a buffer containing 50 mM Hepes–KOH, pH 7.2, 50 mM NaCl, 4 mM MgCl2, 5 mM ATP, 1 mM DTT and various combinations of proteins and DNA substrates, as indicated in the figure legends. Reactions were incubated at 37°C for 60 min, unless otherwise stated, and stopped by the addition of 1% SDS and 50 mM EDTA. Samples were deproteinized by the addition of proteinase K and a further incubation at 37°C for 15 min. Dissolution and unwinding reactions were subjected, respectively, to 8% denaturing and 10% native PAGE. Gels were dried and subjected to PhosphorImage analysis using a STORM 840 scanner and ImageQuant software.
DNA-binding filter assays
DHJ was incubated with varying concentrations of protein, as indicated in the figure legends, in reaction buffer containing 20 mM triethanolamine–HCl, pH 7.5, 4 mM MgCl2, 10 μg/ml BSA, 2 mM ATPγS and 1 mM DTT at 37°C for 20 min. Reactions containing the forked duplex were performed as described above for the DHJ, with the exception that the reaction buffer contained 2 mM MgCl2. Reactions were then adsorbed under vacuum onto a nitrocellulose membrane using a dot-blot manifold apparatus. Each reaction spot was then washed with 500 μl reaction buffer under vacuum before the membrane was air-died and subjected to PhosphorImage analysis using a STORM 840 scanner and ImageQuant software.
DNA-binding EMSA
DHJ was incubated with varying concentrations of protein, as indicated in the figure legends, in reaction buffer containing 20 mM triethanolamine–HCl, pH 7.5, 4 mM MgCl2, 10 μg/ml BSA, 2 mM ATPγS and 1 mM DTT at 37°C for 20 min. Samples were then fixed by the addition of 0.25% glutaraldehyde before being subjected to 5% PAGE. Gels were dried and subjected to PhosphorImage analysis using a STORM 840 scanner and ImageQuant software.
Acknowledgments
We thank members of the Genome Integrity group for useful discussions, and Dr Peter McHugh for comments on the manuscript. This work was funded by Cancer Research UK and The Croucher Foundation (Hong Kong).
References
- Bachrati CZ, Hickson ID (2003) RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J 374: 577–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RJ, Noirot-Gros MF, Wang JC (2000) Interaction between yeast sgs1 helicase and DNA topoisomerase III. J Biol Chem 275: 26898–26905 [DOI] [PubMed] [Google Scholar]
- Bennett RJ, Wang JC (2001) Association of yeast DNA topoisomerase III and Sgs1 DNA helicase: studies of fusion proteins. Proc Natl Acad Sci USA 98: 11108–11113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DA, Keck JL (2003) Domain mapping of Escherichia coli RecQ defines the roles of conserved N- and C-terminal regions in the RecQ family. Nucleic Acids Res 31: 2778–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DA, Zittel MC, Keck JL (2003) High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J 22: 4910–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh RM Jr, Li JL, Kenny MK, Karow JK, Cooper MP, Kureekattil RP, Hickson ID, Bohr VA (2000) Replication protein A physically interacts with the Bloom's syndrome protein and stimulates its helicase activity. J Biol Chem 275: 23500–23508 [DOI] [PubMed] [Google Scholar]
- Brosh RM Jr, Orren DK, Nehlin JO, Ravn PH, Kenny MK, Machwe A, Bohr VA (1999) Functional and physical interaction between WRN helicase and human replication protein A. J Biol Chem 274: 18341–18350 [DOI] [PubMed] [Google Scholar]
- Brosh RM Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA (2001) Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J 20: 5791–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaganti RS, Schonberg S, German J (1974) A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci USA 71: 4508–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA (2004) Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet 36: 877–882 [DOI] [PubMed] [Google Scholar]
- Chen XB, Melchionna R, Denis CM, Gaillard PH, Blasina A, Van de Weyer I, Boddy MN, Russell P, Vialard J, McGowan CH (2001) Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol Cell 8: 1117–1127 [DOI] [PubMed] [Google Scholar]
- Crabbe L, Verdun RE, Haggblom CI, Karlseder J (2004) Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306: 1951–1953 [DOI] [PubMed] [Google Scholar]
- Cui S, Arosio D, Doherty KM, Brosh RM Jr, Falaschi A, Vindigni A (2004) Analysis of the unwinding activity of the dimeric RECQ1 helicase in the presence of human replication protein A. Nucleic Acids Res 32: 2158–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SL, North PS, Dart A, Lakin ND, Hickson ID (2004) Phosphorylation of the Bloom's syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol 24: 1279–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, Pak S, Luo G, Pignolo RJ, DePinho RA, Guarente L, Johnson FB (2004) Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol 24: 8437–8446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J (1995) The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83: 655–666 [DOI] [PubMed] [Google Scholar]
- Ellis NA, Proytcheva M, Sanz MM, Ye TZ, German J (1999) Transfection of BLM into cultured bloom syndrome cells reduces the sister-chromatid exchange rate toward normal. Am J Hum Genet 65: 1368–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Kaliraman V, Brill SJ (2001) Mapping the DNA topoisomerase III binding domain of the Sgs1 DNA helicase. J Biol Chem 276: 8848–8855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu TJ, Tse-Dinh YC, Seeman NC (1994) Holliday junction crossover topology. J Mol Biol 236: 91–105 [DOI] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol 14: 8391–8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia PL, Liu Y, Jiricny J, West SC, Janscak P (2004) Human RECQ5β, a protein with DNA helicase and strand-annealing activities in a single polypeptide. EMBO J 23: 2882–2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulaouic H, Roulon T, Flamand O, Grondard L, Lavelle F, Riou JF (1999) Purification and characterization of human DNA topoisomerase IIIα. Nucleic Acids Res 27: 2443–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon FG, DiGate RJ, Kowalczykowski SC (1999) RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: a conserved mechanism for control of DNA recombination. Mol Cell 3: 611–620 [DOI] [PubMed] [Google Scholar]
- Hickson ID (2003) RecQ helicases: caretakers of the genome. Nat Rev Cancer 3: 169–178 [DOI] [PubMed] [Google Scholar]
- Hu P, Beresten SF, van Brabant AJ, Ye TZ, Pandolfi PP, Johnson FB, Guarente L, Ellis NA (2001) Evidence for BLM and Topoisomerase IIIα interaction in genomic stability. Hum Mol Genet 10: 1287–1298 [DOI] [PubMed] [Google Scholar]
- Huang S, Li B, Gray MD, Oshima J, Mian IS, Campisi J (1998) The premature ageing syndrome protein, WRN, is a 3′ → 5′ exonuclease [letter]. Nat Genet 20: 114–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1–Top3 suppress crossovers during double-strand break repair in yeast. Cell 115: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FB, Lombard DB, Neff NF, Mastrangelo MA, Dewolf W, Ellis NA, Marciniak RA, Yin Y, Jaenisch R, Guarente L (2000) Association of the Bloom syndrome protein with topoisomerase IIIα in somatic and meiotic cells. Cancer Res 60: 1162–1167 [PubMed] [Google Scholar]
- Kamath-Loeb AS, Shen JC, Loeb LA, Fry M (1998) Werner syndrome protein. II. Characterization of the integral 3′ → 5′ DNA exonuclease. J Biol Chem 273: 34145–34150 [DOI] [PubMed] [Google Scholar]
- Karow JK, Chakraverty RK, Hickson ID (1997) The Bloom's syndrome gene product is a 3′–5′ DNA helicase. J Biol Chem 272: 30611–30614 [DOI] [PubMed] [Google Scholar]
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y (1999) Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nat Genet 22: 82–84 [DOI] [PubMed] [Google Scholar]
- Kusano K, Sunohara Y, Takahashi N, Yoshikura H, Kobayashi I (1994) DNA double-strand break repair: genetic determinants of flanking crossing-over. Proc Natl Acad Sci USA 91: 1173–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Masson JY, Shah R, O'Regan P, West SC (2004) RAD51C is required for Holliday junction processing in mammalian cells. Science 303: 243–246 [DOI] [PubMed] [Google Scholar]
- Liu Z, Macias MJ, Bottomley MJ, Stier G, Linge JP, Nilges M, Bork P, Sattler M (1999) The three-dimensional structure of the HRDC domain and implications for the Werner and Bloom syndrome proteins. Struct Fold Des 7: 1557–1566 [DOI] [PubMed] [Google Scholar]
- Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A (2000) Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet 26: 424–429 [DOI] [PubMed] [Google Scholar]
- Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W (2003) A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23: 3417–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohaghegh P, Karow JK, Brosh RM Jr, Bohr VA, Hickson ID (2001) The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res 29: 2843–2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov V, Mushegian AR, Koonin EV, Bork P (1997) A putative nucleic acid-binding domain in Bloom's and Werner's syndrome helicases. Trends Biochem Sci 22: 417–418 [DOI] [PubMed] [Google Scholar]
- Oakley TJ, Goodwin A, Chakraverty RK, Hickson ID (2002) Inactivation of homologous recombination suppresses defects in topoisomerase III-deficient mutants. DNA Repair (Amst) 1: 463–482 [DOI] [PubMed] [Google Scholar]
- Oakley TJ, Hickson ID (2002) Defending genome integrity during S-phase: putative roles for RecQ helicases and topoisomerase III. DNA Repair (Amst) 1: 175–207 [DOI] [PubMed] [Google Scholar]
- Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, May A, Seidman MM, Bohr VA (2004) The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell 14: 763–774 [DOI] [PubMed] [Google Scholar]
- Osman F, Dixon J, Doe CL, Whitby MC (2003) Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81–Eme1 in meiosis. Mol Cell 12: 761–774 [DOI] [PubMed] [Google Scholar]
- Shor E, Gangloff S, Wagner M, Weinstein J, Price G, Rothstein R (2002) Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics 162: 647–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C, Karmakar P, Dawut L, Opresko P, Zeng X, Brosh RM Jr, Hickson ID, Bohr VA (2002) Colocalization, physical, and functional interaction between Werner and Bloom syndrome proteins. J Biol Chem 277: 22035–22044 [DOI] [PubMed] [Google Scholar]
- Wallis JW, Chrebet G, Brodsky G, Rolfe M, Rothstein R (1989) A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58: 409–419 [DOI] [PubMed] [Google Scholar]
- Wang W, Seki M, Narita Y, Nakagawa T, Yoshimura A, Otsuki M, Kawabe Y, Tada S, Yagi H, Ishii Y, Enomoto T (2003) Functional relation among RecQ family helicases RecQL1, RecQL5, and BLM in cell growth and sister chromatid exchange formation. Mol Cell Biol 23: 3527–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt PM, Hickson ID, Borts RH, Louis EJ (1996) SGS1, a homologue of the Bloom's and Werner's syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144: 935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC (2003) Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 4: 435–445 [DOI] [PubMed] [Google Scholar]
- Wu L, Davies SL, North PS, Goulaouic H, Riou JF, Turley H, Gatter KC, Hickson ID (2000) The Bloom's syndrome gene product interacts with topoisomerase III. J Biol Chem 275: 9636–9644 [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID (2001) RecQ helicases and topoisomerases: components of a conserved complex for the regulation of genetic recombination. Cell Mol Life Sci 58: 894–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Hickson ID (2002a) The Bloom's syndrome helicase stimulates the activity of human topoisomerase IIIα. Nucleic Acids Res 30: 4823–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Hickson ID (2002b) RecQ helicases and cellular responses to DNA damage. Mutat Res 509: 35–47 [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874 [DOI] [PubMed] [Google Scholar]
- Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D, Faragher RG, Kipling D (2000) Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat Genet 24: 16–17 [DOI] [PubMed] [Google Scholar]
- Yankiwski V, Noonan JP, Neff NF (2001) The C-terminal domain of the Bloom syndrome helicase is essential for genomic stability. BMC Cell Biol 2: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD (1996) Positional cloning of the Werner's syndrome gene. Science 272: 258–262 [DOI] [PubMed] [Google Scholar]