

Abstract
The c-myc proto-oncogene encodes a transcription factor that promotes cell cycle progression and cell proliferation, and its deficiency results in severely retarded proliferation rates. The ATF3 stress response gene encodes a transcription factor that plays a role in determining cell fate under stress conditions. Its biological significance in the control of cell proliferation and its crosstalk regulation, however, are not well understood. Here, we report that the serum response of the ATF3 gene expression depends on c-myc gene and that the c-Myc complex at ATF/CREB site of the gene promoter plays a role in mediating the serum response. Intriguingly, ectopic expression of ATF3 promotes proliferation of c-myc-deficient cells, mostly by alleviating the impeded G1-phase progression observed in these cells, whereas ATF3 knockdown significantly suppresses proliferation of wild-type cells. Our study demonstrates that ATF3 is downstream of the c-Myc signaling pathway and plays a role in mediating the cell proliferation function of c-Myc. Our results provide a novel insight into the functional link of the stress response gene ATF3 and the proto-oncogene c-myc.
Keywords: ATF3, c-Myc, cell cycle progression, crosstalk
Introduction
The c-myc proto-oncogene regulates cell growth, proliferation, differentiation, and apoptosis, and its deregulation is implicated in the development of numerous human cancers (Henriksson and Luscher, 1996). Since c-Myc is a transcription factor and its expression is induced by serum and a variety of mitogens, it is important to identify its targets or the modulator gene(s) that mediate its effect on cell proliferation. Recently, several studies have used microarray analysis to compare Myc-driven changes in global gene expression. These studies have revealed that candidate c-Myc target genes fall into a broad spectrum of diverse functional categories, ranging from metabolic enzymes, biosynthesis of macromolecules such as RNA, protein and DNA, transcription, and cell signaling (Menssen and Hermeking, 2002; Fernandez et al, 2003; O'Connell et al, 2003). The target genes identified in these studies with respect to cell cycle control partially overlap but there are also non-overlapping outliers, perhaps stemming from the different assay systems used in controlling c-Myc expression. Thus, our knowledge of the functional link between c-myc's role in cell proliferation and those target genes is still limited.
Homozygous deletion of the c-myc gene in mice leads to numerous developmental abnormalities and embryonic lethality at 10.5 days of gestation (Davis et al, 1993). Studies using conditional mutations or incremental reduction of c-Myc expression have shown that c-Myc is required to maintain the proliferation of embryonic fibroblasts, or to regulate the percentage of cells that re-enter the cell cycle (de Alboran et al, 2001; Trumpp et al, 2001). By contrast, a homozygous c-myc knockout in a rat fibroblast cell line is not lethal but results in a severely retarded cell growth phenotype, mainly due to lengthening of the G1 phase (Schorl and Sedivy, 2003). This culture model of c-myc knockout has been used extensively to investigate the roles of c-myc and its related target genes in the regulation of cell cycle progression. For instance, cyclin-dependent kinase 4 (CDK4) has been shown to partially restore the proliferation defect of c-myc-deficient cells (Hermeking et al, 2002). Effectors of the c-Myc signaling pathway and its crosstalk in cell proliferation, however, are still largely unknown.
The stress-inducible transcription factor ATF3 is a member of the ATF/CREB family of basic leucine zipper (b-Zip) type transcription factors. It is induced upon exposure of cells to a variety of physiological and pathological stimuli (Hai et al, 1999, and references therein). The stress response of ATF3 is mediated through the c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) or through a p53-dependent pathway, and stabilization of its mRNA is one mechanism for the control of the rapid induction of ATF3 in HeLa cells treated with anisomycin (Liang et al, 1996). This response is thought to have detrimental effects on the cell, such as cell cycle arrest and apoptosis (Yin et al, 1997; Hai et al, 1999; Cai et al, 2000; Kang et al, 2003). On the other hand, ATF3 is also rapidly induced in regenerating liver (Hsu et al, 1991), or in cells treated with growth-stimulating factors such as serum, epidermal growth factor, or fibroblast growth factor (Mohn et al, 1991; Iyer et al, 1999). ATF3 partially transforms chick embryo fibroblasts by promoting growth factor-independent proliferation (Perez et al, 2001), and induces DNA synthesis and expression of cyclin D1 in hepatocytes (Allan et al, 2001). Recently, we have reported that ATF3 supports cell survival by downregulating p53 transcription in endothelial and cardiac cells (Kawauchi et al, 2002; Nobori et al, 2002). ATF3 also protects neuronal cells from JNK-induced cell death by inducing heat shock protein 27 and Akt activation (Nakagomi et al, 2003). These data support a role for ATF3 in cell proliferation and survival. The transcriptional mechanism and biological significance of ATF3 expression in response to growth stimuli, however, remain elusive.
Here we report that serum response of the ATF3 gene depends on c-myc gene status and that the retarded cell proliferation phenotype of c-myc-deficient cells is partially but significantly restored by ectopic expression of ATF3. This study demonstrates for the first time that ATF3 functionally interacts with the c-Myc signaling pathway, providing a novel insight into the functional coordination between cell cycle control and stress response.
Results
Serum induction of ATF3 is abrogated in c-myc-deficient cells
ATF3 has been shown to be rapidly induced in response to partial hepatectomy in the rat (Hsu et al, 1991) or after serum treatment of HeLa cells and fibroblasts (Mohn et al, 1991; Iyer et al, 1999). We first serum-starved wild-type and c-myc-deficient HO15.19 rat fibroblasts for 48 h, and then stimulated them with serum. ATF3 mRNA was induced in wild-type cells, reaching a maximum level at 3 h after stimulation, whereas ATF3 protein was induced 3 h after stimulation with a peak at 6–12 h (Figure 1A). This rapid induction of ATF3 mRNA may partly be due to its increased stability, as reported (Liang et al, 1996). Under this condition, the c-myc mRNA and protein were more rapidly induced than ATF3, and reached their maximum levels at 1 and 3 h after serum treatment, respectively. In c-myc-deficient cells, on the other hand, ATF3 expression was almost completely abrogated at both the mRNA and protein levels (Figure 1A). ATF3 was induced in response to serum at levels similar to wild-type cells in both c-myc heterozygous cells and c-myc homozygous cells reconstituted with a c-myc transgene (Figure 1B), indicating that the serum induction of ATF3 gene depends on c-myc gene status. We next treated wild-type and c-myc-deficient cells with UV and etoposide, since ATF3 is also induced in response to various cytotoxic stimuli through JNK/MAPK pathway, or p53-dependent and -independent pathways (Yin et al, 1997; Hai et al, 1999; Cai et al, 2000). ATF3 was induced by these stress stimuli in both cell types at similar levels (Figure 1C), indicating that the signaling pathway(s) of ATF3 stress response to UV and etoposide is not c-Myc dependent.
Figure 1.
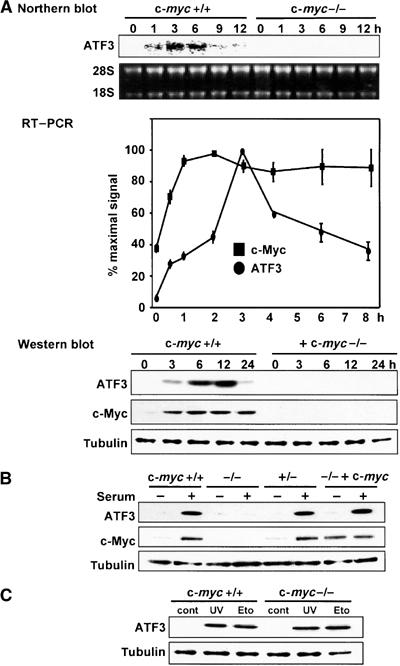
Serum induction of ATF3 in c-myc-deficient cells. (A) Upper panel: Northern blot analysis of ATF3 mRNA in serum-stimulated wild-type TGR1 and c-myc-deficient HO15.19 cells. Middle panel: RT–PCR analysis of ATF3 and c-Myc mRNA in wild-type cells after serum stimulation. Results are means with s.d. of triplicate experiments and shown as percent of the maximal amount of each transcript. Lower panel: Western blot analysis of ATF3 and c-Myc in wild-type and c-myc-deficient cells after serum stimulation. Immunoblot of tubulin is shown as a control of protein loading. (B) Western blot analysis of ATF3 and c-Myc in heterozygous c-myc-knockout +/− cells and homozygous c-myc-knockout −/− cells reconstituted with a c-myc transgene after serum stimulation. Cell extracts were prepared 12 h after serum treatment. (C) Stress-induced expression of ATF3 in wild-type and c-myc-deficient cells in response to UV (40 J/m2) or 20 μM etoposide (Eto). Control (cont) represents untreated cells.
Serum induction of ATF3 is mediated by the ERK/MAPK pathway
Serum treatment causes a rapid phosphorylation of extracellular signal-regulated kinase (ERK) and eventually leads to the activation of its target gene promoters. We thus examined the effect of PD98059, a specific MEK1 kinase inhibitor that suppresses the phosphorylation of ERK, on ATF3 induction. PD98059 almost completely abolished the serum induction of ATF3, whereas JNK inhibitor SP600125 or p38 inhibitor SB203580 had no significant effect. This indicates that the activation of MEK1/ERK is essential for the serum-induced expression of ATF3 (Figure 2A). Serum treatment caused rapid phosphorylation of ERK with similar kinetics in both wild-type and c-myc-deficient cells (Figure 2B), in agreement with a previous report (Mateyak et al, 1999). These data strongly suggest that serum induction of ATF3 is impaired downstream of the ERK/MAPK activation in c-myc-deficient cells.
Figure 2.
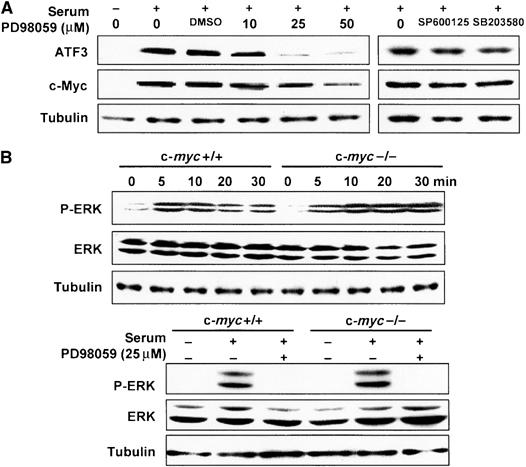
Serum-induced expression of ATF3 via ERK/MAPK signaling pathway. (A) Effect of MAPK inhibitors on serum-induced ATF3 expression in wild-type TGR1 cells determined by Western blot. The inhibitors were added 2 h before adding serum, and cell extracts were prepared 12 h after serum treatment. SP600125 and SB203580 were used at 10 and 25 μM, respectively. DMSO indicates vehicle only. (B) Phosphorylated ERK (P-ERK) and ERK (ERK) were measured at the indicated time after serum treatment of wild-type and c-myc-deficient cells by Western blot. Both 42 and 44 kDa isoforms were detected. In the lower panel, the activity 10 min after serum treatment in the presence of 25 μM PD98059 is shown.
Serum induction of ATF3 promoter activity is suppressed in c-myc-deficient cells
To determine whether the c-myc-dependent serum induction of ATF3 is regulated at the level of transcription, we performed an ATF3 gene reporter assay. Figure 3A illustrates the putative promoter elements of the ATF3 gene promoter, including c-Myc/Max binding sites as well as consensus motifs mediating the serum response (Liang et al, 1996), which are highly conserved between human and rat. As can be seen in Figure 3B, the ATF3 promoter was activated 3.8-fold in wild-type cells after serum treatment, and this activation was suppressed in the presence of PD98059. On the other hand, basal promoter activity in c-myc-deficient cells in the absence of serum was one-fifth the level of wild-type cells, and its induction by serum was significantly abrogated with only 1.1- to 1.3-fold activation. Both the basal and serum-induced promoter activities were fully restored in HO/myc3 cells containing the c-myc transgene. We next measured ATF3 promoter activity in wild-type cells using various deletion mutants. The deletion constructs down to −221 were all induced by serum (Figure 3C). In contrast, a further deletion down to −84 almost completely abolished serum induction. Since the region between −221 and −84 contains the putative ATF/CRE motif at −92 to −85, we performed the assay using the pLucATF3-1850m containing two point mutations at the ATF/CRE site (Cai et al, 2000). This mutation caused a marked reduction of serum-induced activity (Figure 3C). We next examined serum response of the reporter after expressing c-Myc protein in c-myc-deficient cells. c-Myc expression restored the serum response of pLucATF3-1850, -632, and -221, but had no effect on that of pLucATF3-84 and -1850m (Figure 3D), whereas control or LacZ expression had no effect. Combined, these results indicate that the ATF/CRE site in the ATF3 gene promoter represents one of the major elements responsible for the c-Myc-dependent serum response of ATF3.
Figure 3.
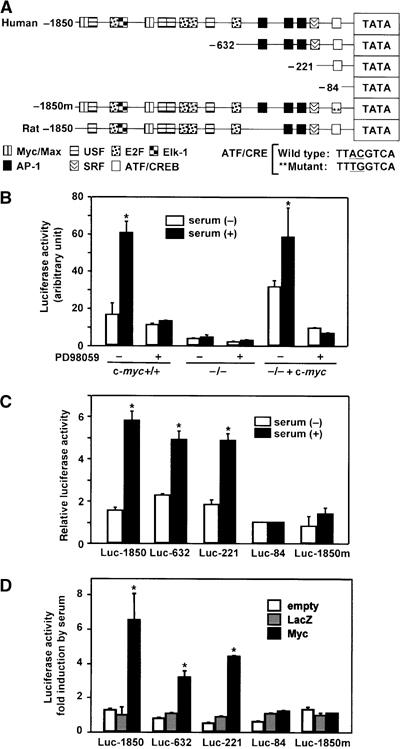
Repression of serum-induced ATF3 reporter activity in c-myc-deficient cells. (A) Human and rat ATF3 gene promoters with putative consensus elements for ATF/CREB, AP1, c-Myc/USF, and other factors are aligned (Liang et al, 1996). Human ATF3 reporter plasmids containing various deletions or two point mutations of ATF/CRE site are also depicted. (B) Serum induction of ATF3 promoter activity was assayed in wild-type TGR1 cells using pATF3Luc1850 in the presence or absence of 25 μM PD98059 for 24 h. Results are mean±s.d. of triplicate assays. Significant induction by serum compared with control, *P<0.05. (C) Wild-type cells were transfected with ATF3 reporters containing various deletions or ATF/CRE point mutations, and assayed for serum induction. Relative activity to that of minimal promoter pATF3Luc-84 was the mean with s.d. of triplicate experiments. Significant induction by serum compared with control, *P<0.05. (D) c-myc-deficient cells were transfected with various reporter plasmids along with empty, LacZ or c-Myc expression vectors and treated with 10% serum. Fold induction by serum is shown and represents the mean with s.d. of triplicate experiments. Significant induction by c-Myc compared with control, *P<0.05.
ATF2/c-Jun complex binds the ATF/CRE motif and c-Myc is recruited to the proximal region of the ATF3 promoter
We next performed a gel mobility shift assay using the ATF/CRE motif as a DNA probe. Incubation of probe with nuclear extracts from serum-starved cells produced bands over a broad size range (Figure 4A). These bands were specific for the ATF/CRE site, since assays in the presence of a wild-type cold probe but not mutant probe completely abolished their formation (data not shown; Cai et al, 2000). The intensity of these bands in serum-stimulated cells was significantly higher than in serum-starved cells. In supershift assays using various antibodies, both anti-ATF2 and anti-c-Jun antibodies resulted in supershifts, indicating that they are components of the protein–DNA complex (Figure 4A). Antibodies against ATF3, ATF4, CREB, JunB, or JunD did not result in any apparent supershifts (data not shown). Anti-c-Myc antibody decreased the intensity of the bands formed. The data indicate that ATF2 and c-Jun are induced to bind to the ATF/CRE motif in response to serum, and c-Myc might be involved in the complex formation (see also Supplementary Figure S1). To further investigate this possibility, we performed a chromatin immunoprecipitation (ChIP) assay to examine the in vivo recruitment of these factors to the ATF3 gene promoter. Both anti-ATF2 and anti-c-Jun antibodies immunoprecipitated the proximal region of the ATF3 gene promoter from −120 to +30, containing the ATF/CRE motif, in both serum-starved and serum-stimulated cells, whereas control IgG did not (Figure 4B). The anti-c-Myc antibody also immunoprecipitated the proximal promoter region after serum treatment, while it immunoprecipitated very little, if any, of this region in serum-starved cells. When ChIP assay was performed using an anti-ATF2 or an anti-c-Jun antibody, the promoter region was less efficiently immunoprecipitated in c-myc-deficient cells than in wild-type cells, indicating that ATF2/c-Jun binding is c-Myc dependent. In the light of these results, we conclude that ATF2 and c-Jun become activated to bind to the ATF/CRE motif in vivo, and c-Myc is also recruited to this region of ATF3 in response to serum.
Figure 4.
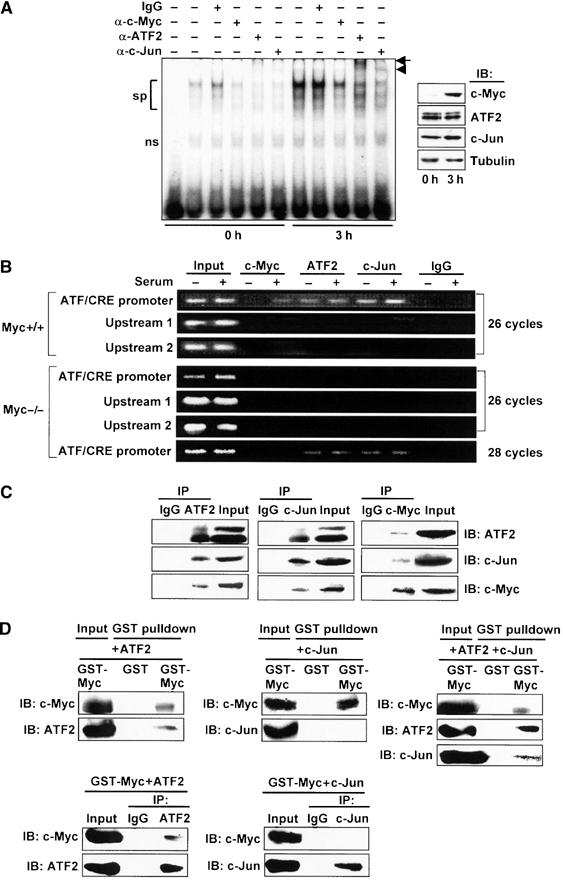
Binding of ATF2/c-Jun to ATF/CRE motif and c-Myc recruitment to the ATF3 gene promoter in response to serum. (A) Nuclear extract from wild-type cells serum starved or serum stimulated for 3 h was assayed for gel shift using radiolabeled DNA probe from −102 to −73 containing the ATF/CRE motif. From left: probe only, extract from the starved or serum-stimulated cells with or without the indicated antibodies. sp: specific bands; ns: nonspecific bands; arrow: supershift by anti-ATF2 antibody; arrowhead: supershift by anti-c-Jun antibody. In the inset, the expressions of c-Myc, ATF2, and c-Jun proteins were measured by Western blot. (B) Upper panel: Serum-starved or serum-stimulated wild-type cells were crosslinked with formaldehyde, and ChIP assay was performed using anti-c-Myc, anti-ATF2, or anti-c-Jun antibodies. ATF/CRE promoter region from −120 to +30, upstream region 1 from −370 to −120, and upstream region 2 from −570 to −370 were amplified by 26 cycles of PCR. Lower panel: c-myc-deficient cells were also subjected to ChIP assay, as above. ATF/CRE promoter region was also amplified by 28 cycles of PCR. (C) Whole-cell extracts from 293T cells overexpressing c-Myc, ATF2, and c-Jun were immunoprecipitated as in Materials and methods using anti-ATF2 (left panel), anti-c-Jun (middle panel), or anti-c-Myc antibody (right panel), respectively, and the resulting immune complex was subjected to Western blot analysis. Input was 10% of total. (D) Recombinant GST-c-Myc was mixed with ATF2 (upper left panel) or c-Jun alone (upper middle panel), or together (upper right panel), and GST pulldown assay was performed as in Materials and methods. In the lower panel, binding of GST-c-Myc with ATF2 or c-Jun was inversely assayed by immunoprecipitation using anti-ATF2 (lower left panel) or anti-c-Jun antibody (lower right panel), respectively. Input was 10% of total.
c-Myc associates with ATF2/c-Jun complex both in vivo and in vitro
To determine the nature of c-Myc recruitment to the proximal region of ATF3 gene promoter, we performed an immunoprecipitation assay using cells overexpressing c-Myc, ATF2, and c-Jun. As illustrated in Figure 4C (see also Supplementary Figure S2), both anti-ATF2 and anti-c-Jun antibodies specifically immunoprecipitated the ATF2/c-Jun complex. Under this condition, c-Myc protein was also co-precipitated by these antibodies, but not by control IgG. When the assay was performed with anti-c-Myc antibody, both ATF2 and c-Jun were co-precipitated, clearly indicating that c-Myc, ATF2, and c-Jun form a complex in vivo. To address whether c-Myc directly interacts with ATF2/c-Jun complex, an in vitro binding assay was performed using recombinant GST-c-Myc, ATF2, and c-Jun proteins. c-Myc was capable of binding to ATF2, while it formed very little, if any, complex with c-Jun (Figure 4D, left panel). This was also observed when the mixture was inversely immunoprecipitated with an anti-ATF2 or an anti-c-Jun antibody (Figure 4D, right panel). In contrast, when GST-c-Myc was mixed with ATF2 and c-Jun together, c-Myc bound to ATF2/c-Jun complex (Figure 4D, left lower panel). These data suggest that c-Myc forms a ternary complex with ATF2/c-Jun through its direct binding to ATF2.
Expression of ATF3 promotes cell cycle progression in c-myc-deficient cells
Homozygous deletion of the c-myc gene in rat fibroblasts significantly impedes G1-phase progression and results in the severe retardation of cell proliferation rates (Schorl and Sedivy, 2003). Since our data indicate that ATF3 is induced downstream of c-myc, we overexpressed the ATF3 protein in c-myc-deficient cells to test whether ATF3 can rescue their proliferation defect. We first employed adenovirus-mediated gene transfer to control the duration and amount of ATF3 expression such that it mimics the in vivo serum induction of ATF3. As illustrated in Figure 5A, ATF3 promoted the proliferation of c-myc-deficient cells, while LacZ expression had no effect. By contrast, ATF3 exerted only a marginal effect on the proliferation of wild-type cells. The level of ATF3 expression at 25 MOI was comparable to that observed in serum-induced wild-type cells while at 50 MOI it was two-to three-fold higher (Figure 5B), indicating that the expression of ATF3 at physiological amounts exerts cell proliferation activity. In the absence of serum, however, ATF3 was not capable of promoting cell proliferation. We also performed retrovirus gene transfers and established c-myc-deficient cell lines stably expressing ATF3 to examine the effect of ATF3 in a long-term assay. As in Figure 5C, these cell lines proliferated faster compared to their parent cells. Moreover, the A2 cell line expressing ATF3 at an amount higher than A1 displayed a higher proliferation rate. A1 cells displayed a roughly circular outline with occasional long processes as parental c-myc-deficient HO15 cells (Mateyak et al, 1997), demonstrating that the ATF3 expression causes no significant alteration of cell shape. Taken together, these results clearly indicate that ATF3 is capable of promoting the proliferation of c-myc-deficient cells.
Figure 5.
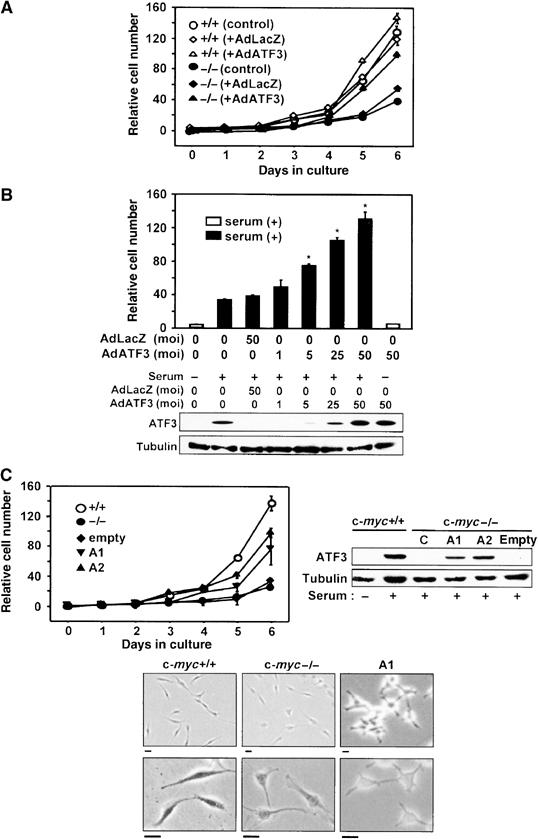
Proliferation of c-myc-deficient cells by ectopic expression of ATF3. (A) Wild-type and c-myc-deficient cells were infected with AdATF3 or AdLacZ at 25 MOI for 48 h and their proliferation rates were assayed. Relative cell number is shown as mean±s.d. of triplicate assays. (B) Dose-dependent cell proliferation by ATF3 in c-myc-deficient cells. Significant proliferation by ATF3 compared with control, *P<0.05. The lower panel shows the ATF3 expression in AdATF3-infected cells. In the left two lanes, the level of ATF3 expression in serum-stimulated wild-type cells is also shown. (C) Upper left panel: Proliferation rates of the c-myc-deficient cell lines A1 and A2 stably expressing ATF3. The relative cell numbers are the mean±s.d. of triplicate assays. Significant cell proliferation in A1 and A2 compared with control, *P<0.05. The upper right panel shows the ATF3 expression in A1 and A2 cells. Lower panel: Morphology of wild-type, c-myc-deficient, and A1 cells is shown. Scale bar, 20 μm.
Cell cycle promoters are upregulated and G1-phase progression defect is alleviated by ATF3 in c-myc-deficient cells
To assess the effect of ATF3 on cell cycle progression, we measured Cdk4 and Cdk2 activities in c-myc-deficient cells exogenously expressing ATF3. While LacZ expression had no effect, ATF3 expression resulted in increased kinase activity by both Cdk4 and Cdk2, providing biochemical evidence that ATF3 promotes cell cycle activation (Figure 6A). Next, we synchronized these cells at G0 by serum starvation, and examined the expression of various cell cycle regulators after stimulating them with serum. As illustrated in Figure 6B, the expressions of cyclins D1, E, and A, and Cdk2 and Cdk4 were all upregulated in these cells compared to control LacZ-expressing cells. We then examined cell cycle progression by flow cytometry. As shown in Figure 7A and Table I (see also Supplementary Figure S3 and Supplementary Tables 1 and 2), the percent cell cycle distribution and the G1-phase duration of c-myc-deficient cells were significantly restored in A2 cells, which stably express ATF3, although S and G2/M phases were less affected. To assess cell cycle profile more accurately, we pulse-labeled the A2 cells with 5-bromodeoxyuridine (BrdU) and corrected the fluorescence-activated cell sorting (FACS) data as in Table I. The result supports a rather selective restoration of G1-phase duration in A2 cells. Alternatively, A2 cells were synchronized in M phase by mitotic shake-off and labeled with BrdU. The cells displayed significantly enhanced incorporation of BrdU and exhibited a shortened G1 phase of 11.7 h, compared to 30.4 h of parental c-myc-deficient cells (Figure 7B and Table I). Overall, ATF3 expression significantly decreased the duration of the cell cycle of c-myc-deficient cells from 46.8 to 26.8 h in A2 cells. These data indicate that ATF3 alleviates the inhibition of cell cycle progression in c-myc-deficient cells by upregulating various cell cycle regulators and selectively augmenting the G1-phase progression of the cell cycle.
Figure 6.
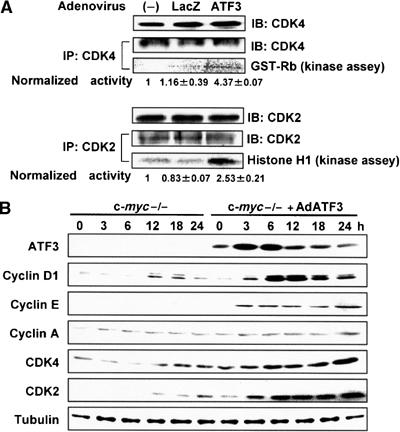
Effect of ATF3 on Cdk activity and the expression of various cell cycle regulators. (A) Cdk4 and Cdk2 kinase activities of c-myc-deficient cells treated with AdATF3 and serum for 48 h were assayed from lysates of exponentially growing cells, as previously reported (Tamamori-Adachi et al, 2004). The activities were normalized to the amount of Cdk4 and Cdk2. (B) Cell cycle regulators were measured by Western blot analysis at each time point after serum stimulation of c-myc-deficient cells infected with AdLacZ or AdATF3 (25 MOI) for 48 h. Immunoblots of tubulin are shown as control.
Figure 7.
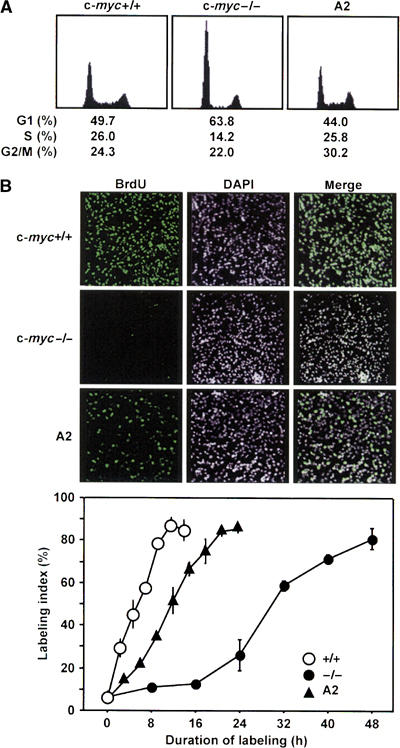
Effect of ATF3 on the cell cycle of c-myc-deficient cells. (A) Asynchronously cycling wild-type, c-myc-deficient, and A2 cells stably expressing ATF3 were analyzed by FACS as in Materials and methods. (B) Each cell line was synchronized by mitotic shake-off in M phase and continuously labeled with 10 μM BrdU as in Materials and methods. Upper panel: Representative data of in situ immunostaining of BrdU-positive cells after 8 h labeling. DAPI denotes nuclear staining. Lower panel: At the indicated time points, BrdU-positive cells were measured by histochemical staining and the percentage of BrdU-positive cells was scored as labeling index. At least 700 cells were scored at each time point.
Table 1.
Cell cycle parameters of wild, c-myc−/−, and A2 cells
| c-myc+/+ |
−/− |
A2 |
|||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G1 | S | G2/M | G1 | S | G2/M | G1 | S | G2/M | |||||||||||||||||||||||||||||||||||||||||
| Cell cycle phase (h)a | 9.2 | 4.8 | 4.6 | 29.9 | 6.6 | 10.3 | 11.8 | 6.9 | 8.1 | ||||||||||||||||||||||||||||||||||||||||
| Total doubling time (h)b | 18.6 | 46.8 | 26.8 | ||||||||||||||||||||||||||||||||||||||||||||||
| Cell cycle distribution (%)c | 33.4 | 58.6 | 8 | 56.7 | 28.4 | 14.9 | 37.3 | 39.2 | 23.5 | ||||||||||||||||||||||||||||||||||||||||
| Cell cycle phase (h)d | 6.2 | 10.9 | 1.5 | 26.5 | 13.3 | 7.0 | 10.0 | 10.5 | 6.3 | ||||||||||||||||||||||||||||||||||||||||
| Mitotic shake-off (h)e |
4.6 |
|
|
30.4 |
|
|
11.7 |
|
|
||||||||||||||||||||||||||||||||||||||||
| aCell cycle phase was calculated from cell cycle distributions based on flow cytometric analysis. | bDoubling time was calculated from cell cycle distributions based on growth rates. | bCell cycle distributions shown in Figure 7A were corrected using BrdU incorporation values of 58.6% (c-myc+/+ cells), 28.4% (c-myc−/− cells), and 39.2% (A2 cells), respectively. These values were determined by pulse labeling of BrdU as in Materials and methods. | dCorrected cell cycle distributions were combined with growth rate values (doubling time). | eG1 phase duration from data in Figure 7B. Values for 50% labeling index are shown. | |||||||||||||||||||||||||||||||||||||||||||||
Serum-induced cell proliferation is inhibited by knockdown of ATF3
We next addressed whether ATF3 is implicated in serum-induced cell proliferation of wild-type cells by employing a loss-of-function approach via an RNA interference-mediated gene knockdown. We isolated wild-type TGR1 cell lines stably expressing siRNAs for ATF3, starved these cells for 48 h, and then stimulated with 10% serum. As illustrated in Figure 8A, ATF3 induction by serum was significantly suppressed in cell lines #1 and #2, while c-Myc induction was not affected. The proliferation of these cells was significantly suppressed compared with control cells (Figure 8B). Moreover, various deletions of ATF3 had inhibitory effect on the proliferation of wild-type cells, possibly acting in a dominant-negative manner (see Supplementary Figure S4). These data clearly indicate that ATF3 plays a crucial role in supporting cell proliferation in response to serum.
Figure 8.
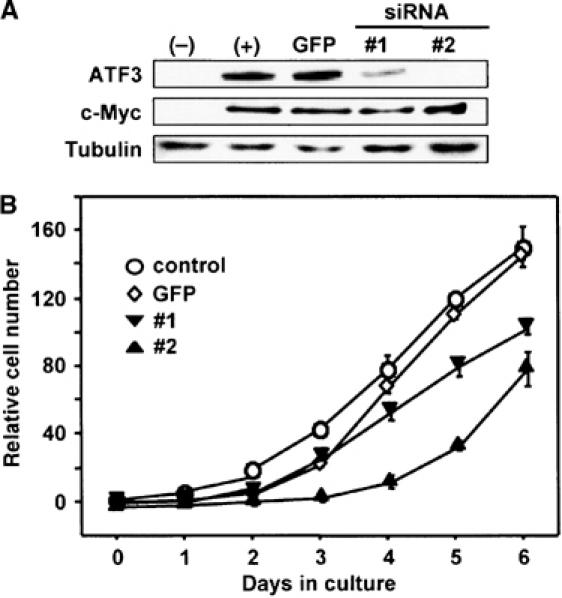
Effect of ATF3 knockdown on proliferation activity of wild-type cells. (A) TGR1 cell lines #1 and #2 stably expressing ATF3 siRNA were established as in Materials and methods. The cells were serum starved for 48 h and stimulated with 10% serum for 8 h, and their expression of ATF3 and c-Myc was assayed by Western blot. TGR1 cell line stably expressing GFP siRNA was used as control. (B) Proliferation of ATF3 siRNA #1, #2, and GFP siRNA cell lines was determined as in Materials and methods.
Discussion
The stress response gene ATF3 is induced by various stress stimuli and its activation is associated with cell death (Yin et al, 1997; Hai et al, 1999; Cai et al, 2000, Kang et al, 2003), survival (Kawauchi et al, 2002; Nobori et al, 2002; Nakagomi et al, 2003), and cell proliferation (Hsu et al, 1991; Mohn et al, 1991; Iyer et al, 1999; Allan et al, 2001; Perez et al, 2001). Thus, ATF3 can function differently depending on cellular context. In this study, we have demonstrated that ATF3 is induced downstream of the c-Myc signaling pathway and plays a crucial role in mediating cell proliferative activity of c-Myc during serum response.
Using rat c-myc-deficient cells, we have shown that ATF3 is induced downstream of the c-Myc signaling pathway in response to serum, as illustrated in our model in Figure 9. This is supported by the following results: (1) ATF3 mRNA and protein were induced at times later than those of c-myc (Figure 1A); (2) ATF3 induction was abrogated in c-myc-deficient cells, and reconstituting these cells with the c-myc transgene restored the serum response (Figure 1A and B); and (3) serum induction of ATF3 reporter activity was severely repressed in c-myc-deficient cells, while coexpression of reporter with c-Myc restored activity (Figure 3). In contrast, ATF3 induction by DNA damage was not c-Myc dependent, suggesting that c-Myc might not be significantly involved in p53-dependent or -independent stress response of ATF3 (Fan et al, 2002; Zhang et al, 2002).
Figure 9.
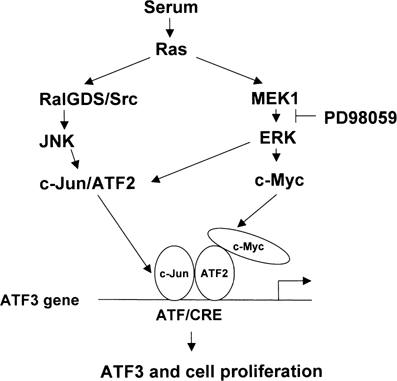
A crosstalk model of ATF3 and c-myc in serum induction. Serum induces c-Myc and ATF2 through MEK1–ERK pathway, and also c-Jun by RALGDS/Src–JNK pathway (de Ruiter et al, 2000; Ouwens et al, 2000). These factors form complex at or around the ATF/CRE motif of ATF3 gene promoter, and thereby activate ATF3 gene expression. Expressed ATF3 protein functions as a positive regulator of cell cycle and mediates cell proliferative activity of c-Myc.
Induction of ATF3 was previously shown to be mediated, at least in part, by increased stability of the message in anisomycin-treated HeLa cells (Liang et al, 1996), whereas TNFα-mediated induction of ATF3 mRNA, but not its stability, is regulated by the JNK and ERK pathways in vascular endothelial cells (Inoue et al, 2004). In this study, we have further demonstrated that the serum induction of ATF3 is also regulated at transcription level, and ATF/CRE motif of the ATF3 gene promoter plays a role in its serum-dependent and c-Myc-dependent activation. In both gel shift and ChIP assays, the ATF2/c-Jun complex was already bound to the ATF/CRE motif in the absence of serum but its binding was significantly enhanced after serum treatment (Figure 4). This is consistent with the idea that the factors bound to the motif are not activated prior to serum treatment, but activated via ERK MAPK after serum stimulation. It has in fact been demonstrated that ATF2 and c-Jun are activated through this pathway in serum-treated cells (de Ruiter et al, 2000; Ouwens et al, 2000), and the coexpression of ATF2 and c-Jun activates the ATF3 promoter, supporting synergistic action of these factors (Liang et al, 1996). c-Myc was involved in the serum-dependent activation of the ATF3 gene, most likely independent of DNA binding, since c-Myc conferred serum response on the pLuc-221 reporter that contains an ATF/CRE site but no apparent E box (Figure 3D). c-Myc was recruited to the proximal promoter region of the ATF3 gene after serum stimulation, and this region, from −120 to +30, does not contain any putative E boxes or other sequences for c-Myc's recruitment such as YY-1 (Shrivastava et al, 1996), SP1, Ap2, or Miz-1 sites (Sakamoto and Prendergast, 1999). Thus, we speculate that the ternary complex of ATF2/c-Jun/c-Myc is formed at or around the ATF/CRE site. This is supported by our data demonstrating that c-Myc interacted with ATF2/c-Jun both in vivo and in vitro (Figure 4C and D). This complex may be recruited to the chromatin remodeling complex or the RNA polymerase II initiation complex, thereby mediating the serum response in a concerted action with ATF2 and c-Jun. Indeed, SWI/SNF (Cheng et al, 1999; McMahon et al, 2000) and some general transcription factors such as TBP and pTEFb (Eberhardy and Farnham, 2001) have been reported to interact with c-Myc. However, the molecular mechanism by which c-Myc activates the ATF3 gene in response to serum currently remains unclear. It should be noted that the c-Myc protein is stabilized by the phosphorylation of its serine 62 residue by the MAPK/ERK signaling pathway (Sears et al, 1999; Sears et al, 2000). This rapid stabilization of c-Myc along with its transcriptional induction may act to efficiently induce ATF3 during the initial stage of cell proliferation. In this study, we could not determine the significance of the binding sites for c-Myc, E2F, SRF, or AP1 in the ATF3 promoter, although the deletion of these sites slightly reduced the serum response of the reporter when the activity of pLuc-1850 was compared with that of pLuc-632 (Figure 3C). Further studies are needed to understand the significance of these sites in serum response.
More intriguingly, ATF3 significantly promoted the proliferation of c-myc-deficient cells that otherwise display a severe retardation of cell cycle progression. This effect of ATF3 may have a role in vivo since enhanced cell proliferation was observed by expression of ATF3 at physiological levels (Figure 5). Furthermore, knockdown of ATF3 by siRNA (Figure 8) or expression of deletion mutants of ATF3 (Supplementary Figure S4) strongly inhibited serum-induced cell proliferation. ATF3 restored the delayed progression of G1 phase in c-myc-deficient cells, as determined by flow cytometry and BrdU labeling (Figure 7 and Table I), and induced the expression of cyclins D1, A, and E, and activated CDK2 and CDK4 (Figure 6). In this regard, it has been shown that cyclin D1 is transcriptionally induced by ATF3 (Allan et al, 2001), which suggests that ATF3 induces cyclin D1, which in turn cooperates with CDK4 to promote G1-phase progression. Furthermore, CDK4 has been shown to be a direct target of c-myc, and overexpression of CDK4 partly alleviates the cell growth defect of c-myc-deficient cells (Hermeking et al, 2002). By contrast, it has also been shown that the overexpression of cyclin D1, E, or A alone does not significantly improve the doubling time of c-myc-deficient cells (Mateyak et al, 1999). Thus, it is possible that not a single but multiple cell cycle regulators of G1 phase are directly or indirectly activated by ATF3, and these combined effects lead to a significant decrease in doubling time. Consistent with this notion, the expressions of cyclins E and A were strongly upregulated in our microarray analysis (data not shown). We have previously shown that ATF3 downregulates p53 gene transcription (Kawauchi et al, 2002), which may result in the downregulation of the cell cycle inhibitor p21. The level of p21, however, was not significantly influenced in c-myc-deficient cells by ATF3 (data not shown). It should be noted here that the cell cycle progression of c-myc-deficient cells was enhanced only in the presence of serum (Figure 5B). Although it is not clear at this moment why serum signal is required in addition to the ectopic expression of ATF3, we speculate that target gene(s) of ATF3 including cell cycle regulators is controlled in a serum-dependent manner. Nevertheless, partial but significant rescue of proliferation of c-myc-deficient cells by ATF3 is noteworthy, since such a remarkable restoration has never been observed by any other target genes of c-Myc. Identification of ATF3 targets, direct or indirect, may uncover the underlying molecular mechanism by which ATF3 controls cell cycle, and such a study is now in progress.
Our study demonstrates for the first time a crosstalk regulation between c-myc and ATF3 in cell proliferation. ATF3 is rapidly induced in the regenerating liver (Hsu et al, 1991; Mohn et al, 1991), very early in which c-myc transcript is also induced (Makino et al, 1984). Thus, c-Myc and ATF3 together may regulate liver regeneration. Moreover, ATF3 is overexpressed in murine melanoma cells with high metastatic potentials (Ishiguro et al, 1996), and the ATF3 gene is amplified in esophageal cancer cells (Prmkhaokham et al, 2000). Since c-Myc expression is commonly deregulated in hepatocellular cancer with poor prognosis (Shachaf et al, 2004), c-Myc/ATF3 crosstalk might play a role in oncogenesis and its progression.
Finally, our results demonstrate for the first time that ATF3 promotes the proliferation of c-myc-deficient cells by alleviating their impeded G1-phase progression. This provides a novel functional interaction between the stress response gene ATF3 and the proto-oncogene c-myc. Further investigations should shed light on the biological implications of ATF3 in cellular stress response, cell proliferation, and oncogenesis.
Materials and methods
Reagents
Antibodies used were anti-ATF3 (C-19), anti-c-Myc (N-262), anti-ATF2 (N-96), anti-c-Jun (H-79), anti-cyclin E (M-20), anti-cyclin A (C-19), anti-CDK4 (C-22), and anti-CDK2 (M2) antibodies from Santa Cruz, anti-c-Myc (R950-25) antibody from Invitrogen, anti-cyclin D1(Ab-3) from Calbiochem, anti-p44/42 MAPK (#9101) and anti-phospho-p44/42 MAP (Thr202/Try204) E10 antibody (#9102) from Cell Signaling Technology, and anti-Flag (F-3165) and anti-β-tubulin (T-4026) antibodies from Sigma. Expression plasmids for c-Myc and LacZ contained mouse c-Myc and LacZ cDNAs, respectively, under the control of the SRα promoter in pcDEBΔ, as described (Nakabeppu et al, 1993). PD98059, SP600125, and SB203580 were from Calbiochem and etoposide was from Sigma. Other biochemicals used were reagents grade.
Cell culture and serum treatment
TGR-1 is a subclone of the immortalized rat embryonic Rat-1 cell line. The HO15.19 and HET15 cell lines contain homozygous or heterozygous deletions of the c-myc gene, respectively, as described previously (Mateyak et al, 1997). HO/myc3 cell line is a c-myc transgene-expressing derivative of the HO15.19 cell line (Mateyak et al, 1999). Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin in a 5% CO2 atmosphere at 37°C. For serum stimulation, cells were first starved in medium containing 0.25% serum for 48 h and subsequently stimulated with 10% serum for the indicated times.
RNA isolation and Northern blot analysis
Total RNA (10 μg) was isolated by an acid-guanidinium method and subjected to Northern blot analysis using cDNA probe for the rat ATF3 gene, as described (Cai et al, 2000). Rat ATF3 cDNA was a generous gift from Dr T Hai of Ohio University.
Whole-cell extract preparation and Western blot analysis
Whole-cell extract was prepared as described (Kawauchi et al, 2002), and the amount of protein was quantitated using bovine serum albumin as a standard (Lowry et al, 1951). Whole-cell protein extract (20 μg) was separated on an SDS–polyacrylamide gel, subjected to Western blot analysis, and visualized by chemiluminescence according to the ECL kit protocol from Amersham.
Luciferase assay
Reporter plasmids containing various lengths of the 5′-upstream region of the human ATF3 gene promoter or mutations of ATF/CRE motif at −92 and −85 are as described (Cai et al, 2000). Approximately 3 × 105 cells at 70–80% confluency in a 35 mm dish were cotransfected with reporter plasmid and the effector expression vector. After serum starvation in media containing 0.25% serum for 48 h, cells were stimulated with 10% serum and incubated for another 24 h. Serum-starved cells were also treated by 25 μM PD98059 for 2 h, and then stimulated by serum. Supernatants of cell extracts were assayed for firefly and seapansy luciferase activity using a dual luciferase reporter assay system (Promega). pRL-CMV (Toyo Ink) containing the seapansy luciferase gene was used as an internal control for transfection and expression.
Electrophoretic mobility shift assay
Nuclear extracts were prepared from the serum-starved and serum-fed TGR1 cells (2 × 107 cells), as described (Cai et al, 2000). Nuclear protein extracts (2 μg) were incubated in 20 μl of binding buffer (10 mM HEPES–KOH, pH 7.9, 60 mM KCl, 0.5 mM EDTA, 5 mM MgCl2, 0.1 mM PMSF, 5 mM β-mercaptoethanol) containing 0.5 μg of poly(dI-dC) and 0.5 ng of radiolabeled DNA probe at room temperature for 30 min. For the supershift assay, 0.1 μg of anti-ATF2, -c-Jun, or -c-Myc antibodies were added and incubated for another 30 min. DNA probe was obtained by annealing 0.1 μg each of sense and antisense oligonucleotides for ATF/CRE site as described previously (Cai et al, 2000), and radiolabeled with 25 μCi of [γ-32P]ATP (6000 Ci/mmol) and polynucleotide kinase. Binding mixture was applied onto a 5% nondenaturing polyacrylamide slab gel in Tris–borate–EDTA buffer. After electrophoresis, the gel was dried on a 3 MM Whatman paper and visualized by Fuji Bas 2500 image analyzer.
Chromatin immunoprecipitation assay
ChIP assays were performed according to the protocol supplied from Upstate. Immunoprecipitations were performed using indicated antibodies. PCR was performed with the following primer pairs for three different regions of ATF3 gene promoter: ATF/CRE promoter region from −120 to +30, 5′-GGCCAGTTCTCCCTGGAAGC-3′ and 5′-AAGCACCTGGCACCGCGCGT-3′; upstream region 1 from −370 to −120, 5′-GCCGGTAACCGTGTGGATTC-3′ and 5′-GACTAGGTGAGGCTGGGAAG-3′; and upstream region 2 from −570 to −370, 5′-TAGCGGAGGGAGAGATGCCA-3′ and 5′-GAGACCGCGGACTTGGTGAT-3′.
Binding assay
For in vivo binding, 293T cells (3 × 106 cells) were transfected with 3 μg each of expression vectors for c-Myc, ATF2, and c-Jun. At 48 h after transfection, whole-cell extracts were prepared and incubated with 0.5 μg of indicated antibody at 4°C for 3 h, followed by incubating with 30 μl Protein G-Sepharose (Pharmacia) for 2 h. The resulting immunocomplex was washed and subjected to Western blot analysis. For in vitro binding, full-length c-Myc was expressed as GST fusion using pGEX-6P1 (Pharmacia), and ATF2 and c-Jun were expressed using pET21a (Novagen). Bacterial extracts containing these recombinant proteins were mixed in a binding buffer (50 mM Tris–HCl, pH 8.0, 0.1 M NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol, 10% glycerol) at 4°C for 60 min. After further incubation with 30 μl of glutathione-Sepharose beads (50% slurry; Pharmacia), or 0.5 μg of anti-ATF2 or anti-c-Jun antibody followed by 30 μl Protein G-Sepharose, protein complex was washed and detected by Western blot using indicated antibody.
Ectopic expression of ATF3 by adenovirus or retrovirus gene transfer
Adenovirus vectors encoding the full-length human ATF3 gene (AdATF3) or β-galactosidase (AdLacZ) were prepared using standard methods (Miyake et al, 1996) as described earlier (Kawauchi et al, 2002). For preparing retrovirus encoding the human ATF3, 543 bp of cDNA was subcloned into the Xho1 site of the retrovirus vector pMXs-neo and packaged in Plat-E cells (Morita et al, 2000). Following the infection of c-myc-deficient HO15.19 cells, colonies were selected with 400 or 600 μg/ml of G418 (nacalai).
Isolation of TGR1 cell lines stably expressing ATF3 siRNA
Oligonucleotides for expressing stem–loop RNAs that target two different regions of rat ATF3 were subcloned into pMXneo under human U6 promoter. The vector was kindly supplied by Dr T Adachi from our institute. Sequences used were:
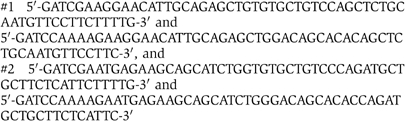
that target amino-acid regions from 176 to 181 and from 137 to 142, respectively. Wild-type TGR1 cells were infected with these retroviruses and cultured in media containing 800 μg/ml of G418.
Assay for serum-induced cell proliferation
Cells treated as indicated were starved in medium containing 0.25% serum. After 48 h, cells were stimulated with 10% serum and cell number was measured at the indicated time intervals. By staining with 0.2% Trypan blue, cell viability was more than 95% in all assays.
Fluorescence-activated cell sorting analysis
Approximately 3 × 105 asynchronously cycling cells were harvested, washed once with PBS, and fixed with 70% ice-cold ethanol. After staining with 50 μg/ml of propidium iodide, DNA content of the cells was analyzed by using FACScan (Becton Dickinson). Cells at each phase of the cell cycle were measured and expressed as percent of total cells.
BrdU incorporation assay
Asynchronously cycling cells were pulse-labeled by BrdU for 15 min using in situ cell proliferation kit (Roche) and its incorporation value was determined by scoring BrdU-positive cells after histochemical staining as described (Schorl and Sedivy, 2003). Cells (1 × 104 cells) were also synchronized in M phase by mitotic shake-off, plated in 24-well plates, and labeled by 10 μM BrdU. At each time point indicated, BrdU incorporation was stopped by adding ascorbic acid to a final concentration of 67 mM, and the labeled cells were counted after staining with anti-BrdU monoclonal antibody from Roche. Percent of BrdU-positive cells was scored as labeling index (Schorl and Sedivy, 2003).
Statistical analysis
Multiple comparisons were evaluated by ANOVA followed by Scheffe's post hoc test. Data are presented as mean±s.d. Statistical significance was assigned at the level of P<0.05.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S5
Supplementary Table S6
Acknowledgments
We thank Dr T Hai at Ohio, USA for her generous gift of rat ATF3 cDNA and Dr S Yokoi from our institute for her technical advice for ChIP assay. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture and Technology of Japan, the Japan Society for the Promotion of Science. IG and JMS were supported in part by NIH grant GM41690 to JMS.
References
- Allan AL, Albanese C, Pestell RG., LaMarre J (2001) Activating transcription factor 3 induces DNA synthesis and expression of cyclin D1 in hepatocytes. J Biol Chem 276: 27272–27280 [DOI] [PubMed] [Google Scholar]
- Cai Y, Zhang C, Nawa T, Aso T, Tanaka M, Oshiro S, Ichijo H, Kitajima S (2000) Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH2 terminal kinase and promoter response element. Blood 96: 2140–2148 [PubMed] [Google Scholar]
- Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV (1999) c-Myc interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet 22: 102–105 [DOI] [PubMed] [Google Scholar]
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A (1993) A null c-myc mutation caused lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev 7: 671–682 [DOI] [PubMed] [Google Scholar]
- de Alboran IM, O'Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW (2001) Analysis of c-Myc function in normal cells via conditional gene-targeted mutation. Immunity 14: 45–55 [DOI] [PubMed] [Google Scholar]
- de Ruiter ND, Wolthuis RMF, van Dam H, Burgering BMT, Bos JL (2000) Ras-dependent regulation of c-Jun phosphorylation is mediated by the Ral guanine nucleotide exchange factor–Ral pathway. Mol Cell Biol 20: 8480–8488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ (2001) c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem 276: 48562–48571 [DOI] [PubMed] [Google Scholar]
- Fan F, Jin S, Amundson SA, Tong T, Fan W, Zhao H, Zhu X, Mazzacurati L, Li X, Petrik KL, Fornace AJ, Rajasekaran B, Zhan Q (2002) ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cell growth. Oncogene 21: 7488–7496 [DOI] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003) Genomic targets of the human c-Myc protein. Genes Dev 17: 1115–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U (1999) ATF3 and stress responses. Gene Expression 7: 321–335 [PMC free article] [PubMed] [Google Scholar]
- Henriksson M, Luscher B (1996) Proteins of the Myc network. Essential regulators of cell growth and differentiation. Adv Cancer Res 68: 109–182 [DOI] [PubMed] [Google Scholar]
- Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O'Connell BC, Mateyak MK, Tam W, Kohlhuber F, Dang CV, Sedivy JM, Erick D, Vogelstein B, Kinzler KW (2002) Identification of CDK4 as a target of c-Myc. Proc Natl Acad Sci USA 97: 2229–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JC, Laz T, Mohn KL, Taub R (1991) Identification of LRF-1, a leucine-zipper protein that is rapidly and highly induced in regenerating liver. Proc Natl Acad Sci USA 88: 3511–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Zama T, Kamimotto T, Aoki R, Ikeda Y, Kimura H, Hagiwara M (2004) TNFα-induced ATF3 expression is bidirectionally regulated by the JNK and ERK pathway in vascular endothelial cells. Genes Cells 9: 59–70 [DOI] [PubMed] [Google Scholar]
- Ishiguro T, Nakajima M, Naito M, Muto T, Tsuruo T (1996) Identification of genes differentially expressed in B6 murine melanoma sublines with different metastatic potentials. Cancer Res 56: 875–879 [PubMed] [Google Scholar]
- Iyer VR, Eisen MB, Ross DT, Schuler G., Moore T, Lee JCF, Trent JM, Staudt LM, Hudson J, Boguski MS, Lashkari D, Shalon D, Botstein D, Brown PO (1999) The transcriptional program in the response of human fibroblasts to serum. Science 283: 83–87 [DOI] [PubMed] [Google Scholar]
- Kang Y, Chen CR, Massague J (2003) A self-enabling TGFβ response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 11: 915–926 [DOI] [PubMed] [Google Scholar]
- Kawauchi J, Zhang C, Nobori K, Hashimoto Y, Adachi TM, Noda A, Sunamori M, Kitajima S (2002) Transcriptional repressor activating transcription factor 3 protects human umbilical vein endothelial cells from tumor necrosis factor-alpha-induced apoptosis through down-regulation of p53 transcription. J Biol Chem 277: 39025–39034 [DOI] [PubMed] [Google Scholar]
- Liang G, Wolfgang CD, Chen BPC, Chen TH, Hai T (1996) ATF3 gene. Genomic organization, promoter, and regulation. J Biol Chem 271: 1695–1701 [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193: 265–275 [PubMed] [Google Scholar]
- Makino R, Hayashi K, Sugimura T (1984) c-myc transcript is induced in rat liver at a very early stage of regeneration or by cycloheximide treatment. Nature 310: 697–698 [DOI] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Adachi S, Sedivy JM (1997) Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ 8: 1039–1048 [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Sedivy JM (1999) c-Myc regulates cyclin D-cdk4 and -cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol 19: 4672–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, Cole MD (2000) The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol 20: 556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menssen A, Hermeking H (2002) Characterization of the c-Myc-regulated transcriptome by SAGE: identification and analysis of c-Myc target genes. Proc Natl Acad Sci USA 99: 6247–6279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake S, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I (1996) Efficient generation of recombinant adenoviruses using adenovirus DNA–terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci USA 93: 1320–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn KL, Laz TM, Hsu JC, Melby AE, Bravo R, Taub R (1991) The immediate-early growth response in regenerating liver and insulin-stimulated H-35 cells: comparison with serum-stimulated 3T3 cells and identification of 41 novel immediate-early genes. Mol Cell Biol 11: 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Kojima T, Kitamura T (2000) Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Therapy 7: 1063–1066 [DOI] [PubMed] [Google Scholar]
- Nakabeppu Y, Oda S, Sekiguichi M (1993) Proliferative activation of quiescent Rat-1A cells by ΔFosB. Mol Cell Biol 13: 4157–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi S, Suzuki Y, Namikawa K, Kiryu-Seo S, Kiyama H (2003) Expression of the activating transcription factor ATF3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and akt activation. J Neurochem 23: 5187–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobori K, Ito H, Adachi TM, Adachi S, Ono Y, Kawauchi J, Kitajima S, Marumo F, Isobe M (2002) ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: a novel cardioprotective role of ATF3. J Mol Cell Cardiol 34: 1387–1397 [DOI] [PubMed] [Google Scholar]
- O'Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, Sedivy JM (2003) A large scale genetic analysis of c-Myc-regulated gene expression patterns. J Biol Chem 278: 12563–12573 [DOI] [PubMed] [Google Scholar]
- Ouwens DM, de Ruiter ND, van der Zon GCM, Carter AP, Schouten J, van der Burgt C, Kooistra K, Bos JL, Maassen JA, van Dam H (2000) Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras–MEK–ERK pathway and of Thr69 through RalGDS–Src–p38. EMBO J 21: 3782–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prmkhaokham A, Shimada Y, Fukuda Y, Kurihara N, Imoto I, Yang Z-Q, Imamura M, Nakamura Y, Amagasa T, Inazawa J (2000) Nonrandom chromosomal imbalances in esophageal sequamous cell carcinoma cell lines: possible involvement of the ATF3 and CENPE gene in the 1q32 amplicon. Jpn J Cancer Res 91: 1126–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez S, Vial E, van Dam H, Castellazzi M (2001) Transcription factor ATF3 partially transforms chick embryo fibroblasts by promoting growth factor-independent proliferation. Oncogene 20: 1135–1141 [DOI] [PubMed] [Google Scholar]
- Sakamoto D, Prendergast GC (1999) New Myc-interacting proteins: a second Myc network emerges. Oncogene 18: 2942–2954 [DOI] [PubMed] [Google Scholar]
- Schorl C, Sedivy JM (2003) Loss of protooncogene c-Myc function impedes G1 phase progression both before and after the restriction point. Mol Biol Cell 14: 823–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R, Leone G., DeGregori J, Nevins JR (1999) Ras enhances Myc stability. Mol Cell 3: 169–179 [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR (2000) Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH, Felsher DW (2004) MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431: 1112–1117 [DOI] [PubMed] [Google Scholar]
- Shrivastava A, Yu J, Artandi S, Calame K (1996) YY1 and c-Myc associate in vivo in a manner that depends on c-Myc levels. Proc Natl Acad Sci USA 93: 10638–10641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamori-Adachi M, Hayashida K, Nobori K, Omizu C, Yamada K, Sakamoto N, Kamura T, Fukuda K, Ogawa S, Nakayama KI, Kitajiam S (2004) Down-regulation of p27 promotes cell proliferation of rat neonatal cardiomyocytes induced by nuclear expression of cyclin D1 and CDK4. J Biol Chem 279: 50429–50436 [DOI] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM (2001) c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768–773 [DOI] [PubMed] [Google Scholar]
- Yin T, Sandhu G, Wolfgang CD, Burrier A, Webb RL, Rigel DF, Hai T, Whelan J (1997) Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J Biol Chem 272: 19943–19950 [DOI] [PubMed] [Google Scholar]
- Zhang C, Gao C, Kawauchi J, Hashimoto Y, Tsuchida N, Kitajima S (2002) Transcriptional activation of the human stress-inducible transcriptional repressor ATF3 gene promoter by p53. Biochem Biophys Res Commun 297: 1302–1310 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S5
Supplementary Table S6