

Abstract
Human tumors frequently present heat shock protein 70 (Hsp70) on their cell membranes, whereas corresponding normal tissues fail to do so. Therefore, an Hsp70 membrane-positive phenotype provided a tumor-specific marker. Moreover, membrane-bound Hsp70 provides a target structure for the cytolytic attack mediated by natural killer (NK) cells. Vitamin A derivatives 13-cis retinoic acid (13-RA) and all-trans retinoic acid (ATRA) and sodium-butyrate (SBU) are known for their redifferentiating capacity. Therefore, we asked the question whether loss in tumorigenicity might be associated with a reduced Hsp70 membrane expression. For our studies we used epithelial colon (CX+/CX−) and thyroid (ML-1) cancer cells, with initially different Hsp70 cell surface expression pattern. After treatment up to 7 weeks with freshly prepared 13-RA, ATRA, and SBU at nonlethal concentrations of 10 μM, 1 μM, and 0.5 mM, respectively, growth morphology, Hsp70 levels, and sensitivity toward Hsp70-specific NK cells were compared with that of untreated tumor cells. Significant growth delay was determined in CX+ tumor cells after 6 weeks treatment with 13-RA. Concomitantly, growth morphology changed from spheroid cell clusters to monolayers. Despite a weak increase in cytosolic Hsp70, the percentage of Hsp70 membrane-positive cells dropped significantly after repeated treatments with 13-RA and ATRA in CX+ and ML-1 but not in CX− tumor cells. Similar results were observed with SBU. Functionally, the decrease in Hsp70 membrane-positive CX+ and ML-1 cells correlated with a reduced sensitivity to lysis mediated by NK cells. In summary, redifferentiating agents predominantly affected Hsp70 membrane-positive tumors. The decrease in Hsp70 membrane positivity correlated with a lower sensitivity to NK lysis, growth delay, and altered growth morphology.
INTRODUCTION
Heat shock proteins (HSPs) are highly conserved molecules mediating protection against lethal damage after various stress stimuli in prokaryotic and eukaryotic cells. Also, under physiological conditions, they support folding of nonnative or misfolded proteins and prevent aggregation during proliferation and cellular differentiation (Hartl and Hayer-Hartl 2002). Cell surface localization of several HSPs including Hsp70, the major stress-inducible member of the HSP70 group, has been documented for tumor cells by selective cell surface proteomics (Shin et al 2003) and flow cytometry (Multhoff et al 1995). In contrast, corresponding normal tissues were found to be Hsp70 membrane negative, thus indicating that surface-bound Hsp70 serves as a tumor marker. Several clinically applied reagents including alkyl-lysophospholipids (Botzler et al 1999), cytostatic drugs (Gehrmann et al 2002), and anti-inflammatory reagents (Gehrmann et al 2004) have been found to enhance Hsp70 membrane expression. Also UV and γ-irradiation resulted in an upregulated Hsp70 expression (Suzuki and Watanabe 1992; Sierra-Rivera et al 1993; Matsumoto et al 1995). Functionally, a tumor-specific Hsp70 membrane localization could be associated with an increased sensitivity to the cytolytic attack mediated by natural killer (NK) cells (Multhoff et al 1997; Gastpar et al 2004), although these tumors were found to be highly resistant to radio- and chemotherapy.
Another approach to cure cancer is based on retinoids representing natural and synthetic derivatives of vitamin A, regulating cell growth (Nagpal et al 1996; Zhang et al 1996), apoptosis (Massaro and Massaro 2000), and homeostasis (Wan et al 2000). Apart from these effects, 13-cis retinoic acid (13-RA) and all-trans retinoic acid (ATRA) have been found to promote differentiation of tumors into a more benign cell type. Clinically, retinoids are frequently used in the therapy of acute promyelocytic leukemia (Park et al 2003) and myelodysplastic syndrome (Kuendgen et al 2004). Partial redifferentiation of promyelocytes carrying the reciprocal translocation t(15;17), coding for the fusion protein promyelocytic leukemia-retinoic acid receptor alpha (PML-RARa) into mature granulocytes, was demonstrated by the group of Huang (1988). Retinoic acid reverts the PML-RARa–induced inhibition in transcription and thus initiates granulocytic differentiation (Miller et al 1992; Grignani et al 1998). The inhibitory effects of retinoic acid on RAR promoter also resulted in growth delay in solid tumors (Altucci and Gronemeyer 2001). In combination with interferon-l alpha, 13-RA exerts beneficial effects in the treatment of epithelial cancers including squamous cell skin cancer and cervical carcinomas (Moore et al 1994; Berg et al 2000). As a single reagent, 13-RA was also effective in premalignancies, including oral leukoplakia and xeroderma pigmentosum (Freemantle et al 2003), and in poorly differentiated thyroid cancer. In follicular thyroid tumors, 13-RA has been found to inhibit tumor growth and to enhance iodine uptake by the induction of type I iodothyronine-5′-deiodinase and alkaline phosphatase receptors and intracellular adhesion molecule–1 (Bassi et al 1995; Schmutzler et al 1996). Furthermore, treatment of follicular thyroid tumor cells with 13-RA resulted in loss of tumorigenicity in athymic nude mice.
With respect to the finding that Hsp70 membrane positivity is selectively detectable on tumors but not on normal cells, we asked the question whether a retinoid-induced loss in tumorigenicity might be associated with a reduced Hsp70 membrane expression. Therefore, we analyzed the differentiating capacity of sublethal concentrations of 13-RA and ATRA on Hsp70 membrane levels and correlated them with the effects on the cytolytic capacity of NK cells. Moreover, retinoid-induced effects were compared with that of sodium-butyrate (SBU) also known to exhibit redifferentiating capacity. For our investigations, we used epithelial cancer cell lines that differ in their capacity to present Hsp70 on the plasma membrane. We demonstrated that long-term incubation with 13-RA and ATRA predominantly affects tumor cells with initially high Hsp70 membrane levels. Apart from a significant growth delay, retinoids promote altered growth morphology. Functionally, the retinoid- and SBU-induced decrease in Hsp70 membrane-positive tumor cells correlated with a reduced sensitivity to NK cell–mediated lysis. These data confirmed previous findings showing that membrane-bound Hsp70 acts as a tumor-selective danger signal for NK cells.
MATERIALS AND METHODS
Cells and cell culture
The human colon carcinoma sublines CX+ and CX−, generated from the parental cell line CX-2 (Tumorzellbank, DKFZ Heidelberg, Germany) by cell sorting using the Hsp70-specific monoclonal antibody (cmHsp70.1, multimmune GmbH, Regensburg, Germany), were cultured in Roswell Park Memorial Institute 1640 medium (GIBCO, Eggenstein, Germany) supplemented with 10% heat-inactivated fetal calf serum (FCS) (GIBCO), 1% antibiotics (penicillin-streptomycin, GIBCO), and 2 mM l-glutamine (GIBCO). ML-1 thyroid carcinoma cells were cultured under identical conditions, as described above. Exponential cell growth was maintained by regular cell passages. An amount of 2 pM triiodothyronine (T3), a characteristic marker of thyroid function, was constantly produced by either untreated or 13-RA–treated ML-1 cancer cells (Schonberger et al 2000).
All adherent tumor cells were trypsinized for less than 1 minute with trypsin-ethylenediamine-tetraacetic acid (EDTA) (GIBCO), and single cell suspensions were seeded at constant cell densities of 0.5 × 106 cells in 5 mL fresh medium in T-25 ventilated small culture flasks (Greiner, Nuertingen, Germany).
Plating efficiency, doubling-time (20 hours), and protein content in Hsp70 high- and low-expressing CX+ and CX− tumor sublines and ML-1 cells were comparably low under physiological conditions at 37°C.
Treatment with 13-RA, ATRA, SBU, and γ-irradiation
Exponentially growing, adherent tumor cells were treated either with freshly prepared 13-RA and ATRA (Biomol Research Lab Inc, PA, USA) (Gendimenico and Mezick 1993) at indicated concentrations ranging from 0.5 up to 20 μM or with the nontoxic concentration of 0.5 mM SBU (Sigma-Aldrich, Steinheim, Germany) for 48 hours. Alternatively, tumor cells were γ-irradiated at 10 Gy followed by a recovery period of 12 hours. Untreated controls were passaged identically but in the absence of 13-RA, ATRA, and SBU.
Viability testing
Viability was determined weekly in untreated, retinoid- and SBU-treated CX+/CX− and ML-1 cells by trypan blue exclusion assay on a light microscope and by 7-amino-actinomycin D (7-AAD) and Annexin-V fluorescein isothiocyanat (FITC) staining on a FACSCalibur instrument (Becton Dickinson, Heidelberg, Germany). In brief, the supernatants (5 mL) of untreated and treated cells were collected. Then adherent cells were washed in 5 mL phosphate-buffered saline (PBS), which was removed and combined with the harvested cell culture supernatant. After a 1-minute incubation period of the adherent cells with trypsin-EDTA (500 μL) at 37°C, culture flasks were shaken gently and inspected carefully for adherent cells by light microscopy. Only if all cells were found to be detached, the flasks were washed with the collected supernatant and PBS fraction and centrifuged at 800 × g for 8 minutes. After resuspension of the cell pellet in 0.5 mL PBS-10% FCS, absolute cell counts of viable and dead cells were determined by trypan blue exclusion assay. Cell viability and apoptosis were also determined by 7-AAD and Annexin-V FITC staining. The percentage of viable cells was determined on a FACSCalibur instrument counting 10 000 events.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot analysis
Cell pellets of either untreated or treated cells (1.0 × 106) were washed in PBS and solubilized in lysis buffer (120 mM sodium chloride, 40 mM Tris pH 8.0, 0.5% NP40). Samples were denatured in sample buffer (25 mM Tris hydrochloride pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, 10% mercaptoethanol, and bromphenol blue) by boiling for 10 minutes. Equal protein amounts (10 μg) were administered to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis under reducing conditions (Laemmli 1970). After electrophoresis, proteins were transferred onto nitrocellulose membranes and blotted following a standard protocol of Towbin et al (1979). Antibodies for Western blotting were used at dilutions of 1:1000, mouse anti-human Hsp70 (cmHsp70.2, multimmune GmbH) and mouse anti-human tubulin (DM1A, Oncogene, Boston, MA, USA). After incubation with horseradish peroxidase–conjugated secondary antibodies (DAKO, Glostrup, Denmark), also diluted 1:1000, protein bands were observed by chemiluminescence (ECL detection-kit, Amersham Biosciences, Little Chalfont, UK). Autoradiographs were recorded on X-omat films (Kodak, Stuttgart, Germany). Prestained standard protein markers (Amersham Biosciences) were used for estimating the molecular weights of each protein.
Flow cytometry
Tumor cells, kept either untreated or treated with retinoids or SBU, were trypsinized, counted, and 0.1 × 106 cells were incubated with an FITC-conjugated Hsp70 monoclonal antibody (IgG1, cmHsp70.1, multimmune GmbH), FITC-conjugated major histocompatibility complex (MHC) class I–specific antibody (IgG2a, W6/32, CBL139F, Cymbus Biotech, Hants, UK), human leukocyte antigen class E (HLA-E)–specific antibody (IgG1, MEM-E/06, EXBIO, Praha, Czech Republic), and the corresponding isotype-matched control antibodies (Dianova, Hamburg, Germany) for 15 minutes at 4°C. After washing in PBS 10% FCS (heat inactivated), 7-AAD-negative (Becton Dickinson Pharmingen, Heidelberg, Germany), viable cells were analyzed on a FACSCalibur flow cytometer (Becton Dickinson, Heidelberg, Germany).
Light microscopy
Adherent growing tumor cells, either untreated or treated with 13-RA for 6 cell passages or with ATRA for 5 cell passages, were photographed at a magnification of 100× on a Zeiss model Axioscope 2 scanning microscope (Zeiss, Jena, Germany) equipped with a 100× (planar). Representative images were treated by multiplicative shading correction using the software Axiovision (Zeiss Vision, Jena, Germany).
51Cr-release assay
NK cell–mediated cytotoxicity was measured in a standard 4-hour 51Cr-release assay (MacDonald et al 1974). NK cells were separated from peripheral blood lymphocytes of healthy human volunteers by using a standard CD3-CD19 depletion protocol of miltenyi (miltenyi biotech, Bergisch Gladbach, Germany). CD3- and CD19-negative NK cells were stimulated with Hsp70 peptide TKD (2 μg/mL) together with 100 IU/mL interleukin-2 for 4 days, at 37°C and after 2 washing steps used as effector cells. As target cells, either untreated or long-term retinoic acid–treated or SBU-treated tumor cells with reduced Hsp70 membrane expression were used. In brief, 51Cr-labeled target cells were coincubated in u-bottom, 96-well plates at different effector to target cell (E:T) ratios, ranging from 20:1 to 2:1 or 10:1 to 1:1, in a total volume of 200 μL medium. After a 4-hour coincubation period at 37°C, supernatants and cell suspensions (40 μL each) were harvested, transferred onto Luma Plates-96 (Perkin-Elmer, Boston, MA, USA), and counted in a gamma-counter (Topcount, Packard Instruments, Meriden, CT, USA). The percentage specific lysis was calculated as: ([experimental release − spontaneous release]/[maximal release − spontaneous release]) × 100. Spontaneous release in all experiments was always less than 20%.
Miscellaneous
Chemicals were from Sigma (Munich, Germany), Merck (Darmstadt, Germany), or Roth (Karlsruhe, Germany), if not indicated otherwise.
Statistics
For statistical analysis, Student's t-test was used. A confidence level above 95% (P < 0.05) was defined as significant.
RESULTS
Direct toxic effects of retinoids on viability of different tumor cells
Retinoids are clinically applied in cancer therapy and chemoprevention. In this study, we focused on immunological effects induced by 13-RA and ATRA in human epithelial cancer cells that are related to Hsp70. By cell sorting of the parental CX-2 colon carcinoma cell line using an Hsp70-specific monoclonal antibody, stably Hsp70 high- (CX+ 82% ± 5) and low-expressing (CX− 22% ± 12) tumor sublines were generated. In addition, the thyroid cancer cell line ML-1, expressing Hsp70 on 39% ± 9 of the cells, was analyzed. At each cell passage, CX-2, CX+, CX−, and ML-1 were incubated with freshly prepared 13-RA at different concentrations ranging from 1 to 20 μM for 1 week and with ATRA at concentrations of 0.5 and 1 μM. Up to a concentration of 10 μM of 13-RA and a 1 week treatment, cell viability of CX-2, CX+, and CX− colon carcinoma sublines was not significantly affected; however, a retinoid concentration of 20 μM induced cell death, as determined by 7-AAD staining of 10 000 cells in a flow cytometer (Table 1). Moreover, a concentration of 0.5 mM SBU, another agent with redifferentiating capacity, was also found to be nontoxic for CX+ and CX− tumor sublines after an incubation period of 48 hours (data not shown).
Table 1.
a) Definition of the nontoxic dose of 13-RA and ATRA for the original colon carcinoma cell line CX-2, the sublines CX+ and CX−, and the thyroid cancer cell line ML-1. Cell viability was determined in each cell type by 7-AAD staining of 10 000 cells on a FACSCalibur after 1 week of treatment with different concentrations of 13-RA and ATRA. The data represent mean values of 3 to 4 independent experiments ± SD; values shown are percentage viable cells (mean values ± SD)a

ML-1 thyroid cancer cells were found to be more sensitive to 13-RA, as compared with the colon carcinoma sublines. A concentration of 10 μM 13-RA for 1 week already was toxic; a concentration of 1 μM was determined as the nontoxic concentration for ML-1 thyroid cancer cells (Table 1). For ATRA, a concentration of 1 μM was found to be nontoxic for all tumor cell types (Table 1). Viability assays of each cell type were repeated 3 to 4 times.
Effects of long-term retinoid treatment on growth rates of different tumor cells
After 6 weekly repeated treatments with freshly prepared 13-RA (10 μM), absolute cell counts of CX+ tumor cells (black bars) were found to be significantly reduced as compared with untreated cells and CX− cells (Fig 1). This reduction in Hsp70-positive CX+ cells after long-term retinoid treatment is most likely due to growth delay because trypan blue–positive dead cells were not detectable either in the supernatant or in the adherent cell population. Annexin V-FITC staining also revealed that treatment of CX+/CX− tumor sublines with 10 μM 13-RA and of ML-1 tumor cells with 1 μM 13-RA for 6 weeks did not cause significant apoptosis; in contrast, γ-irradiation (10 Gy) initiated apoptosis in CX− and ML-1 tumor cells with initially low Hsp70 membrane expression (Table 2). These findings are in line with previous data showing that CX+ cells are more resistant to γ-irradiation–induced cell death as compared with CX− cells (Gehrmann et al 2003).
Fig 1.

CX+ tumor cell growth was reduced by treatment with 13-cis retinoic acid (13-RA). Adherent growing CX+ and CX− tumor cells were incubated with freshly prepared 13-RA (10 μM) at each individual cell passage (twice a week). After each passage, equal cell counts (0.5 × 106 cells in 5 mL medium) were seeded in T-25 flasks. Absolute counts of viable cells were determined by trypan blue staining after 1 to 6 weeks of treatment in single cell suspensions of adherent and nonadherent cells after trypsinization. The percentage of trypan blue–positive, dead tumor cells before and after treatment was always less than 5%. Growth inhibition data represented mean values of 6 independent experiments ± SD; *marks values significantly different from control (P < 0.05)
Table 2.
Definition of apoptotic cell death after treatment of colon carcinoma sublines CX+/CX− with 10 μM 13-RA and of thyroid cancer cell line ML-1 with 1 μM 13-RA for 6 weeks. In addition, tumor cells were exposed to γ-irradiation (1 × 10 Gy) followed by a recovery period of 24 hours. Apoptosis was determined by Annexin V-FITC staining of 10 000 cells on a FACSCalibur. The data represent mean values of 3 independent experiments ± SD; values shown are cell counts (mean values ± SD)a

A concentration of 1 μM ATRA even after 5 repeated treatments affected neither cell growth nor apoptosis in CX+ and CX− tumor cells (data not shown). Similar results were observed after a 7-week treatment of ML-1 thyroid cancer cells with 13-RA and ATRA at a concentration of 1 μM each (data not shown).
Effects of a long-term retinoid treatment on growth morphology of tumor cells
Under physiological conditions, doubling-time of CX+/ CX− sublines and the original cell line CX-2 cells was comparable at about 20 hours. Already 2 hours after seeding of trypsinized single-cell suspensions in T-25 tissue culture flasks, colon carcinoma cells form spheroid cell clusters that become larger after each cell division. After long-term treatment with 13-RA (10 μM) for 6 weeks and with ATRA (1 μM) for 5 weeks, doubling-time increased up to 25 hours in CX+ tumor cells with initially high Hsp70 membrane expression levels. Doubling-time of CX− cells remained unaffected. Photographs were taken from representative parts of either untreated (ctrl, upper graph) or long-term 13-RA–treated (6 weeks, middle graph) and ATRA-treated (5 weeks, lower graph) CX+ tumor cell cultures on day 2 after the last cell passage. As illustrated in Figure 2, the typical 3-dimensional cell clusters (ctrl) of CX+ colon carcinoma cells changed to monolayer after long-term treatment with 13-RA (10 μM). The spheroid cell morphology changed to a spindle-shaped morphology. Treatment of CX+ cells with a concentration of 1 μM ATRA for 5 weeks induced similar, but less pronounced, effects. Retinoids are documented to initiate redifferentiation processes in tumor cells. Similar results were observed after a 48-hour treatment of CX+ tumor cells with a nontoxic concentration of SBU (5 mM; data not shown). The observed retinoid-induced changes in growth morphology and behavior of CX+ tumor cells might be a first hint for a process reverting loss of contact inhibition, which is typical for malignant tumors.
Fig 2.

Growth morphology of CX+ tumor sublines changed from cell clusters to monolayers after treatment with 13-cis retinoic acid (13-RA) and all-trans retinoic acid (ATRA). Under physiological conditions, CX+ tumor sublines showed spheroid cell cluster growth (magnification 100×). After treatment with 13-RA (10 μM) for 6 weeks and with ATRA (1 μM) for 5 weeks, cells changed their growth behavior to monolayers. On a single cell level it became obvious that the shape of CX+ tumor cells changed from spheroid to spindle shaped. Scale bar, 200 μm
Effects of long-term retinoid treatment on Hsp70 levels in different tumor cells
Under physiological conditions, cytosolic Hsp70 content was comparably low in CX+ and CX− tumor sublines, as determined by comparative quantitative Western blot analysis. A mild heat shock at 41.8°C for 2 hours resulted in a strong induction of the synthesis of Hsp70 (about 10-fold) in both tumor sublines (Multhoff et al 1997). In this study, we were interested in testing 13-RA on the induction of Hsp70 synthesis, the major stress-inducible member of the HSP70 family. Even after 6 repeated treatments with 13-RA (10 μM), the increase in cytosolic Hsp70 was only 1.31-fold in CX+ and 1.27-fold in CX− tumor cells, if related to tubulin (Fig 3). Next, we studied cell surface expression of Hsp70 after long-term treatment with retinoids. As mentioned earlier, under physiological conditions, the tumor cell lines CX+ (82% ± 5), CX− (22% ± 12), and ML-1 (39% ± 9) differed significantly in their capacity to present Hsp70 on their cell membranes. After a 4- and 6-week treatment interval with 13-RA (10 μM), the percentage of Hsp70 membrane-positive cells dropped significantly in CX+ tumor cells from 82% ± 5 to 54% ± 3 and 32% ± 6, respectively (Fig 4A). No significant changes in the Hsp70 cell surface expression pattern were found in CX− tumor cells (22% ± 12, 23% ± 8, and 32% ± 5) after an identical treatment (Fig 4A). Similar effects were observed if CX+ and CX− tumor sublines were treated with a concentration of 1 μM ATRA for 5 weeks (Fig 4B; Table 3). Again, a significant decrease in Hsp70 membrane-positive cells was only detected in CX+ tumor cells. With respect to ML-1 thyroid cancer cells, a 4- and 7-week treatment interval with a concentration of 1 μM 13-RA was sufficient to decrease the percentage of Hsp70-positive cells from 39% ± 9 to 26% ± 5 and 21% ± 8, respectively (Fig 4C). The expression of classical MHC I molecules, as determined with the antibody W6/32, in CX+ and CX− tumor sublines was always greater 95% before and after long-term treatment with 13-RA (10 μM) and ATRA (1 μM), as summarized in Table 3. Neither before nor after treatment with retinoids, HLA-E was expressed on the tumor sublines (data not shown). However, the expression of the adhesion molecule neuronal-cellular adhesion molecule (N-CAM) (CD56) in CX+ and CX− colon carcinoma cells dropped significantly after long-term treatment with 13-RA (10 μM) and ATRA (1 μM) (Table 3). This finding might provide an explanation for the modulated growth morphology from 3-dimensional cell clusters to spindle-shaped monolayers. Two (data not shown) and 4 weeks after removal of the retinoids from cell culture, the phenotype remained unaltered (Table 3). Moreover, γ-irradiation at 10 Gy did not significantly enhance Hsp70 membrane expression on retinoid pretreated CX+ cells (data not shown).
Fig 3.

13-cis retinoic acid (13-RA) increased cytosolic heat shock protein 70 (Hsp70) levels in CX+ and CX− tumor sublines. Adherent growing CX+ and CX− tumor cells were kept either untreated (ctrl) or were incubated with13-RA (10 μM) at each cell passage. After 6 weeks, cell lysates were prepared and equal protein amounts (10 μg) were run on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Western blots were stained with an Hsp70 and a tubulin-specific antibody, and protein bands were analyzed by densitometry. Increase in cytosolic Hsp70 was related to tubulin. The data represent mean values of 7 independent experiments ± SD. One representative Western blot was shown in the upper part of the graph
Fig 4.

The 13-cis retinoic acid (13-RA) reduced the percentage of Hsp70 membrane-positive cells in the CX+ and ML-1 tumor cells. The percentage of Hsp70 positively stained cells was determined by flow cytometry in untreated (ctrl), 13-RA-treated, all-trans retinoic acid (ATRA)-treated CX+/CX− and ML-1 tumor cells. CX+/CX− tumor cells were treated with 10 μM 13-RA for 4 and 6 weeks (A) and with 1μM ATRA for 5 weeks (B); ML-1 tumor cells were treated with 1 μM 13-RA for 2, 4, and 7 weeks (C). Data represent mean percentages of Hsp70-positively stained cells ± SD of 6 (A,B) and 3 (C) independent experiments; *marks values significantly different from control (P < 0.05)
Table 3.
Flow cytometric analysis of cell surface markers on CX+ and CX− tumor cells after long-term treatment with +13-RA and +ATRA and 4 weeks after removal of 13-RA (−13-RA). Exponen tially growing tumor cells were maintained either untreated or treated with freshly prepared 13-RA (10 μM) for 6 weeks and ATRA (1 μM) for 5 weeks at each cell passage. Percentage positively stained cells minus the number of cells stained with an isotype-matched control antibody are indicated. Data represent mean values of at least 3 independent experimentsa

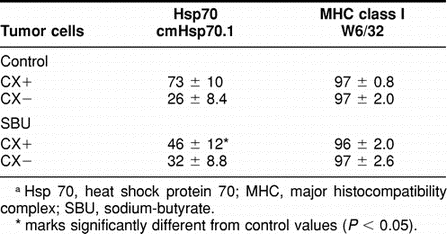
Interestingly, SBU, a short-chain lipid with redifferentiating capacity, exhibits similar effects like retinoids. As summarized in Table 4, Hsp70 membrane positivity shrinked drastically after a 48-hour incubation period with SBU (5 mM) in CX+ (73% ± 10 vs 46% ± 12) but not in CX− (26% ± 8.4 vs 32% ± 8.8) tumor sublines.
Table 4.
Flow cytometric analysis of the Hsp70 and MHC class I membrane expression on CX+ and CX− tumor cells either untreat ed (Control) or after a 48 hour incubation period with SBU at the nontoxic concentration of 5 mM. Data represent mean values of at least 3 independent experiments a
Immunological effects of long-term retinoid treatment on tumor cells
As previously reported, the amount of Hsp70 membrane-positive tumor cells determined the sensitivity to lysis mediated by NK cells (Gehrmann et al 2003). Hsp70 low-expressing tumor sublines CX− were lysed significantly less efficient as compared with their higher-expressing counterparts CX+ (Multhoff et al 1997). In this study, we addressed the question as to whether a retinoid-induced decrease in Hsp70 membrane-positive cells corresponded with a reduced sensitivity to lysis mediated by CD3-/ CD19-depleted NK cells that had been stimulated with Hsp70 peptide TKD (Multhoff et al 1999). As summarized in Figure 5A, the decreased amount of membrane-bound Hsp70 in CX+ tumor cells after a 4- and 6-week treatment interval with 13-RA (10 μM) correlated with a reduced sensitivity to lysis mediated by NK cells that had been stimulated with TKD, as described earlier (Multhoff et al 1995). In contrast, the weak lysis of CX− tumor cells by NK cells remained weak at the level of 13-RA–treated CX+ cells after treatment with 13-RA (Fig 5B). This finding is in line with the comparable percentages of Hsp70 membrane-positive cells in untreated (22% ± 12), 6–week 13-RA–treated (32% ± 7) CX− cells, and 6-week 13-RA– treated CX+ (32% ± 6) tumor cells (Table 3). In thyroid ML-1 cancer cells, a reduced Hsp70 membrane expression after a 4- and 7-week treatment interval with 13-RA (1 μM) also corresponded with a decreased sensitivity to the cytolytic attack mediated by Hsp70-activated NK cells (Fig 5C). Similar results were obtained after long-term (5 weeks) treatment of Hsp70 membrane-positive tumor cells with 1 μM ATRA (data not shown).
Fig 5.

13-cis retinoic acid (13-RA) reduced the sensitivity of CX+ and ML-1 tumor cells toward the lytic capacity of NK cells. CX+ (A) and CX− (B) tumor sublines were kept either untreated or were treated with 13-RA (10 μM) for 4 and 6 weeks; ML-1 tumor cells were kept either untreated or were treated with 13-RA (1 μM) for 4 and 7 weeks (C). Untreated and treated tumor cells were used as target cells in a standard 4-hour 51Cr release assay. CD3-/CD19-depleted natural killer (NK) cells, stimulated for 4 days with Hsp70 peptide TKD (2 μg/ mL) and low dose interleukin-2 (100 IU/mL), were used as effector cells at different effector to target cell (E:T) ratios ranging from 20:1 to 2:1. Purity of CD3-/CD19-negative NK cells was greater 95%. Mean values of 3 independent experiments ± SD were illustrated
SBU, at the nontoxic concentration of 0.5 mM, also reduced Hsp70 membrane expression on CX+ tumor cells and caused a reduced lysability mediated by NK cells. Lysis of CX− tumor sublines either untreated or SBU treated remained unaltered low (Fig 6).
Fig 6.

Sodium-butyrate (SBU) reduced the sensitivity of CX+ tumor cells toward the lytic capacity of natural killer (NK) cells. CX+ and CX− tumor sublines were kept either untreated or were treated with SBU (0.5 mM for 48 hours). Untreated and treated tumor cells were used as target cells in a standard 4-hour 51Cr release assay. CD3-/CD19-depleted NK cells, stimulated for 4 days with heat shock protein 70 (Hsp70) peptide TKD (2 μg/mL) and low dose interleukin-2 (100 IU/mL), were used as effector cells at different effector to target cell (E:T) ratios ranging from 10:1 to 1:1. Purity of CD3-/CD19-negative NK cells was greater 95%. Mean values of 3 independent experiments ± SD were illustrated
DISCUSSION
Retinoids, synthetic derivatives of vitamin A, were frequently used in the therapy of premalignant and malignant cells, either alone or in combination with cytostatic drugs. Despite promising results of ATRA in the redifferentiation of acute promyelocytic leukemia, intrinsic and acquired mechanisms of resistance to retinoids limit their therapeutic efficiency. Moreover, the exact biological mechanisms that are affected by retinoids are not completely understood for solid tumors. Here we studied immunological effects of nontoxic concentrations of retinoids 13-RA and ATRA on Hsp70 expression in different tumor cell types including colon and thyroid cancer cells. Although highly conserved, Hsp70 is critical in eliciting an efficient antitumor immune response. Hsp70 plays an important role in antigen presentation and acts as a carrier for tumor-derived peptides (Srivastava 2001). For the innate immune system, membrane-bound Hsp70 was found to serve as a tumor-selective recognition structure for NK cells (Multhoff et al 1995, 1997). The Hsp70 epitope exposed to the extracellular milieu of tumor cells was identified (Multhoff et al 1999) and defined as immunogenic for NK cells. The immunostimulatory capacity of Hsp70 is in line with observations of other laboratories. Recently, several HSP receptors, including CD40 on B cells (Becker et al 2002), CD14 and Toll-like receptors on antigen presenting cells (APC) such as monocytes and dendritic cells (Asea et al 2000), CD91 on macrophages (Binder et al 2000), and CD94 on NK cells (Gross et al 2003), have been characterized for different cell types with immunoregulatory capacity. Contact of APCs with Hsp70 via Toll-like receptors resulted in the secretion of proinflammatory cytokines stimulating the innate immune system (Asea 2003). On the other hand, overexpression of Hsp70 has been found to protect tumor cells from apoptosis induced by exogenous stress including cytostatic drugs and radiation (Nylandsted et al 2004). However, Hsp70-reactive NK cells exerted beneficial effects in a clinical phase I trial in therapy-refractory patients (Krause et al 2004).
Long-term incubation of colon carcinoma cells with a nontoxic concentration of 13-RA only moderately increased cytosolic Hsp70 levels in colon carcinoma cells. In comparison, nonlethal heat shock, as a classical stressor, resulted in rapid and 10-fold higher increase in Hsp70 levels (Multhoff et al 1997).
In contrast to the cytosol, tumor cells with initially high Hsp70 membrane expression levels showed a significant reduction in Hsp70 membrane-positive cells after treatment with nontoxic doses of 13-RA and ATRA. Concomitantly, doubling-time increased from 20 to 25 hours. Because nearly no Annexin V–positive cells were detectable in retinoid-treated cell cultures, we speculated about growth inhibition rather than induction of apoptosis in Hsp70 membrane-positive tumor cells. Morphologically, growth of colon cancer cells altered from 3-dimensional cell clusters to monolayers. Loss of cell-to-cell contact might be a first hint for a retinoid-induced redifferentiation. This finding was further supported by the fact that the expression of the neuronal adhesion molecule N-CAM was downregulated on tumor cells after retinoid treatment. In contrast, other cell surface markers including classical and nonclassical MHC molecules remained unaltered after identical treatment conditions.
Screening of a large panel of different tumor cell lines (Multhoff et al 1995), primary tumor biopsies, metastases, and corresponding normal tissues (unpublished observation) revealed that Hsp70 was frequently expressed on tumors and metastases but not on the corresponding normal tissues. A comparison of primary tumors and metastases revealed higher Hsp70 expression levels on metastases (S. Stangl, L. Rossbacher, G. Multhoff, in preparation) and on tumors with unfavorable prognosis (Gehrmann et al 2003). Because Hsp70 membrane expression was found to be reduced by nontoxic concentrations of 13-RA and ATRA, we speculated about a retinoid-induced redifferentiation process to a less aggressive tumor stage. Moreover, SBU, another agent with redifferentiating capacity, caused similar effects.
Thyroid and colon cancer cells differ in their sensitivity toward direct toxic effects induced by 13-RA. For colon cancer cells a concentration of 10 μM and for thyroid cancer cells a concentration of 1 μM of 13-RA was found to be nontoxic. However, in both tumor cell systems, the nontoxic concentration of retinoids and also of SBU induced a reduction in Hsp70 cell surface–positive tumor cells correlating with a decreased sensitivity to lysis mediated by NK cells. Patients with poorly differentiated, therapy-resistant thyroid cancer were frequently treated with retinoids. A major achievement of this therapeutic approach was the induction of an increased sensitivity toward radioactive iodine 131I (Schmutzler et al 1996). As previously reported by Gehrmann et al (2005), elevated Hsp70 membrane expression levels correlated with protection against radiation-induced effects. The retinoid-induced downregulation of Hsp70 membrane expression might provide a rationale for the clinically observed effects induced by retinoids in thyroid cancer.
With respect to immunological functions, we could demonstrate that tumor cells with diminished Hsp70 cell surface levels are less sensitive toward the cytolytic attack mediated by Hsp70-reactive NK cells. This reduction in membrane-bound Hsp70 was of special interest because other exogenous factors including γ-irradiation, tubulin-interacting agents including taxol and vincristinsulfate (Gehrmann et al 2002), alkyl-lysophospholipids (Botzler et al 1999), and anti-inflammatory drugs (Gehrmann et al 2004) at nonlethal doses consistently resulted in an upregulated Hsp70 membrane expression corresponding with an augmented sensitivity to the cytolytic attack of NK cells.
The decreased sensitivity to NK cell–mediated lysis could be attributed to the fact that retinoids as well as SBU, at nontoxic concentrations, were able to induce redifferentiation in tumors into a less malignant cell type. This corresponds with the fact that normal cells, lacking Hsp70 membrane expression, did not provide targets for Hsp70-reactive NK cells. In line with these observations was the finding that the lower Hsp70 membrane expression levels remained low after removal of retinoids and that γ-irradiation was unable to enhance Hsp70 membrane expression on retinoid-treated tumor cells.
Acknowledgments
The work was supported in part by an EU-grant (TRANS-EUROPE, QLK3-CT-2002-01936) and by multimmune GmbH.
REFERENCES
- Altucci L, Gronemeyer H. Nuclear receptors in cell life and death. Trends Endocrinol Metab. 2001;12(10):460–468. doi: 10.1016/s1043-2760(01)00502-1.1043-2760(2001)012[0460:NRICLA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Asea A. Chaperokine-induced signal transduction pathways. Exerc Immunol Rev. 2003;9:25–33.1077-5552(2003)009[0025:CSTP]2.0.CO;2 [PMC free article] [PubMed] [Google Scholar]
- Asea A, Kraeft SK, and Kurt-Jones EA. et al. 2000 HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 6(4):435–442. [DOI] [PubMed] [Google Scholar]
- Bassi V, Vitale M, Feliciello A, De Riu S, Rossi G, Fenzi G. Retinoic acid induces intercellular adhesion molecule-1 hyperexpression in human thyroid carcinoma cell lines. J Clin Endocrinol Metab. 1995;80(4):1129–1135. doi: 10.1210/jcem.80.4.7714081.0021-972X(1995)080[1129:RAIIAM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Becker T, Hartl FU, Wieland F. CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. J Cell Biol. 2002;158(7):1277–1285. doi: 10.1083/jcb.200208083.0021-9525(2002)158[1277:CAERFB]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg WJ, Divgi CR, Nanus DM, Motzer RJ. Novel investigative approaches for advanced renal cell carcinoma. Semin Oncol. 2000;27(2):234–239.0093-7754(2000)027[0234:NIAFAR]2.0.CO;2 [PubMed] [Google Scholar]
- Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol. 2000;1(2):151–155. doi: 10.1038/77835.1529-2908(2000)001[0151:CARFHS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Botzler C, Ellwart J, Gunther W, Eissner G, Multhoff G. Synergistic effects of heat and ET-18-OCH3 on membrane expression of Hsp70 and lysis of leukemic K562 cells. Exp Hematol. 1999;27(3):470–478. doi: 10.1016/s0301-472x(98)00055-1.0301-472X(1999)027[0470:SEOHAE]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene. 2003;22(47):7305–7315. doi: 10.1038/sj.onc.1206936.0950-9232(2003)022[7305:RICTAC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Gastpar R, Gross C, Rossbacher L, Ellwart J, Riegger J, Multhoff G. The cell surface-localized heat shock protein 70 epitope TKD induces migration and cytolytic activity selectively in human NK cells. J Immunol. 2004;172(2):972–980. doi: 10.4049/jimmunol.172.2.972.0022-1767(2004)172[0972:TCSHSP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Gehrmann M, Brunner M, Pfister K, Reichle A, Kremmer E, Multhoff G. Differential up-regulation of cytosolic and membrane-bound heat shock protein 70 in tumor cells by anti-inflammatory drugs. Clin Can Res. 2004;10:3354–3364. doi: 10.1158/1078-0432.CCR-03-0382.1078-0432(2004)010[3354:DUOCAM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Gehrmann M, Marienhagen J, and Eichholtz-Wirth H. et al. 2005 Dual function of membrane-bound heat shock protein 70 (Hsp70), Bag-4, and Hsp40: protection against radiation-induced effects and target structure for Natural Killer (NK) Cells. Cell Death Differ. 12:38–51. [DOI] [PubMed] [Google Scholar]
- Gehrmann M, Pfister K, Hutzler P, Gastpar R, Margulis B, Multhoff G. Effects of antineoplastic agents on cytoplasmic and membrane-bound heat shock protein 70 (Hsp70) levels. Biol Chem. 2002;383(11):1715–1725. doi: 10.1515/BC.2002.192.1431-6730(2002)383[1715:EOAAOC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Gehrmann M, Schmetzer H, Eissner G, Haferlach T, Hiddemann W, Multhoff G. Membrane-bound heat shock protein 70 (Hsp70) in acute myeloid leukemia: a tumor specific recognition structure for the cytolytic activity of autologous NK cells. Haematologica. 2003;88(4):474–476.0390-6078(2003)088[0474:MHSPHI]2.0.CO;2 [PubMed] [Google Scholar]
- Gendimenico GJ, Mezick JA 1993 Pharmacological effects of retinoids on skin cells. Skin Pharmacol. 6(Suppl 1). 24–34. [DOI] [PubMed] [Google Scholar]
- Grignani F, De Matteis S, and Nervi C. et al. 1998 Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 391:815–818. [DOI] [PubMed] [Google Scholar]
- Gross C, Hansch D, Gastpar R, Multhoff G. Interaction of heat shock protein 70 peptide with NK cells involves the NK receptor CD94. Biol Chem. 2003;384(2):267–279. doi: 10.1515/BC.2003.030.1431-6730(2003)384[0267:IOHSPP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408.0193-4511(2002)295[1852:MCITCF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Huang ME, Ye YC, and Chen SR. et al. 1988 Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 72(2):567–572.3165295 [Google Scholar]
- Krause S, Gastpar R, Andreesen R, Gross C, Ullrich H, Thonigs G, Pfister K, Multhoff G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous Natural Killer cells: a clinical phase I trail. Clin Canc Res. 2004;10:3699–3707. doi: 10.1158/1078-0432.CCR-03-0683.1078-0432(2004)010[3699:TOCALC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Kuendgen A, Strupp C, and Aivado M. et al. 2004 Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid. Blood. 104(5):1266–1269. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0.0028-0836(1970)227[0680:COSPDT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- MacDonald HR, Engers HD, Cerottini JC, Brunner KT. Generation of cytotoxic T lymphocytes in vitro. II. Effect of repeated exposure to alloantigens on the cytotoxic activity of long-term mixed leukocyte cultures. J Exp Med. 1974;140(3):718–730. doi: 10.1084/jem.140.3.718.0022-1007(1974)140[0718:GOCTLI]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaro GD, Massaro D. Retinoic acid treatment partially rescues failed septation in rats and in mice. Am J Physiol Lung Cell Mol Physiol. 2000;278(5):955–960. doi: 10.1152/ajplung.2000.278.5.L955.1040-0605(2000)278[0955:RATPRF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Wang X, Ohnishi T. Binding between wild-type p53 and hsp72 accumulated after UV and gamma-ray irradiation. Cancer Lett. 1995;92(2):127–133. doi: 10.1016/0304-3835(95)03769-s.0304-3835(1995)092[0127:BBWPAH]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Miller WH Jr., Kakizuka A, and Frankel SR. et al. 1992 Reverse transcription polymerase chain reaction for the rearranged retinoic acid receptor alpha clarifies diagnosis and detects minimal residual disease in acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 89(7):2694–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DM, Kalvakolanu DV, and Lippman SM. et al. 1994 Retinoic acid and interferon in human cancer: mechanistic and clinical studies. Semin Hematol. 31(4 Suppl 5). 31–37. [PubMed] [Google Scholar]
- Multhoff G, Botzler C, Jennen L, Schmidt J, Ellwart J, Issels R. Heat shock protein 72 on tumor cells: a recognition structure for natural killer cells. J Immunol. 1997;158(9):4341–4350.0022-1767(1997)158[4341:HSPOTC]2.0.CO;2 [PubMed] [Google Scholar]
- Multhoff G, Botzler C, Wiesnet M, Eissner G, Issels R. CD3- large granular lymphocytes recognize a heat-inducible immunogenic determinant associated with the 72-kD heat shock protein on human sarcoma cells. Blood. 1995;86(4):1374–1382.0006-4971(1995)086[1374:CLGLRA]2.0.CO;2 [PubMed] [Google Scholar]
- Multhoff G, Botzler C, and Wiesnet M. et al. 1995 A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int J Cancer. 61(2):272–279. [DOI] [PubMed] [Google Scholar]
- Multhoff G, Mizzen L, and Winchester CC. et al. 1999 Heat shock protein 70 (Hsp70) stimulates proliferation and cytolytic activity of natural killer cells. Exp Hematol. 27(11):1627–1636. [DOI] [PubMed] [Google Scholar]
- Nagpal S, Thacher SM, and Patel S. et al. 1996 Negative regulation of two hyperproliferative keratinocyte differentiation markers by a retinoic acid receptor-specific retinoid: insight into the mechanism of retinoid action in psoriasis. Cell Growth Differ. 7(12):1783–1791. [PubMed] [Google Scholar]
- Nylandsted J, Gyrd-Hansen M, and Danielewicz A. et al. 2004 Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 200(4):425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DJ, Vuong PT, de Vos S, Douer D, Koeffler HP. Comparative analysis of genes regulated by PML/RAR alpha and PLZF/RAR alpha in response to retinoic acid using oligonucleotide arrays. Blood. 2003;102(10):3727–3736. doi: 10.1182/blood-2003-02-0412.0006-4971(2003)102[3727:CAOGRB]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Schmutzler C, Brtko J, Bienert K, and Kohrle J 1996 Effects of retinoids and role of retinoic acid receptors in human thyroid carcinomas and cell lines derived therefrom. Exp Clin Endocrinol Diabetes. 104(Suppl 4). 16–19. [DOI] [PubMed] [Google Scholar]
- Schonberger J, Bauer J, Spruss T, Weber G, Chahoud I, Eilles C, Grimm D. Establishment and characterization of the follicular thyroid carcinoma cell line ML-1. J Mol Med. 2000;78(2):102–110. doi: 10.1007/s001090000085.0946-2716(2000)078[0102:EACOTF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Shin BK, Wang H, and Yim AM. et al. 2003 Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 278(9):7607–7616. [DOI] [PubMed] [Google Scholar]
- Sierra-Rivera E, Voorhees GJ, Freeman ML. Gamma irradiation increases hsp-70 in Chinese hamster ovary cells. Radiat Res. 1993;135(1):40–45. doi: 10.2307/3578394.0033-7587(1993)135[0040:GIIHIC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Srivastava PK. A central role for heat shock proteins in host deficiency. Adv Exp Med Biol. 2001;495:121–126. doi: 10.1007/978-1-4615-0685-0_16.0065-2598(2001)495[0121:ACRFHS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Watanabe M. Augmented expression of HSP72 protein in normal human fibroblasts irradiated with ultraviolet light. Biochem Biophys Res Commun. 1992;186(3):1257–1264. doi: 10.1016/s0006-291x(05)81541-4.0006-291X(1992)186[1257:AEOHPI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350.0027-8424(1979)076[4350:ETOPFP]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan YJ, Cai Y, Cowan C, Magee TR. Fatty acyl-CoAs inhibit retinoic acid-induced apoptosis in Hep3B cells. Cancer Lett. 2000;154(1):19–27. doi: 10.1016/s0304-3835(00)00341-4.0304-3835(2000)154[0019:FAIRAA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Zhang L, Nadzan AM, and Heyman RA. et al. 1996 Discovery of novel retinoic acid receptor agonists having potent antiproliferative activity in cervical cancer cells. J Med Chem. 39(14):2659–2663. [DOI] [PubMed] [Google Scholar]